
Abstract
The aim of this study was to determine the clinicopathological significance of programmed cell death ligand 1 (PD-L1) expression in glioblastoma (GBM). In a retrospective cohort of 115 consecutive patients with GBM, PD-L1 expression was determined using immunohistochemistry (IHC). Membranous and fibrillary PD-L1 staining of any intensity in > 5% neoplastic cells and tumour infiltrating immune cells (TIIs) was considered positive staining. In addition, isocitrate dehydrogenase-1 (IDH-1) (R132H) expression and cluster of differentiation 3 (CD3)-positive T-cell infiltration were investigated using IHC. O(6)-methylguanine-DNA methyltransferase (MGMT) promoter methylation assay and fluorescence in situ hybridization (FISH) for the assessment of 1p/19q deletion were performed. Expression of PD-L1 in tumour cells and TIIs was found in 37 (32.2%) and 6 (5.2%) patients, respectively. Kaplan–Meier analysis indicated that PD-L1 expression in tumour cells was significantly associated with poor overall survival (OS) (P = 0.017), though multivariate Cox analysis did not confirm this association (hazard ratio 1.204; P = 0.615). PD-L1 expression in TIIs did not correlate with the patient prognosis (P = 0.545). In addition, MGMT methylation and IDH-1 (R132H) expression were associated with a better prognosis (P < 0.001 and P = 0.024, respectively). The expression of PD-L1 was associated with CD3-positive T-cell infiltration (P < 0.001), and IDH-1 wild type status (P = 0.008). A deeper insight into PD-L1 expression could help to ensure the success of future immunotherapy in GBM. Our study suggested that PD-L1 target therapy might be beneficial for PD-L1-expressing GBM patients with a poor prognosis.
Similar content being viewed by others

Background
The remarkable therapeutic efficacy of immune checkpoint inhibitors has been proven in various cancers [1, 2]. In particular, targeting of programmed cell death 1 (PD-1) and its ligand, programmed cell death ligand 1 (PD-L1), has delivered excellent anti-tumour efficacy in patients with metastatic melanoma, non-small-cell lung cancer (NSCLC), urothelial carcinoma, and microsatellite instability-high (MSI-H) colorectal cancer [3,4,5,6]. Anti-PD-1/PD-L1 antibodies (nivolumab, pembrolizumab, and atezolizumab) have been approved by the US Food and Drug Administration (FDA) for the treatment of melanoma, NSCLC, and urothelial carcinoma. Physiologically, PD-L1 is expressed to protect tissues from excessive inflammation [7], but malignant tumours express PD-L1 to escape from host immune surveillance [8]. The PD-1/PD-L1 axis is believed to play a key role in the immune evasion mechanism of tumours [8]. Therefore, an increased understanding of the role of PD-L1 expression will aid in the development of immunotherapies.
Glioblastoma (GBM) is the most malignant and most common primary brain tumour, and patients usually survive for < 15 months after diagnosis [9]. Conventional strategies for the treatment of GBM include a combination of surgery, radiation therapy, and chemotherapy, although their efficacies have remained limited. Therefore, novel and more effective treatment strategies for this tumour are desirable. Recent studies have increased the expectations regarding immunotherapy for GBM, which has previously proven effective for several other malignancies. Current immunotherapies for GBM include the use of cancer vaccines, checkpoint inhibitors, oncolytic viruses, adoptive cell therapy, and chimeric T-cell receptors [10, 11]. The noticeable success of PD-1/PD-L1 inhibitors for cancer treatment has led to their application in neuro-oncology. Various clinical trials of PD-1/PD-L1 inhibitors for GBM are ongoing; Durvalumab (NCT02336165) and Pembrolizumab (NCT02337491) are currently in phase II trials involving patients with primary GBM, while nivolumab is in 2 phase III trials involving newly-diagnosed GBM patients presenting with a methylated MGMT Promoter Status (NCT02667587), as well as with an un-methylated MGMT Promoter Status (NCT02617589). Unfortunately, nivolumab treatment has not achieved a better overall survival (OS) outcome than bevacizumab treatment in patients with recurrent GBM (NCT02017717) [12].
Increased PD-L1 expression is a highly specific and effective indicator for the use of PD-1/PD-L1 inhibitors in patients with NSCLC and urothelial carcinoma [4, 13]. PD-1/PD-L1 blockade was associated with an improved response in patients with advanced urothelial carcinoma exhibiting PD-L1 expression in tumour infiltrating immune cells (TIIs) [13]. Therefore, evaluation of PD-L1 expression in TIIs, as well as in tumour cells in patients with GBM, would be valuable.
