
Abstract
Natural-abundance isotopic substitution in isotopically prochiral groups of otherwise achiral molecules can provide stochastically formed enantiomeric excesses which exceed the sensitivity threshold of sensitive asymmetric autocatalytic (Soai-type) reactions. This kind of induction of chirality should be taken into consideration in in vitro model experiments and offer a new kind of entry into primary prebiotic or early biotic enantioselection in the earliest stages of molecular evolution.
Similar content being viewed by others

Introduction
Chirality due to different isotopes of an element, bound to the same stereocenter, which becomes chiral only through the difference between these isotopes (Scheme 1) was studied primarily with the H/D pair (Arigoni and Eliel 1969; Barth and Djerassi 1981; Pracejus 1966; Galimov 1985; Griffith 1998; Schmidt 2003; Tcherkez and Farquhar 2005). These studies were mostly aimed at the exploration of reaction mechanisms. To the best of our knowledge, the aspect of chirality in the context of biological isotope fractionation phenomena was not investigated (Galimov 1985, 2004, 2006a, b; Galimov and Polyakov 1990; Nakashima et al. 2001; Pizzarello et al. 2004; Schidlowski 2002; Schmidt 2003; Schmidt et al. 2003; Verbit 1970). In the present paper we point out another neglected aspect of this field: the chirality due to isotopes, by the spontaneous (stochastic) formation of enantiomeric excesses from natural-abundance quantities of stable isotopes. Some consequences of this phenomenon will also be discussed, with particular attention to the not yet resolved problem(s) of the origin of biological chirality (Avalos et al. 1998; Avetisov 2002; Avetisov and Goldanskii 1996; Berger et al. 2005; Blackmond and Matar 2008; Bushman et al. 2000; Caglioti et al. 2006a, b; Cline 1996; Compton and Pagni 2002; Fujii and Saito 2004; Keszthelyi 1995; Kondepudi and Asakura 2001; Pályi et al. 1999, 2003, 2004).

Example of isotope chirality: enantiomers of benzaldichloride by different positions of the chlorine 35 and 37 isotopes
Natural Abundance Isotope Chirality
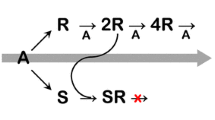
Several molecules contain achiral groups, which constitute a potentially chirogenic center with two atoms of the same isotope of an element bounded to this central atom. Such groups could be regarded as prochiral in the sense, that these groups become configurationally chiral by substitution of one of the two equal isotopic atoms by another isotope of the same element (Scheme 1). Compounds of this type can be prepared by routine preparative techniques from isotopically enriched reagents. A neglected aspect of this chemistry is, however, the spontaneous formation of such centers of configurational chirality in substances which are usually regarded as achiral or in achiral groups of otherwise chiral compounds. This phenomenon does not lead to effects which could be observed by conventional instrumentation or reactivity, and thus can be classified justly as a case of “cryptochirality”, using the denomination ingeniously coined by Mislow (Mislow 2003; Mislow and Bickart 1976/1977). Because of the absence of observable effects, natural abundance isotope chirality is not considered in common chemical thinking. The discovery of asymmetric autocatalysis (Soai 2002; Soai and Sato 2001, 2002; Soai et al. 1995, 2004) and its extremely sensitive variant, which can be used (Caglioti et al. 2006a, b; Kawasaki et al. 2006a; Singleton and Vo 2002, 2003; Soai et al. 2003) for “absolute enantioselective synthesis” (AES) in the most rigorous sense of the word (Avalos et al. 1998; Bredig et al. 1923; Feringa and van Delden 1999; Frank 1953; Mislow 2003; Pályi et al. 2005) fundamentally changed this picture. It has been shown (Micskei et al. 2006a, b; Sato et al. 2003), that the AES by the Soai-autocatalysis is the consequence of the chiral induction of a very low number of molecules (in the order of e.e. 10–10–10–12%). Even more important, such calculations indicate that macroscopically observable chirality could be achieved in the Soai system by the induction of an enantiomeric excess of only one molecule (Caglioti et al. 2005, 2007). The Soai-autocatalysis was documented for the C-alkylation of N-heterocyclic carboxaldehydes by dialkylzinc reagents (Scheme 2) (Soai 2002; Soai and Sato 2001, 2002; Soai et al. 2004). Even if generalizations of this particularly important discovery have not yet been reported, its existence proves that such sensitive autocatalytic reactions are an experimental reality. The sensitivity of this reaction was proved not only by the realization of AES, but also by its responsiveness to very low amounts of chiral additives (Kawasaki et al. 2006b; Lutz et al. 2004, 2005; Sato et al. 2003, 2007). Thus it has been reported that chiral induction can be initiated by low amounts of an isotopically chiral (deuterated) additive (Sato et al. 2000).

