
Abstract
Terbium fluoride nanocrystals, covered by a shell, composed of cerium fluoride were synthesized by a co-precipitation method. Their complex structure was formed spontaneously during the synthesis. The surface of these core/shell nanocrystals was additionally modified by silica. The properties of TbF3@CeF3 and TbF3@CeF3@SiO2 nanocrystals, formed in this way, were investigated. Spectroscopic studies showed that the differences between these two groups of products resulted from the presence of the SiO2 shell. X-ray diffraction patterns confirmed the trigonal crystal structure of TbF3@CeF3 nanocrystals. High resolution transmission electron microscopy in connection with energy-dispersive X-ray spectroscopy showed a complex structure of the formed nanocrystals. Crystallized as small discs, ‘the products’, with an average diameter around 10 nm, showed an increase in the concentration of Tb3+ ions from surface to the core of nanocrystals. In addition to photo-physical analyses, cytotoxicity studies were performed on HSkMEC (Human Skin Microvascular Endothelial Cells) and B16F0 mouse melanoma cancer cells. The cytotoxicity of the nanomaterials was neutral for the investigated cells with no toxic or antiproliferative effect in the cell cultures, either for normal or for cancer cells. This fact makes the obtained nanocrystals good candidates for biological applications and further modifications of the SiO2 shell.
Graphical Abstract
.

Similar content being viewed by others

Introduction
Luminescent nanocrystals (NCs) activated by lanthanide ions (Ln3+) have been the object of intense research in recent years as a direct consequence of their unique properties (Grzyb and Lis 2011; Grzyb et al. 2012a, b; Wang et al. 2006a, 2010, 2011a; Wiglusz et al. 2009; Yan and Yan 2008; Yang et al. 2011). Taking advantage of phenomenon such as energy transfer (ET) and the characteristic features of f-electronic transitions, it is possible to obtain efficient luminescence from Ln3+-doped materials even when the materials are prepared at nanodimensions. This property is necessary for their potential applications in many areas. The current development of high technologies is one example of domains requiring appropriately designed systems (Bedekar et al. 2009; Boulon 2012). Furthermore, advanced fields of science, such as medicine and biology, use characteristic properties of Ln3+-doped NCs for bioimaging or cancer therapy (Bao et al. 2011; Di et al. 2011a; Diamente et al. 2006; Kang et al. 2011; Ren et al. 2012). Lanthanide-doped nanomaterials are attractive in connection with their exceptional optical and chemical properties, i.e. large effective Stokes shifts, high resistance to photobleaching, blinking and photochemical degradation. These excellent luminescent properties of the lanthanide ions, long luminescence lifetimes and the ability of ET processes occurring between dopants, make it possible to obtain an intense, visible emission under UV or IR (up-conversion) irradiation. In addition, the emission (luminescence lifetime) from Ln3+ is usually longer than the fluorescence of tissue (background) and differs in the spectral region, which allows for time-resolved fluorescence lifetime imaging (TRFI) (Cubeddu et al. 2002; Väisänen et al. 2000). This avoids disadvantages of UV excitation resulting in auto-fluorescence of cells. Hence, they present interesting alternatives for dye-based markers. Functionalization and coating of Ln3+-doped materials could increase their impact on fundamental biomedical research and clinical practice. Also receiving nanoparticles with mesoporous structure allows for designing multifunctional systems for simultaneous drug delivery and cell imaging (Di et al. 2011b; Janowski et al. 2012; Park et al. 2012).
Rare earth (RE) fluorides are important hosts for luminescent Ln3+ ions (Chen et al. 2010a; Diamente et al. 2006; Evanics et al. 2006; Kang et al. 2011; Wang et al. 2006a; Yan and Yan 2008). The low phonon energy of these compounds (e.g. LaF3, phonon energy is close to 350 cm−1) is the reason for their high effectiveness as matrices for displaying the luminescence of Ln3+ ions. The known methods of RE fluoride synthesis consist of a precipitation reaction in various systems, a thermal decomposition reaction of trifluoroacetate precursors in high boiling solvents, or a combination of these methods (Diamente et al. 2006; Evanics et al. 2006; Kang et al. 2011; Wang et al. 2006a; Yan and Yan 2008). Cerium fluoride is an especially important host for Tb3+ ions promoting efficient ET between Ce3+ and Tb3+ (Chai et al. 2009; Grzyb et al. 2012a; Wang et al. 2006b). ET results in an increased luminescence intensity which is important for bio-photonic applications. In this way lowering of the concentration of NCs used in in vitro imaging is possible; however, the luminescent signal can still be satisfactory. The presence of trivalent lanthanide ions in NCs could also change their magnetic properties. Ln3+ ions are known for their paramagnetic properties which can be used for magnetic resonance imaging (Grzyb et al. 2012c; Johnson et al. 2011). Also Ce3+ ions are known for their paramagnetic properties (Chen et al. 2010b).