Studies have indicated that PD-L1-expression is associated with a poor prognosis in cancer [1, 14]. In GBM, however, the frequency and prognostic value of PD-L1 expression remain to be determined. PD-L1 expression was observed in 88.0% of newly-diagnosed and 72.2% of relapsed patients with GBM, although it was not related to patient outcomes [15]. In contrast, Nduom et al. reported that increased expression of both PD-1 and PD-L1 based on The Cancer Genome Atlas (TCGA) datasets predicts a poor prognosis [16]. Another recent study reported PD-L1 expression in 49.2, 53.7, and 68.8% patients with grade II, III, and IV gliomas, respectively, and concluded that immunohistochemical PD-L1 expression was significantly associated with a poor OS in patients with GBM (grade IV) only [17]. Since the literature shows no consensus, further studies on PD-L1 expression in GBM are necessary.
Considering the above perspectives, we aimed to verify the prognostic significance of PD-L1 expression, and to determine the correlation between PD-L1 expression and other molecular alterations in patients with GBM.
Materials and methods
Patients and tumour samples
This study included 115 patients with newly-diagnosed GBM who underwent radical surgery or stereotactic biopsy at the Seoul National University Bundang Hospital between May 2003 and February 2013. In total, 52 patients underwent stereotactic biopsy or partial resection and 63 underwent gross total resection. Two neuropathologists (K.S.L. and G.C.) reviewed the histopathology and confirmed GBM diagnoses for all lesions. The lesions were re-classified according to the 2016 World Health Organization (WHO) classification of central nervous system tumours [18]. Oligodendroglial tumours were excluded using 1p19q fluorescence in situ hybridization (FISH) testing. Even though IDH gene mutations were not evaluated using sequencing, the IDH-1 R132H mutation status was assessed using immunohistochemistry (IHC). Clinicopathological information was obtained from hospital medical reports.
The Institutional Review Board of the Seoul National University Bundang Hospital approved the use of medical record data and tissue samples for this study (Reference: B-1612/374- 304).
Tissue array method
Two tissue microarrays (TMA) were constructed with formalin-fixed paraffin-embedded (FFPE) donor GBM tissues. For each patient, two cores (each of 3 mm diameter) from representative regions of the GBM tissues were punched out (ensuring > 30% cancer cells in each sample), and transferred to a TMA recipient block.
Immunohistochemistry
Immunostaining using PD-L1 (E1L3N, 1:50, Cell Signaling Technology, Danvers, MA, USA), cluster of differentiation 3 (CD3) (1:100, Dako, Glostrup, Denmark), and isocitrate dehydrogenase 1-R132H (IDH-1; 1:100, DIANOVA, Hamburg, Germany) antibodies was performed with 2 TMA slides per sample, using the Ventana Benchmark XT autostainer (Ventana, Tucson, AZ, USA) with the ultraView Universal DAB kit (Ventana) as per the manufacturer’s recommendations.
Normal glial tissues served as internal negative controls. PD-L1 expression was considered positive when > 5% neoplastic cells showed membranous or fibrillary staining of any intensity (Fig. 1a, b) [15]. The percentage of TIIs with PD-L1 staining was counted; positivity was recorded when immune cells showed > 5% staining [13]. The CD3+ Tumour-infiltrating lymphocytes (TILs) were analysed by counting the number of positive nuclei in the core region (×40). We arbitrarily defined the cut-off value for CD3+ TILs as > 30 positive cells/microscopic view (×40). Positive IDH-1 status was attributed to samples showing cytoplasmic staining in neoplastic cells.

Representative figures showing immunohistochemical staining for programmed cell death ligand 1 (PD-L1) (a, b and c), cluster of differentiation 3 (CD3) (d), and isocitrate dehydrogenase 1 (IDH-1) (e) antibodies and the findings of fluorescence in-situ hybridization (FISH) for 1p36 (f) in glioblastoma samples. a Membranous PD-L1 expression in tumour cells (×400). b Fibrillary PD-L1 expression in tumour cells (× 400). c PD-L1 expression in immune cells (× 400). d CD3 expression in tumour-infiltrating lymphocytes (× 400). e IDH-1 (R132H) expression (× 400). f Absence of chromosome 1p36 deletion (×1000)
O(6)-methylguanine-DNA methyltransferase (MGMT) promoter methylation assay
Deoxyribose-nucleic acid (DNA) was extracted from GBM tissues and treated with bisulfite using the EZ DNA methylation kit (Zymo Research, Freiburg, Germany), according to the manufacturer’s protocol. Methylation-specific polymerase chain reaction (PCR) was performed as previously described [19]. The predicted fragment size was 80 bp for methylated samples and 92 bp for non-methylated samples. The PCR products were visualized on 4% agarose gels.
Fluorescence in situ hybridization for the assessment of 1p/19q deletion
The Locus-Specific Identifier (LSI) 1p36/LSI 1q25 and LSI 19q13/LSI 19p13 DNA probes (Vysis, IL, USA) were used for 1p/19q FISH, which was performed according to the manufacturer’s instructions. Briefly, 4-µm thick paraffin slides were deparaffinized, dehydrated, and incubated in solution at 80 °C for 30 min. The slides were immersed in protease solution at 37 °C for 20 min, and then incubated in 10% buffered formalin. The slides were hybridized with Hybrite (ThermoBrite™, Vysis). After drying in darkness, 10 µl of counterstain was applied to the target region on the slide, and a cover slip was placed. The FISH results were interpreted according to the guidelines defined by the European Society for Paediatric Oncology (SIOP Europe) Neuroblastoma Pathology and Biology and Bone Marrow Group [20].