Example of Soai-autocatalysis
The observations and considerations described above prompted us to devote attention to quantitative aspects of isotopically chiral compounds, where the chirality is derived from natural abundance of isotopes. We considered mostly organic compounds, where usually the natural abundance isotope chirality derives from 1H-to-2D or 12C-to-13C or 14N-to-15N or 16O-to-17O + 18O substitution, but evidently also other stable natural isotope pairs as e.g. 35Cl/37Cl could lead to similar stereochemical consequences.
The calculations were based on the standard list of natural isotope abundances (Weast 1989/1990) and elementary principles of probability theory and combinatorics (Reimann 1989; Rényi 1970). Calculated e.e. (%) values, which can be expected with at least 50% probability, were obtained for 1 mol quantities by the Pars–Mills formula (Mills 1932) (Scheme 3 and Appendix I.). The results are listed in Table 1 for some common organic compounds, including a few molecules with eminent biological importance. It should be noted that the expected e.e. values increase with decreasing sample size. (Some examples of this effect are also shown in Table 1).

Scheme of calculation. P Probability of the formation of an isotope chiral isomer, p,q natural abundances, x atoms of the more abundant isotope in the molecule in prochiral groups, N Avogadro number (6.022 × 1023), n number of isotope chiral molecules, e.e. enantiomeric excess expected with ≥50% probability (Mills 1932) [%], e.e. 100|R−S|/(R+S), R,S molar quantities of the R or S enantiomers of a chiral molecule
The e.e. values reported in Table 1 are relatively high, with respect to the sensitivity threshold of the Soai-autocatalysis. This has a practical consequence, that is: in AES experiments it appears to be recommendable to avoid molecules as solvents (or any other kind of additives) which could lead to isotope chirality by natural abundance. To the best of our knowledge natural-abundance isotope-free solvents or other chemical reagents are not commercially available and if such substances could be reliably prepared, the costs would be extremely high.
A general and important consequence of the above, is that this effect could have been one of the influences which ultimately triggered the evolution of biological (or prebiological) enantioselection (Fujii and Saito 2004; Pályi et al. 1999, 2003, 2004).
One should note at this point, that due to the mathematical structure of the Pars–Mills formula, the expected stochastic enantiomeric excesses (e.e.1/2) in isotope substituted species (for example in natural abundance chiral molecules derived from H-to-D substitution) are higher than enantiomeric excesses obtained by stochastic fluctuations in achiral-to-chiral reactions conducted in the same sample size (for the apha-methylene group in CH3CH2R, for example, by a factor of ∼60). On the other hand, the stochastic fluctuations in the latter example yield higher absolute enantiomeric excess quantities. The difference of the absolute quantities decreases considerably by decreasing sample size. (An example is shown in Appendix II.)
A very important biologically active molecule which is included in Table 1 is glycine (Gly), which is generally regarded as the only natural (proteinogenic) alpha-amino acid which is achiral. The possibility of natural abundance isotope chirality in Gly could lead to speculations as to whether the isotope-chiral forms of Gly were also involved in events of the “primary enantioselection”. This possibility is important also in the light of suggestions that Gly is one of the most “archaic” amino acids (Wong and Xue 2002).