Recently, the core/shell structures have been intensely investigated, and much valuable information such as the changed luminescence intensity or biological compatibility was obtained (Abel et al. 2009; Fu and Sun 2009; Ghosh et al. 2008; Kang et al. 2011; Zhu et al. 2008). The increased interest concerning core/shell structures is an effect of their improved physicochemical properties attributed to the surface coating, higher thermal stability and enhanced water solubility (Ghosh et al. 2008; Wang et al. 2009).
Silica coating is a popular method for improving water solubility and protecting NC cores from the surrounding environment (Runowski et al. 2012). Inexpensive and easy methods of obtaining spherical particles with narrow size distributions, chemical inertness and optical transparency are some of the advantages of using SiO2 as shells in NCs. Increased optical response, higher luminescence and longer decay times have been observed when the cores of luminescent Ln3+-doped materials are covered by SiO2 shells (Wang et al. 2009). As a result, biological effects of silica nanoparticles have become the subject of growing interest in recent years (Wang et al. 2012).
Developing core/shell nanostructures is also important for optoelectronic applications. Moreover, silica-coated NCs have become increasingly important in biomedical research (Bao et al. 2011; Hu et al. 2011). Because of the low chemical activity of SiO2, the coating process seems to be an effective way of lowering the cytotoxicity of the cores. However, it must nonetheless be mentioned that SiO2-based NCs could still be toxic, especially if their size is small (Lin et al. 2006; Yang et al. 2010). The HaCaT cells viability could be significantly decreased after exposure for SiO2 nanoparticles, which also induced apoptosis in a size-dependent manner (Yang et al. 2010). The mechanism underlying the toxic effects of SiO2 nanoparticles is based on the abnormal expression of oxidative stress-associated molecules, which inhibit cell growth and cause apoptosis (Lin et al. 2006; Yang et al. 2010). Effects of particle size on the cytotoxicity were also noticed in the case of silver nanoparticles. Smaller nanoparticles (20 nm) were more toxic than larger ones (80 and 110 nm) (Park et al. 2011). The most pronounced effects of these nanoparticles were a changed cellular metabolic activity and membrane damage (Park et al. 2011).
Covering NCs with a SiO2 shell allows for their further modification. The presence of the –OH groups in the SiO2 shell is an advantage over unmodified cores. These groups could be used for chemical modifications and conjugation of biomolecules such as biotin, antibodies and oligonucleotides (Selvan et al. 2009). The silica shell also improves the water solubility of the NCs, and is known as an anti-agglomerating agent. Therefore, it is important to study the effect of SiO2 coatings on the spectroscopic properties of NC cores. The aim of this research was to investigate this effect. Our experiments have been additionally extended to be able to study the cytotoxicity of the prepared materials.
Experimental
Reagents: Tb4O7 (99.99 %, Stanford Materials) was dissolved in ultra-pure nitric acid, HNO3 (POCh, Gliwice, Poland) in order to obtain Tb(NO3)3. Cerium chloride, CeCl3·6H2O (Sigma-Aldrich, 99.9 %), ammonium fluoride NH4F (POCh S.A., ACS grade, 98 %), Triton X-100 (POCh, Gliwice, Poland, reagent grade), TEOS (Sigma-Aldrich, 98 % reagent grade), 25 % NH4OH aqueous solution (Chempur, pure p.a.) and ethanol C2H5OH (99.98 % reagent grade) were used without further purification. For all experiments distilled water was used.