Statistical analyses
Chi square tests or Fisher’s exact tests were used for the comparison of categorical variables as appropriate. Kaplan–Meier analysis with a log-rank test was used to evaluate survival curves. Univariate and multivariate regression analyses were performed using Cox proportional hazards models to determine the hazard ratios (HRs) and 95% confidence intervals (CIs) for each factor. Separate analyses were conducted to evaluate OS. A P value of < 0.05 was considered statistically significant. All statistical analyses were performed using the IBM SPSS statistics version 21 software (IBM, Armonk, NY, USA).
Results
Clinicopathological characteristics of GBM patients
Table 1 demonstrates the clinicopathological characteristics of the patients included in this study. The average age was 59.3 (20–84) years. More than half of the GBM lesions originated from the temporoparietal lobe (56.5%). At the time of diagnosis, 11 GBM lesions had already multiple involved sites. One patient exhibited primary cerebellar GBM (rare in adults, accounting for approximately 1% of all GBMs) [21]. Secondary GBM, which originates from a low-grade glioma, was observed in 11 (9.6%) patients. The incidence rate of secondary GBM was similar to that in previous studies [22, 23]. In total, 63 (54.8%) patients underwent gross total resection. Post-operative therapy including temozolomide (TMZ) and radiotherapy was performed in 93 (80.9%) and 105 (91.3%) patients, respectively. All patients who received TMZ also received radiotherapy. Fifty-three (46.1%) patients received gross total resection plus TMZ and radiotherapy. Ten of the patients who underwent gross total resection had no further treatment. Among them, eight patients were in poor condition and were too old to receive chemotherapy or radiotherapy, and the other two patients refused for economic reasons. Recurrence after gross total resection was observed in 44 patients. Twenty-four of the relapsed patients received Irinotecan/Avastin therapy.
Frequency of PD-L1 expression and other molecular alterations in GBM patients
We immunohistochemically investigated PD-L1 and IDH-1 expression and CD3+ TIL infiltration in GBM tissues. All 115 GBM tissues were stained for PD-L1 and IDH-1. PD-L1 expression was primarily represented by a membranous or fibrillary staining pattern in tumour cells (Fig. 1a, b), and positive staining was observed in 37 (32.2%) samples. CD3+ TILs were present in 21 (18.3%) samples (Fig. 1d). Immune cell PD-L1 expression was observed in 6 (5.2%) samples (Fig. 1c). Expression of IDH-1, a specific marker for the detection of IDH-1 (R132H) mutations [24], was observed in 13 (11.3%) samples (Fig. 1e), in agreement with previously published data [23]. All secondary GBM samples expressed IDH-1. MGMT methylation was observed in 23 (31.9%) of 72 GBM samples. Co-deletion of 1p/19q was not observed in any of the GBM samples.
Correlation between PD-L1 expression and other molecular alterations in GBM patients
Table 2 presents the correlations between PD-L1 expression and other molecular alterations in GBM. Tumour cell PD-L1 expression correlated negatively with IDH-1 (R132H) alterations (P = 0.008), and positively with CD3+ T-cell infiltration (P < 0001), although this is not a molecular alteration per se.
Prognostic impact of PD-L1 expression and other molecular alterations in GBM patients
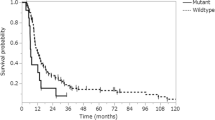
Survival analysis was successfully conducted for all 115 patients (Fig. 2). The mean follow-up duration was 21 months (range 1–134 months), and 76 (66.1%) patients died during this period. Kaplan–Meier analysis indicated that PD-L1 expression in tumour cells correlated significantly with poor survival in all 115 patients (P = 0.017). Immune cell PD-L1 expression did not correlate with patient prognosis (P = 0.545). Additionally, Multivariate Cox proportional hazards analysis (Table 3) revealed that PD-L1 expression in GBM did not predict OS (HR 1.204; P = 0.615). Patients who received gross total resection plus TMZ and radiotherapy had better survival than other treatments (P = 0.003).

Kaplan–Meier survival curves illustrating the prognostic effects of programmed cell death ligand 1 (PD-L1) expression and other molecular alterations in glioblastoma (GBM) a PD-L1 expression in tumour cells. b PD-L1 expression by tumour infiltrating immune cells (TII). c Gross total resection plus TMZ and radiotherapy. d O(6)-methylguanine-DNA methyltransferase (MGMT) promoter methylation. e Isocitrate dehydrogenase 1 (IDH-1) (R132H) expression. f CD3-positive T-cell infiltration
While CD3+ TIL infiltration was not associated with survival (P = 0.557), IDH-1 (R132H) expression was significantly associated with improved survival (P = 0.024), in agreement with a previous study [23]. Methylation of the MGMT promoter is known to have prognostic and predictive value in GBM [25]. In the present study, there was a significant correlation between MGMT promoter methylation and a better prognosis (Fig. 2d; Table 3).