The above considerations relate to 1H/2D substitutions in H-containing molecules. Such molecules are important because of the large (relative) mass difference (100%) between hydrogen isotopes, on the other hand the natural abundance of deuterium is fairly low (0.0156 atom %). The stable isotopes of carbon (12C and 13C) show different features. Here the natural (“inorganic”) abundance of the heavy isotope is much higher (1.10 atom %), but the relative mass difference is much lower (8.3%). Relative abundances of the pairs 14N/15N or 16O/17O + 18O are intermediate, while the mass differences are similar to that at carbon: 7.1, 6.3 and 12.5% respectively. Strangely, biological systems show more important isotope effects with carbon than with deuterium. The non-chiral aspects of biological isotope effects have been abundantly documented, especially in the light of identification of archaic carbon-containing materials of suspected biological origin (Galimov 1985, 2004, 2006a, b; Galimov and Polyakov 1990; Schidlowski 2002; Schmidt 2003).
Some biologically important molecules however can generate new centers of chirality through 12C-to-13C substitution. A simple example of this is iso-propanol (or its esters) (Table 1), where 13C substitution in one of the methyl groups generates a new center of chirality on 2-C. An example of exchange of either carbon or oxygen isotopes, leading to a chiral derivative is represented by the simplest “achiral” carbohydrate glycerol. Chirality by N isotope exchange was exemplified by derivatives of methylmalonic acid in Table 1.
Some achiral biologically important molecules, as citric acid (Table 1) can show both effects, either exchanging one of its 1H atoms on one of the methylene groups for deuterium, or exchanging one 12C either in one methylene group or in one of its terminal carboxyl groups for 13C.
At almost all proteinogenic amino acids (except Gly, Ala and Thr) the isotope exchange at certain points of the molecule results in diastereomers. There are various possibilities for this: a deuterium atom in methylene groups yield one (Leu, Ile, Ser, Cys, Phe, Tyr, Trp, His, Asp, Asn), two (Met, Glu, Gln), three (Pro, Arg) or even four (Lys) new centers of chirality, introduction of 13C into terminal methyl groups of branched chain structures yield diastereomers at two amino acids, Val and Leu. Leu is the only one where both effects can develop. The importance of the formation of such diastereomeric compounds derives from the fact that in this manner mono-chiral molecules can “escape” the thermodynamic limitation of equal energies of enantiomeric pairs. Whether these effects can also have an enantioselective influence on oligo- and polymerization (Green et al. 1999) and other reactions of these amino acids is not yet documented experimentally, but it appears to be certainly an attractive research goal.
The formation of diastereomers through natural abundance isotope substitution is obviously possible for a very large number of biologically active chiral molecules. Here we would like to point out only one highly interesting example: 2-desoxy-D-ribose is one of the fundamental components of the genetic information carrier DNA. If the 2-methylene group in this molecule gets mono-deuterated the 2-C atom becomes chiral. If the structure of D-ribose was already (enantioselectively) synthesized, the “rest” of the molecule (three chiral C atoms) may certainly accomplish a certain degree of chiral induction. Neglecting eventual inductive effects, by pure stochastic formalism e.e.1/2∼7.1 × 10–2% can be calculated for one molecule of human DNA (3 × 109 base) where 9.36 × 105 nucleotides are expected to bear mono-D substitution in their 2-methylene groups. Whether the concentration of these “new” stereocenters have any effect on the genetic behaviour of DNA is still to be cleared up. It should be added, that if one of the two non-esterified oxygens is substituted by one of the heavier isotopes of oxygen, the phosphorous atom also becomes chiral, which yields similar quantities of P-chirality in DNA (or RNA) with consequences not yet fully explored (Potter et al. 1983).
There have been very few studies on isotope-generated chirality in biochemistry (Battersby 1985; Stevens et al. 2005). The above results indicate that such studies would be highly interesting both from theoretical and practical viewpoints.
It should be noted that the calculations reported above were made considering average (“inorganic”) isotope abundance values (Weast 1989/1990). These data could be refined by taking into consideration isotope fractionation effects due to thermodynamic (Bottinga 1969; Galimov 1985, 2006a, b; Urey 1947) or kinetic (Schmidt 2003) factors. Biological fractionation appears to be controlled by thermodynamics (Galimov 1985, 2006a, b). Here the equations deduced by Urey (1947) and Galimov (1971, 1985, 2006a, b) can be used (Appendix III). These calculations have shown that the symmetry combinations do not influence substantially the fractionation of isotopes (therefore also not the chirality), but do affect the strengths of the corresponding bonds (considered in the β-factor values in these equations). These differences are highly important for biochemical (e.g. metabolic) studies but do not influence the orders of magnitude of the e.e.1/2 values shown in Table 1.