Synthesis of TbF3@CeF3 nanoparticles
In our experiment, the core/shell, TbF3@CeF3 nanocrystals were formed spontaneously from Tb3+- to Ce3+-containing solutions. This self-organization resulted in the synthesis of trigonal nanoplates with a Tb3+-rich core and a Ce3+-rich shell.
In order to synthesize TbF3@CeF3, solution A was prepared as follows: an aqueous solution of Tb(NO3)3 and CeCl3 was mixed in the molar ratio 15:85, respectively. The obtained solution was mixed with 1.25 g of Triton X-100 (surfactant) and filled with water up to 50 mL. Subsequently, solution B containing the fluoride ion source was prepared as follows: 0.83 g of NH4F (50 % excess) was dissolved in water and transferred onto a hot-plate magnetic stirrer. Then, 5 g of Triton X-100 was added to this solution and filled with water up to 100 mL. The whole system was stirred and heated up to 50 °C. This temperature was maintained over the entire process of synthesis. Afterwards, solution A was added drop by drop to the continuously stirred and heated solution B. Addition of solution A lasted approximately 30 min. After this time the reaction was finished, and a white precipitate of TbF3@CeF3 NCs was obtained. The precipitate was purified by centrifugation and washed several times with water and ethanol. The product exhibited a bright green luminescence under UV lamp irradiation (λ ex = 254 nm).
Synthesis of TbF3@CeF3@SiO2
Twenty milligram of the previously synthesized TbF3@CeF3 nanocrystals was dispersed in 10 mL of water. Subsequently, the colloid was mixed with 180 mL of ethanol and 10 mL of 25 % aqueous solution of ammonia, NH4OH. The final colloid was ultrasonicated in order to fully homogenise the whole system. The homogeneous colloidal solution was transferred onto the magnetic stirrer and vigorously stirred, at ambient conditions. Afterwards, 1.5 mL of TEOS was injected into this system. The reaction (TEOS hydrolysis) was carried out over 2 h. When the reaction was finished, the clear and transparent solution became white and turbid. The trigger of this was the formation of the silica shell. The hybrid, core/shell type product was purified by centrifugation and washed several times with water and ethanol. The final complex, core/shell type product, exhibited also a bright green luminescence under UV lamp irradiation (λ ex = 254 nm); however, its intensity was less than in the case of TbF3@CeF3 NCs. The scheme of synthesis is presented in Fig. 1.

Schematic presentation of the formation process of TbF3@CeF3 and TbF3@CeF3@SiO2 nanoparticles
Characterisation
X-ray diffraction patterns (XRD) were measured with a Bruker AXS D8 advance diffractometer using Cu Kα radiation (λ = 1.541874 Å) in the 2θ ranges from 6° to 60°. The IR absorption spectrum was recorded between 500 and 4,000 cm−1 on an FTIR spectrophotometer, Bruker FT-IR IFS 66/s. The material was mixed with KBr and then pressed into discs. The hydrodynamic diameter and ζ-potential of the obtained nanoparticles in water were measured with a Malvern Nano Zeta Sizer, Dynamic Light Scattering (DLS) instrument (He–Ne laser 633 nm, Max 5 mW). The excitation and emission spectra as well as luminescence lifetime measurements were performed on a Hitachi F-7000 fluorescence spectrophotometer at room temperature. Excitation and emission spectra were corrected for the instrumental response. The measured luminescence decays showed nonexponential character, which is a result of cross-relaxation between Tb3+ ions. The kinetics of the cross-relaxation is complex, and the model explaining this process has been previously reported (Ricci et al. 2010). However, the decay profiles can be fitted with an exponential function as a first approximation to the reported kinetics model. Therefore, to calculate luminescence lifetimes of TbF3@CeF3 NCs, luminescence decays were fitted to an exponential function:
where I represents the intensity at any time, I 0 is the intensity at t = 0 and τ is the luminescence lifetime. Nanocrystals covered by SiO2 shells, TbF3@CeF3@SiO2 showed different characteristics in their luminescence decays which were fitted by biexponential functions:
Luminescence lifetimes were calculated with help of OriginLab 8.5 software. The goodness of fit to the time traces was not lower than R 2 = 0.996, and errors were not higher than τ err < 0.01 ms. Transmission electron microscopy (TEM) was performed with a Philips CM200FEG electron microscope operating at 200 kV, equipped with an EDAX analyzer for qualitative and quantitative materials identification. The samples were dispersed on a Cu grid covered with 550-nm thick amorphous carbon film. Low-dose electron-beam imaging was used to prevent any beam damage of the sample and any additional heating effect of the electron beam (Henglein and Giersig 1999). The quality of the lattice images was improved using the conditions of minimum phase contrast according to Kunath (1987).