Discussion
Though the brain was earlier believed to be an immune privileged organ [26] and the blood brain barrier (BBB) was thought to restrict brain parenchymal access to immune cells, the brain is now considered an immunocompetent organ containing a plethora of immune cell types, including microglia, dendritic cells (DCs), macrophages, and T-cells [11, 27]. Microglial cells have phagocytic and antigen presenting functions, and can recruit other immune cells [28]. Intratumoural macrophages and DCs mainly act as antigen presenting cells (APCs) [29]. The aggressive tumour growth and invasiveness of GBM generally lead to breakdown of the BBB [30], thereby facilitating migration and recruitment of various immune cells into the tumour [31]. A previous study found that microglia and macrophages occupy > 30% of GBM tissue [32], and GBM utilizes several mechanisms of immune evasion to survive. Communication between the GBM cell and microglia can decrease the expression of major histocompatibility complex (MHC) I and II [33]. Furthermore, GBM can produce immune inhibitory molecules such as transforming growth factor- (TGF- ), prostaglandin E2, and interleukin 10 (IL-10), while also downregulating inflammatory cytokines such as IL-12, IL-18, and interferon-γ (IFN-γ) [34]. Similar to the expression of other immune regulating molecules, that of PD-L1 is increased in GBM. Wilmotte et al. demonstrated that human glioma cell lines express PD-L1, which downregulates IFN-γ expression by T-lymphocytes [35]. Jacobs et al. indicated that the PD-1/PD-L1 axis controls regulatory T-lymphocytes (Tregs), which promote the immunosuppressive property of GBM [36]. The results of the present study are in agreement with those of these studies, and show that PD-L1 expression correlates significantly with CD3+ T-cell infiltration in GBM (Table 2).
The PD-L1 positivity rate in GBM is highly variable and controversial. Berghoff et al. used a non-commercial anti-PD-L1 antibody 5H1 for staining full slide sections [15], and showed membranous PD-L1 expression in 37.6% of newly-diagnosed and 16.7% of recurrent GBMs. Additionally, they observed diffuse/fibrillary PD-L1 expression in 84.4% of newly-diagnosed and 72.2% of recurrent GBMs. Nduom et al. used the clone EPR1161(2) in TMA [16], and showed ≥ 1, ≥ 5, ≥ 25, and ≥ 50% PD-L1 expression in 60.6, 38.3, 17.0 and 5.3% GBM patients, respectively. Zeng et al. performed TMA analysis using a rabbit polyclonal anti-PD-L1 antibody (Envision, Dako, Denmark) [17], and showed cytoplasmic or membranous PD-L1 expression in ≥ 5% of tumour cells in 68.8% of GBMs. As is apparent, there are methodological discrepancies between studies. In addition, differences in the size of the tissue samples (assessment of a full slide section or TMA analysis), antibody manufacturers, evaluation of staining patterns, and cut-off criteria, may have introduced variability in the results. Recently, regional heterogeneity in PD-L1 expression was reported in various cancers [37,38,39]. Thus, evaluation of various regions of the tumour is recommended to enhance the validity of findings. However, TMAs can evaluate only a small portion (our study: 3 mm and Berghoff et al.: 1 mm) of the tumour samples, which may result in sampling bias.
Immunohistochemical assays for PD-L1 have received FDA approval in conjunction with counterpart anti-PD-1/PD-L1 immuno-therapeutics. For example, pembrolizumab is the recommended drug when > 50% tumour cells exhibit positive PD-L1 staining (Agilent, PD-L1 IHC 22C3 pharmDx) in lung cancer patients [40]. Interestingly, the Blueprint PD-L1 IHC Assay Comparison Project revealed differences in staining results between various PD-L1 IHC assays in lung cancer [41]. A standardized PD-L1 antibody for use in GBM, and well accepted cut-off criteria are as yet lacking, since clinical trials with PD-L1 antibodies are ongoing. Although the E1L3N antibody test is a laboratory developed test (LDT) and not an FDA-cleared assay, IHC data utilizing the antibody used in our study are available in the literature [39, 42]. Moreover, PD-L1 expression in tumour cells showed high concordance between 22c3 and 28-8 (FDA-cleared assays), and E1L3N (LDT), as described by pathologists [43]. Several other studies have suggested cut-off criteria of 5–10% for PD-L1 positivity in various cancers [2, 44], though none have been able to determine the cut-off criteria in GBM [15,16,17]. In the present study, membranous or fibrillary PD-L1 staining of any intensity in > 5% of neoplastic cells was arbitrarily considered positive staining.