Conclusions
The spontaneous emergence of enantiomeric chiral excesses as a result of the effects of natural abundance isotope chirality was analyzed quantitatively for stable hydrogen and carbon isotopes in simple organic and biologically active molecules. The expected excesses are in the range of sensitivity of the only experimentally documented example of asymmetric autocatalysis and its absolute enantioselective synthesis variant. Several biologically important molecules, for example 17 from the 20 proteinogenic natural amino acids or 2-desoxy-D-ribose, yield diastereomers by the same mechanism. The results prompt us to speculate that this kind of spontaneous chirality could have had a role in the initial phase of the development of chirality in prebiotic or early biological evolution. The possibility of testing experimentally these suggestions should also be considered.
References
Arigoni D, Eliel E (1969) Chirality due to the presence of hydrogen isotopes in noncyclic positions. Top Stereochem 4:127–243
Avalos M, Babiano R, Cintas P, Jimenez JL, Palacios JC, Barron LD (1998) Absolute asymmetric synthesis under physical fields: facts and fictions. Chem Rev 98:2391–2404
Avetisov VA (2002) Emergence of biomolecular homochirality: complexity, hierarchicity and dynamics. In: Palyi G, Zucchi C, Caglioti L (eds) Fundamentals of life. Elsevier, Paris, pp 361–367
Avetisov VA, Goldanskii VI (1996) Mirror symmetry breaking at the molecular level. Proc Natl Acad Sci USA 93:11435–11442
Barth G, Djerassi C (1981) Circular dichroism of molecules with isotopically engendered chirality. Tetrahedron 37:4123–4142
Battersby AR (1985) Enzymic synthesis of isotopically labeled substances. Ciba Found Symp 111:22–30 Enzymes Org Synth
Berger L, Laubender G, Quack M, Sieben A, Stohner J, Willeke M (2005) Isotopic chirality and molecular parity violation. Angew Chem Int Ed Engl 44:3623–3626
Blackmond DG, Matar OK (2008) Re-examination of reversibility in reaction models for the spontaneous emergence of homochirality. J Phys Chem B 112:5098–5104
Bottinga Y (1969) Calculated fractionation factors for carbon and hydrogen isotope exchange in the system calcite–carbon dioxide–graphite–methane–hydrogen–water vapor. Geochim Cosmochim Acta 33:49–64
Bredig G, Mangold P, Williams TG (1923) Absolute asymmetric synthesis. Z Angew Chem 36:456–458
Bushman H, Thede R, Setter D (2000) New developments in the origins of the homochirality of biologically relevant molecules. Angew Chem Int Ed 39:4033–4036
Caglioti L, Zucchi C, Palyi G (2005) Single-molecule chirality. Chem Today 23(5):38–43
Caglioti L, Hajdu C, Holczknecht O, Zekany L, Zucchi C, Micskei K, Palyi G (2006a) The concept of racemates and the Soai reaction. Viva Orig 34:62–80
Caglioti L, Holczknecht O, Fujii N, Zucchi C, Palyi G (2006b) Astrobiology and biological chirality. Orig Life Evol Biosph 36:459–466
Caglioti L, Micskei K, Palyi G (2007) Chirality of the very first molecule in absolute enantioselective synthesis. Viva Orig 35:82–84
Cline DB (ed) (1996) Physical origin of the homochirality of life. AIP, Woodburg (NY, USA)
Compton RN, Pagni RM (2002) Chirality of biomolecules. Adv At Mol Opt Phys 48:219–261
Feringa BL, van Delden RA (1999) Absolute asymmetric synthesis: the origin, control and amplification of chirality. Angew Chem Int Ed 38:3418–3438
Frank FC (1953) On spontaneous asymmetric synthesis. Biochim Biophys Acta 11:459–463
Fujii N, Saito T (2004) Homochirality and life. Chem Rec 4:267–278
Galimov EM (1971) On the relationship of the fractionation coefficient of isotopes to the equilibrium constants of the isotope exchange reactions of carbon in hydrocarbon systems. Zh Fiz Khim 45:118701191