PD-L1 is an immature predictive biomarker, and PD-L1 positivity in GBM warrants further evaluation. Nduom et al. indicated that positive PD-L1 expression, as assessed using IHC, was associated with a poor prognosis, though this result has limited significance [16]. Only two recent studies found that positive PD-L1 immunostaining in human GBM tissue signifies a poor prognosis [17, 45]. Though this finding is in line with our results, in our study, PD-L1 expression did not appear to be an independent factor for poor survival according to multivariate analysis (Table 3). Furthermore, we attempted to evaluate PD-L1 expression in TIIs in GBM because elevated immune cell PD-L1 expression was linked to an augmented response to atezolizumab in urothelial carcinoma [13]. However, the incidence of PD-L1 expression in TIIs in GBM was low and did not correlate with patient survival.
Expression of PD-L1 in cancer cells can be influenced by oncogenic driver mutations. A recent study reported that PD-L1 expression correlated with EGFR mutations in NSCLC patients [46], and with BRAF mutations in melanoma and MSI-H colorectal cancer [47]. Primary GBM develops de novo without clinical or histological evidence of a low-grade precursor lesion, while secondary GBM develops from a low-grade glioma (WHO grades II and III) [22]. These GBM types depend on mutually exclusive oncogenic pathways. Alterations in IDH-1 are well-known molecular indicators of secondary GBM [23]. Studies have reported a significant correlation between IDH-1 wild-type status and PD-L1 expression in glioma, including GBM. Interestingly, IDH-1 mutated glioma shows hypermethylation of the PD-L1 gene promoter [48, 49], reduced expression of STAT1, and decreased cytotoxic T lymphocyte accumulation at tumour sites [50]. Published evidence implies that IDH-1 mutated glioma evades the immune system by mechanisms independent of PD-L1. In agreement with this contention, our data demonstrated that PD-L1 expression is associated with IDH-1 wild-type status, and may be unrelated to the pathogenesis of secondary GBM. Diverse oncogenic mutations correlated with PD-L1 expression in GBM should be investigated.
The current study has several limitations. Firstly, previous studies reported that regional heterogeneity of PD-L1 expression in cancers is not uncommon [37,38,39]. Since this study used the TMA technique, sampling bias cannot be ruled out. Secondly, this is a retrospective single institutional study and the statistical power of the analysis might be low due to the small sample size and possible selection bias. Thirdly, because of the nature of GBM, our study included a heterogeneous patient population treated with a variety of treatment regimens (Table 1). This can affect the patients’ outcomes and does not guarantee that the correlation with OS in the multivariate analysis is completely reliable (Table 3). Nevertheless, it does not affect the PD-L1 expression status because assessments were performed at the time of new diagnosis. Finally, there are no FDA-cleared assays for PD-L1 test in GBM.
In conclusion, we comprehensively evaluated PD-L1 expression in human GBM tissue and found that GBM patients with PD-L1 expression had poor OS according to univariate analysis only. Although we could not evaluate whether the PD-L1 expression status correlates with PD-1/PD-L1 targeted immunotherapy, our findings show that PD-L1 might be a logical therapeutic target for blockade in GBM patients with increased PD-L1 expression.
References
Sznol M, Chen L (2013) Antagonist antibodies to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human cancer. Clin Cancer Res 19(5):1021–1034. https://doi.org/10.1158/1078-0432.CCR-12-2063
Patel SP, Kurzrock R (2015) PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther 14(4):847–856. https://doi.org/10.1158/1535-7163.MCT-14-0983
Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, Brahmer JR, Lawrence DP, Atkins MB, Powderly JD, Leming PD, Lipson EJ, Puzanov I, Smith DC, Taube JM, Wigginton JM, Kollia GD, Gupta A, Pardoll DM, Sosman JA, Hodi FS (2014) Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 32(10):1020–1030. https://doi.org/10.1200/JCO.2013.53.0105
Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, Molina J, Kim JH, Arvis CD, Ahn MJ, Majem M, Fidler MJ, de Castro G Jr, Garrido M, Lubiniecki GM, Shentu Y, Im E, Dolled-Filhart M, Garon EB (2016) Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 387(10027):1540–1550. https://doi.org/10.1016/S0140-6736(15)01281-7
Asaoka Y, Ijichi H, Koike K (2015) PD-1 blockade in tumors with mismatch-repair deficiency. New Engl J Med 373(20):1979. https://doi.org/10.1056/NEJMc1510353#SA1
Sonpavde G (2017) PD-1 and PD-L1 inhibitors as salvage therapy for urothelial carcinoma. New Engl J Med 376(11):1073–1074. https://doi.org/10.1056/NEJMe1701182
He J, Hu Y, Hu M, Li B (2015) Development of PD-1/PD-L1 pathway in tumor immune microenvironment and treatment for non-small cell lung cancer. Sci Rep 5:13110. https://doi.org/10.1038/srep13110
Azuma T, Yao S, Zhu G, Flies AS, Flies SJ, Chen L (2008) B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood 111(7):3635–3643. https://doi.org/10.1182/blood-2007-11-123141
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO, European Organisation for R, Treatment of Cancer Brain T, Radiotherapy G, National Cancer Institute of Canada Clinical Trials G (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Engl J Med 352 (10):987–996. https://doi.org/10.1056/NEJMoa043330
Dunn-Pirio AM, Vlahovic G (2017) Immunotherapy approaches in the treatment of malignant brain tumors. Cancer 123(5):734–750. https://doi.org/10.1002/cncr.30371
Tivnan A, Heilinger T, Lavelle EC, Prehn JH (2017) Advances in immunotherapy for the treatment of glioblastoma. J Neurooncol 131(1):1–9. https://doi.org/10.1007/s11060-016-2299-2
Reardon DA, Brandes AO,AA, Rieger J, Wick A, Sepulveda J, Phuphanich S, de Souza P, Ahluwalia MS, Lim M, Vlahovic G, Sampson J (2017) Randomized phase 3 study evaluating the efficacy and safety of nivolumab vs bevacizumab in patients with recurrent glioblastoma: checkMate 143. Paper presented at the 5th Quadrennial Meeting of the World Federation of Neuro-Oncology Societies, Abstract (OS10.3)
Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, Dawson N, O’Donnell PH, Balmanoukian A, Loriot Y, Srinivas S, Retz MM, Grivas P, Joseph RW, Galsky MD, Fleming MT, Petrylak DP, Perez-Gracia JL, Burris HA, Castellano D, Canil C, Bellmunt J, Bajorin D, Nickles D, Bourgon R, Frampton GM, Cui N, Mariathasan S, Abidoye O, Fine GD, Dreicer R (2016) Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 387(10031):1909–1920. https://doi.org/10.1016/s0140-6736(16)00561-4
Wu P, Wu D, Li L, Chai Y, Huang J (2015) PD-L1 and survival in solid tumors: a meta-analysis. PLoS ONE 10(6):e0131403. https://doi.org/10.1371/journal.pone.0131403
Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wohrer A, Dieckmann K, Filipits M, Brandstetter A, Weller M, Kurscheid S, Hegi ME, Zielinski CC, Marosi C, Hainfellner JA, Preusser M, Wick W (2015) Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neur-Oncology 17(8):1064–1075. https://doi.org/10.1093/neuonc/nou307
Nduom EK, Wei J, Yaghi NK, Huang N, Kong LY, Gabrusiewicz K, Ling X, Zhou S, Ivan C, Chen JQ, Burks JK, Fuller GN, Calin GA, Conrad CA, Creasy C, Ritthipichai K, Radvanyi L, Heimberger AB (2016) PD-L1 expression and prognostic impact in glioblastoma. Neur-Oncology 18(2):195–205. https://doi.org/10.1093/neuonc/nov172
Zeng J, Zhang XK, Chen HD, Zhong ZH, Wu QL, Lin SX (2016) Expression of programmed cell death-ligand 1 and its correlation with clinical outcomes in gliomas. Oncotarget 7(8):8944–8955. https://doi.org/10.18632/oncotarget.6884
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW (2016) The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131(6):803–820. https://doi.org/10.1007/s00401-016-1545-1
Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG (2000) Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. New Engl J Med 343(19):1350–1354. https://doi.org/10.1056/NEJM200011093431901
Ambros PF, Ambros IM, Siop Europe Neuroblastoma Pathology B, Bone Marrow G (2001) Pathology and biology guidelines for resectable and unresectable neuroblastic tumors and bone marrow examination guidelines. Med Pediatr Oncol 37(6):492–504
Babu R, Sharma R, Karikari IO, Owens TR, Friedman AH, Adamson C (2013) Outcome and prognostic factors in adult cerebellar glioblastoma. J Clin Neurosci 20(8):1117–1121. https://doi.org/10.1016/j.jocn.2012.12.006
Ohgaki H, Kleihues P (2007) Genetic pathways to primary and secondary glioblastoma. Am J Pathol 170(5):1445–1453. https://doi.org/10.2353/ajpath.2007.070011