Galimov EM (1985) The biological fractionation of isotopes. Academic, New York
Galimov EM (2004) Phenomenon of life: between equilibrium and non-linearity. Orig Life Evol Biosph 34:599–613
Galimov EM (2006a) Phenomenon of life: between equilibrium and non-linearity. Origin and principles of evolution. Geochem Int 44(Suppl. 1):S1–S95
Galimov EM (2006b) Isotope organic geochemistry. Org Geochem 37:1200–1262
Galimov EM, Polyakov VB (1990) On thermodynamically ordered distribution of isotopes of carbon in biogenic geochemical objects. Geokhimiya 9:1232–1240
Green MM, Park J-W, Sato T, Takahiro T, Teramoto A, Lifson S, Selinger RLB, Selinger JV (1999) The macromolecular route to chiral amplification. Angew Chem Int Ed 38:3139–3154
Griffith H (ed) (1998) Stable isotopes. BIOS Sci Publisher Ltd, Oxford (GB)
Kawasaki T, Suzuki K, Shimizu M, Ishikawa K, Soai K (2006a) Spontaneous absolute asymmetric synthesis in the presence of achiral silica gel in conjunction with asymmetric autocatalysis. Chirality 18:479–482
Kawasaki T, Tanaka H, Tsutsumi T, Kasahara I, Sato I, Soai K (2006b) Chiral discrimination of cryptochiral saturated quaternary and tertiary hydrocarbons by asymmetric autocatalysis. J Am Chem Soc 128:6032–6033
Keszthelyi L (1995) Origin of the homochirality of biomolecules. Quart Rev Biophys 28:473–507
Kondepudi DK, Asakura K (2001) Chiral autocatalysis, spontaneous symmetry breaking and stochastic behaviour. Acc Chem Res 34:946–954
Lutz F, Sato I, Soai K (2004) The asymmetric power of chiral ligands determined by competitive asymmetric autocatalysis. Org Lett 6:1613–1616
Lutz F, Igarashi T, Kawasaki T, Soai K (2005) Small amounts of achiral beta-amino alcohols reverse the enantioselectivity of chiral catalysts in cooperative asymmetric autocatalysis. J Am Chem Soc 127:12206–12207
Micskei K, Maioli M, Zucchi C, Caglioti L, Palyi G (2006a) Generalization possibilities of autocatalytic absolute enantioselective synthesis. Tetrahedron Asymmetry 17:2960–2962
Micskei K, Pota G, Caglioti L, Palyi G (2006b) Empirical description of chiral autocatalysis. J Phys Chem A 110:5982–5984
Mills WH (1932) Some aspects of stereochemistry. Chem Ind (Lond) 750–759
Mislow K (2003) Absolute asymmetric synthesis. A commentary. Coll Czech Chem Commun 68:849–864
Mislow K, Bickart P (1976/1977) An epistemological note on chirality. Isr J Chem 15:1–6
Nakashima S, Muruyama S, Brack A, Widley BF (eds) (2001) Geochemistry and the origin of life. Universal Academy Press, Tokyo
Pályi G, Zucchi C, Caglioti L (eds) (1999) Advances in biochirality. Elsevier, Amsterdam
Pályi G, Micskei K, Bencze L, Zucchi C (2003) Biological chirality. Magy Kem Lapja 58:218–223
Pályi G, Zucchi C, Caglioti L (eds) (2004) Progress in biological chirality. Elsevier, Oxford (GB)
Pályi G, Micskei K, Zekany L, Zucchi C, Caglioti L (2005) Racemates and the Soai reaction. Magy Kem Lapja 60:17–24
Pizzarello S, Huang Y, Fuller M (2004) The carbon isotopic distribution of Murchinson amino acids. Geochim Cosmochim Acta 68:4963–4969
Potter BVL, Connolly BA, Eckstein F (1983) Synthesis and configurational analysis of a nucleoside phosphate isotopically chiral at phosphorus. Stereochemical course of Penicillium citrinum nuclease P1 reaction. Biochemistry 22:1369–1377
Pracejus H (1966) Steric isotope effect as cause of a catalytic asymmetric synthesis. Tetrahedron Lett 32:3809–3813
Reimann J (1989) Mathematical statistics with application in flood hydrology. Akademiai Kiadó, Budapest
Rényi A (1970) Probability theory. Akadémiai Kiadó, Budapest
Sato I, Omiya D, Saito T, Soai K (2000) Highly enantioselective synthesis induced by chiral primary alcohols due to deuterium substitution. J Am Chem Soc 122:11739–11740
Sato I, Urabe H, Ishiguro S, Shibata T, Soai K (2003) Amplification of chirality from extremely low level to greater than 99.5% ee. Angew Chem Int Ed 42:315–317
Sato I, Ohgo Y, Igarashi H, Nishiyama D, Kawasaki T, Soai K (2007) Determination of absolute configurations of amino acids by asymmetric autocatalysis of 2-alkynylpyrimidyl alkanol as a chiral sensor. J Organomet Chem 692:1783–1787
Schidlowski M (2002) Sedimentary carbon isotope archives as recorders of early life: implications for extraterrestrial scenarios. In: Palyi G, Zucchi C, Caglioti L (eds) Fundamentals of life. Elsevier, Paris, pp 307–329
Schmidt H-L (2003) Fundamentals and systematics of the non-statistical distribution of isotopes in natural compounds. Naturwissenschaften 90:537–552
Schmidt H-L, Werner RA, Eisenreich W (2003) Systematics of 2H patterns in natural organic compounds and its importance to elucidation of biosynthetic pathways. Phytochem Rev 2:61–85
Singleton DA, Vo LK (2002) Commentary on enantioselective synthesis without discrete optically active additives. J Am Chem Soc 124:10010–10011
Singleton DA, Vo LK (2003) A few molecules can control enantiomeric outcome. Evidence supporting absolute enantioselective synthesis using the Soai asymmetric autocatalysis. Org Lett 5:4337–4339
Soai K (2002) Asymmetric autocatalysis and the origin of chiral homogeneity of biologically relevant molecules. In: Palyi G, Zucchi C, Caglioti L (eds) Fundamentals of life. Elsevier, Paris, pp 427–435
Soai K, Sato I (2001) Application of asymmetric autocatalysis to the determination of absolute configurations of amino acids with low enantiomeric excesses. Enantiomer 6:189–192
Soai K, Sato I (2002) Asymmetric autocatalysis and the homochirality of biomolecules. Viva Orig 30:186–198
Soai K, Shibata T, Morioka H, Choji K (1995) Asymmetric autocatalysis and the amplification of enantiomeric excess of a chiral molecule. Nature 378:767–768
Soai K, Sato I, Shibata T, Komiya S, Hayashi M, Matsueda Y, Imamura H, Hayase T, Morioka H, Tabira H, Yamamoto J, Kowata Y (2003) Asymmetric synthesis of pyrimidyl alkanol without adding chiral substances by the addition of diisopropylzinc to pyrimidine-5-carboxaldehyde in conjunction with asymmetric synthesis. Tetrahedron Asymmetry 14:185–188
Soai K, Shibata T, Sato I (2004) Discovery and development of chiral autocatalysis. Bull Chem Soc Japan 77:1063–1073
Stevens SM Jr, Prokai-Tatrai K, Prokai L (2005) Screening of combinatorial libraries for substrate preference by mass spectrometry. Anal Chem 77:698–701
Tcherkez G, Farquhar GD (2005) Carbon isotope effect predictions for enzymes involved in the primary carbon metabolism of plant leaves. Funct Plant Biol 32:277–291
Urey HG (1947) The thermodynamic properties of isotopic substances. J Chem Soc 562–581
Verbit L (1970) Optically active deuterium compounds. Prog Phys Org Chem 7:51–127
Weast RC (ed) (1989/1990) Handbook of Chemistry and Physics. CRC, Boca Raton (FL, USA), table of isotopes, pp. B227–B448
Wong JT-F, Xue H (2002) Self-perfecting evolution of heteropolymer building blocks and sequences as the basis of life. In: Pályi G, Zucchi C, Caglioti L (eds) Fundamentals of life. Elsevier, Paris, pp 473–494
Acknowledgment
Support of the present research is acknowledged to the Italian Ministry of University and Research (Contract No. RBPR05NWWC). The authors acknowledge with thanks the advice of an anonymous Referee and discussions with Profs. I. Shiina and K. Soai (Tokyo).