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD (2009) IDH1 and IDH2 mutations in gliomas. New Engl J Med 360(8):765–773. https://doi.org/10.1056/NEJMoa0808710
Takano S, Tian W, Matsuda M, Yamamoto T, Ishikawa E, Kaneko MK, Yamazaki K, Kato Y, Matsumura A (2011) Detection of IDH1 mutation in human gliomas: comparison of immunohistochemistry and sequencing. Brain Tumor Pathol 28(2):115–123. https://doi.org/10.1007/s10014-011-0023-7
Rivera AL, Pelloski CE, Gilbert MR, Colman H, De La Cruz C, Sulman EP, Bekele BN, Aldape KD (2010) MGMT promoter methylation is predictive of response to radiotherapy and prognostic in the absence of adjuvant alkylating chemotherapy for glioblastoma. Neur-Oncology 12(2):116–121. https://doi.org/10.1093/neuonc/nop020
Barker CF, Billingham RE (1977) Immunologically privileged sites. Adv Immunol 25:1–54
Ponomarev ED, Shriver LP, Maresz K, Dittel BN (2005) Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. J Neurosci Res 81(3):374–389. https://doi.org/10.1002/jnr.20488
Aloisi F (2001) Immune function of microglia. Glia 36(2):165–179
Hambardzumyan D, Gutmann DH, Kettenmann H (2016) The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci 19(1):20–27. https://doi.org/10.1038/nn.4185
Dubois LG, Campanati L, Righy C, D’Andrea-Meira I, Spohr TC, Porto-Carreiro I, Pereira CM, Balca-Silva J, Kahn SA, DosSantos MF, Oliveira Mde A, Ximenes-da-Silva A, Lopes MC, Faveret E, Gasparetto EL, Moura-Neto V (2014) Gliomas and the vascular fragility of the blood brain barrier. Front Cell Neurosci 8:418. https://doi.org/10.3389/fncel.2014.00418
Atai NA, Bansal M, Lo C, Bosman J, Tigchelaar W, Bosch KS, Jonker A, De Witt Hamer PC, Troost D, McCulloch CA, Everts V, Van Noorden CJ, Sodek J (2011) Osteopontin is up-regulated and associated with neutrophil and macrophage infiltration in glioblastoma. Immunology 132(1):39–48. https://doi.org/10.1111/j.1365-2567.2010.03335.x
Streit WJ, Conde JR, Fendrick SE, Flanary BE, Mariani CL (2005) Role of microglia in the central nervous system’s immune response. Neurol Res 27(7):685–691. https://doi.org/10.1179/016164105X49463
Zagzag D, Salnikow K, Chiriboga L, Yee H, Lan L, Ali MA, Garcia R, Demaria S, Newcomb EW (2005) Downregulation of major histocompatibility complex antigens in invading glioma cells: stealth invasion of the brain. Lab Invest 85(3):328–341. https://doi.org/10.1038/labinvest.3700233
Constam DB, Philipp J, Malipiero UV, ten Dijke P, Schachner M, Fontana A (1992) Differential expression of transforming growth factor-beta 1, -beta 2, and -beta 3 by glioblastoma cells, astrocytes, and microglia. J Immunol 148(5):1404–1410
Wilmotte R, Burkhardt K, Kindler V, Belkouch MC, Dussex G, Tribolet N, Walker PR, Dietrich PY (2005) B7-homolog 1 expression by human glioma: a new mechanism of immune evasion. Neuroreport 16(10):1081–1085
Jacobs JF, Idema AJ, Bol KF, Nierkens S, Grauer OM, Wesseling P, Grotenhuis JA, Hoogerbrugge PM, de Vries IJ, Adema GJ (2009) Regulatory T cells and the PD-L1/PD-1 pathway mediate immune suppression in malignant human brain tumors. Neur-Oncology 11(4):394–402. https://doi.org/10.1215/15228517-2008-104
Madore J, Vilain RE, Menzies AM, Kakavand H, Wilmott JS, Hyman J, Yearley JH, Kefford RF, Thompson JF, Long GV, Hersey P, Scolyer RA (2015) PD-L1 expression in melanoma shows marked heterogeneity within and between patients: implications for anti-PD-1/PD-L1 clinical trials. Pigment Cell Melanoma Res 28(3):245–253. https://doi.org/10.1111/pcmr.12340
Ilie M, Long-Mira E, Bence C, Butori C, Lassalle S, Bouhlel L, Fazzalari L, Zahaf K, Lalvee S, Washetine K, Mouroux J, Venissac N, Poudenx M, Otto J, Sabourin JC, Marquette CH, Hofman V, Hofman P (2016) Comparative study of the PD-L1 status between surgically resected specimens and matched biopsies of NSCLC patients reveal major discordances: a potential issue for anti-PD-L1 therapeutic strategies. Ann Oncol 27(1):147–153. https://doi.org/10.1093/annonc/mdv489
Lee KS, Kwak Y, Ahn S, Shin E, Oh HK, Kim DW, Kang SB, Choe G, Kim WH, Lee HS (2017) Prognostic implication of CD274 (PD-L1) protein expression in tumor-infiltrating immune cells for microsatellite unstable and stable colorectal cancer. Cancer Immunol Immunother 66(7):927–939. https://doi.org/10.1007/s00262-017-1999-6
Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, Carcereny E, Ahn MJ, Felip E, Lee JS, Hellmann MD, Hamid O, Goldman JW, Soria JC, Dolled-Filhart M, Rutledge RZ, Zhang J, Lunceford JK, Rangwala R, Lubiniecki GM, Roach C, Emancipator K, Gandhi L, Investigators K (2015) Pembrolizumab for the treatment of non-small-cell lung cancer. New Engl J Med 372(21):2018–2028. https://doi.org/10.1056/NEJMoa1501824
Hirsch FR, McElhinny A, Stanforth D, Ranger-Moore J, Jansson M, Kulangara K, Richardson W, Towne P, Hanks D, Vennapusa B, Mistry A, Kalamegham R, Averbuch S, Novotny J, Rubin E, Emancipator K, McCaffery I, Williams JA, Walker J, Longshore J, Tsao MS, Kerr KM (2017) PD-L1 immunohistochemistry assays for lung cancer: results from phase 1 of the blueprint PD-L1 IHC assay comparison project. J Thorac Oncol 12(2):208–222. https://doi.org/10.1016/j.jtho.2016.11.2228
Krabbe LM, Heitzplatz B, Preuss S, Hutchinson RC, Woldu SL, Singla N, Boegemann M, Wood CG, Karam JA, Weizer AZ, Raman JD, Remzi M, Rioux-Leclercq N, Haitel A, Rapoport LM, Glybochko PV, Roscigno M, Bolenz C, Bensalah K, Sagalowsky AI, Shariat SF, Lotan Y, Xylinas E, Margulis V (2017) Prognostic Value of PD-1 and PD-L1 expression in patients with high-grade upper tract urothelial carcinoma. J Urol. https://doi.org/10.1016/j.juro.2017.06.086
Rimm DL, Han G, Taube JM, Yi ES, Bridge JA, Flieder DB, Homer R, West WW, Wu H, Roden AC, Fujimoto J, Yu H, Anders R, Kowalewski A, Rivard C, Rehman J, Batenchuk C, Burns V, Hirsch FR, Wistuba II (2017) A prospective, multi-institutional, pathologist-based assessment of 4 immunohistochemistry assays for PD-L1 expression in non-small cell lung cancer. JAMA Oncol 3(8):1051–1058. https://doi.org/10.1001/jamaoncol.2017.0013
Wang X, Teng F, Kong L, Yu J (2016) PD-L1 expression in human cancers and its association with clinical outcomes. Onco Targets Ther 9:5023–5039. https://doi.org/10.2147/OTT.S105862
Han J, Hong Y, Lee YS (2017) PD-l1 expression and combined status of PD-L1/PD-1-positive tumor infiltrating mononuclear cell density predict prognosis in glioblastoma patients. J Pathol Transl Med 51(1):40–48. https://doi.org/10.4132/jptm.2016.08.31
Azuma K, Ota K, Kawahara A, Hattori S, Iwama E, Harada T, Matsumoto K, Takayama K, Takamori S, Kage M, Hoshino T, Nakanishi Y, Okamoto I (2014) Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann Oncol 25(10):1935–1940. https://doi.org/10.1093/annonc/mdu242
Rosenbaum MW, Bledsoe JR, Morales-Oyarvide V, Huynh TG, Mino-Kenudson M (2016) PD-L1 expression in colorectal cancer is associated with microsatellite instability, BRAF mutation, medullary morphology and cytotoxic tumor-infiltrating lymphocytes. Mod Pathol 29(9):1104–1112. https://doi.org/10.1038/modpathol.2016.95
Berghoff AS, Kiesel B, Widhalm G, Wilhelm D, Rajky O, Kurscheid S, Kresl P, Wohrer A, Marosi C, Hegi ME, Preusser M (2017) Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro Oncol. https://doi.org/10.1093/neuonc/nox054
Heiland DH, Haaker G, Delev D, Mercas B, Masalha W, Heynckes S, Gabelein A, Pfeifer D, Carro MS, Weyerbrock A, Prinz M, Schnell O (2017) Comprehensive analysis of PD-L1 expression in glioblastoma multiforme. Oncotarget 8(26):42214–42225. https://doi.org/10.18632/oncotarget.15031
Kohanbash G, Carrera DA, Shrivastav S, Ahn BJ, Jahan N, Mazor T, Chheda ZS, Downey KM, Watchmaker PB, Beppler C, Warta R, Amankulor NA, Herold-Mende C, Costello JF, Okada H (2017) Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J Clin Invest 127(4):1425–1437. https://doi.org/10.1172/JCI90644
Funding
This research was funded by the Seoul National University Bundang Hospital research fund (Grant Number: 11-2011-021).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in this study involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. The Institutional Review Board of Seoul National University Bundang Hospital approved the use of medical record data and tissue samples for this study (Reference: B-1612/374-304).
Informed consent
For this type of study, formal consent is not required.
Rights and permissions
About this article

Cite this article
Lee, K.S., Lee, K., Yun, S. et al. Prognostic relevance of programmed cell death ligand 1 expression in glioblastoma. J Neurooncol 136, 453–461 (2018). https://doi.org/10.1007/s11060-017-2675-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-017-2675-6