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
-
I.
The Pars–Mills equation (Mills 1932; Mislow 2003; Pályi et al. 2005) was published only in its final form. Here we describe its mathematical deduction.
The problem is analogous to that of the well-known ‘coin tossing’ experiment.
Let us define u as the number of outcomes by one enantiomer exceeding the 1:1 ratio and let us calculate the probability (P) of the outcomes u ≥ 0, that is:
where R (or analogously S) is the total number of R (or S) outcomes (number of molecules) in the experiment and n = R+S.
If \(R >\frac{{1 + u}}{2}n\) then \(S <\frac{{1 - u}}{2}n\) and \(\frac{{R - S}}{{R + S}} \geqslant u\) consequently
The distribution of R (or S) is a binomial distribution with n, p = 0.5 parameters. This could be approximated, if n is ‘sufficiently large’ with a normal distribution, characterized by m = np and \(\sigma = \frac{{\sqrt n }}{2}\) parameters, and one obtains
where Φ is the distribution function of the standard normal distribution. Consequently:
which is equivalent to:
The values of Φ can be obtained from tables (e.g. Reimann 1989 or Microsoft Excel). Thus for the case where the probability (P) is expected to be ≥50% confidence one obtains Φ(0.675) = 3/4 and thus \(P\left( {\left| {\frac{{R - S}}{{R + S}}} \right| >\frac{{0.675}}{{\sqrt n }}} \right) = \frac{1}{2}\) which is equivalent to the Pars–Mills formula \(e.e._{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-\nulldelimiterspace} 2}} = \frac{{0.675}}{{\sqrt n }}\) or \( = \frac{{67.5}}{{\sqrt n }}\left( \% \right)\). Narrowing the confidence range to 5%, one obtains Φ(1.96) = 0.975 thus: \(P\left( {\left| {\frac{{R - S}}{{R + S}}} \right| >\frac{{1.96}}{{\sqrt n }}} \right) = 0.05\) which yields an analogous formula:
This latter operation shows qualitatively that if the confidence range is lowered from 50% to 5% one obtains approximately three-fold higher e.e. values, or in other words, to obtain three-fold higher e.e. than e.e.1/2 the confidence decreases by a factor of 10.
-
II.
Comparison of the expected relative and absolute enantiomeric excesses yielded in a stoichiometric achiral-to-chiral chemical reaction (neglecting the effect of isotopic substitution) and in a natural-abundance isotopically chiral molecule (Table 2).
-
III.
Urey–Galimov equations for calculation of the thermodynamic isotope fractionation (Urey 1947; Galimov 2006b). The equilibrium constant (K) of an isotope exchange reaction between compounds A and B is:
$$K = \frac{{\left( {{{ * Q} \mathord{\left/ {\vphantom {{ * Q} Q}} \right. \kern-\nulldelimiterspace} Q}} \right)_{\text{A}} }}{{\left( {{{ * Q} \mathord{\left/ {\vphantom {{ * Q} Q}} \right. \kern-\nulldelimiterspace} Q}} \right)_{\text{B}} }} = \frac{{\left( {{s \mathord{\left/ {\vphantom {s { * s}}} \right. \kern-\nulldelimiterspace} { * s}}} \right)_{\text{A}} \beta _{\text{A}} }}{{\left( {{s \mathord{\left/ {\vphantom {s { * s}}} \right. \kern-\nulldelimiterspace} { * s}}} \right)_{\text{B}} \beta _{\text{B}} }}$$
Where
- Q :
-
partition function
- *:
-
denotes the less abundant isotope
- s :
-
symmetry number
- β :
-
so called β-factor.
The β-factor can be related to the bond stretching vibrational frequencies (ν i) of the isotopic species:
where
- h :
-
Plank constant
- k :
-
Boltzmann constant
- T :
-
temperature (K)
Rights and permissions
About this article
Cite this article
Barabás, B., Caglioti, L., Micskei, K. et al. Isotope Chirality and Asymmetric Autocatalysis: A Possible Entry to Biological Chirality. Orig Life Evol Biosph 38, 317–327 (2008). https://doi.org/10.1007/s11084-008-9138-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11084-008-9138-1