The cytotoxicity of TbF3@CeF3 and TbF3@CeF3@SiO2 nanoparticles
The cytotoxicity of the investigated nanocrystals was tested in proliferation assays and by cell imaging. Both cancer cells and normal cells were used. The HSkMEC (Human Skin Microvascular Endothelial Cells) cell line was obtained from Cell Culture Collection of the IIET (Wroclaw, Poland) (Crola Da Silva et al. 2009). The B16F0 mouse melanoma cancer cell line was obtained from the American Type Culture Collection (ATCC, USA). Both lines are maintained in the Cell Culture Collection of the IIET (Wroclaw, Poland). Cells were cultured in OptiMEM (Invitrogen, Cergy Pontoise, France) supplemented with 5 % foetal bovine serum, 40 μg/mL gentamycin (Invitrogen) and 0.05 μg/mL fungizone (Invitrogen). Cells were seeded at 2 × 104 cells/cm2 at 96-well plate or 6-well plate 24 h before experiments and maintained at 37 °C in a 5 % CO2/95 % air atmosphere during the pre-culture and the experiment. Final concentrations of the nanoparticles were: 0.05 mg/mL (0.029 mg/cm2) and 0.005 mg/mL (0.0029 mg/cm2) for TbF3@CeF3@SiO2 and its mass-equivalents for TbF3@CeF3 (the mass-equivalents represent the dose of uncoated-TbF3@CeF3 contained in TbF3@CeF3@SiO2). Higher concentrations of NCs caused mechanical damage to cell cultures and difficulties in interpreting the cytotoxicity analysis. The formed colloids were not stable, and the NCs tended to agglomerate. Before cell incubation, the prepared colloids were sterilized. Control cultures were supplemented with PBS.
The effect of nanoparticles on the cells was assessed as follows: (i) cell condition and morphology was assessed by optical microscopy in sequential imaging, (ii) total cell production was assessed by SRB assay (sulphorodamine B assay). The details of this technique were described by Skehan et al. (1990). The assay was performed after 72-h exposures of the cultured cells to the tested agents. The cells attached to the plastic were fixed by gently layering cold 50 % TCA (trichloroacetic acid, Aldrich-Chemie, Germany) on the top of the culture medium in each well. The plates were incubated at 4 °C for 1 h and then washed five times with tap water. The background optical density was measured in the wells filled with the culture medium, without the cells. The cellular material fixed with TCA was stained with 0.4 % sulforhodamine B (SRB, Sigma, Germany) dissolved in 1 % acetic acid (POCh, Gliwice, Poland) for 30 min. The unbound dye was removed by rinsing with 1 % acetic acid. The protein-bound dye was extracted with 100 μL 10 mM unbuffered Tris base (POCh, Gliwice, Poland) for the determination of the optical density (at 540 nm) in a computer-interfaced, 96-well microtiter plate reader Multiskan RC photometer (Labsystems, Helsinki, Finland). Each nanoparticle type and concentration in each cell line culture was tested two times in six-well groups (N = 6).
Results and discussion
Structure and morphology
Synthesis of the nanocrystalline rare earth fluorides is usually based on a precipitation reaction. It can be carried out under various conditions, such as reverse micelles formed in surfactants containing organic solvents, where the source of F− ions can be a salt such as NaF or NH4F (Karbowiak et al. 2005; Qiu 2000) There are also other well-known methods, e.g. a decomposition of trifluoroacetates in high boiling solvents or solvo- and hydrothermal methods. However, these methods usually require an advanced laboratory, specific reagents and a long purification process (Guo 2006; Zhang et al. 2005). A simpler way of synthesizing monodispersed NCs is the co-precipitation method. In the presence of the chosen organic compounds, the growth and nucleation process of the precipitated crystals occurred, which results in specific nanodimensions. The role of a capping agent is often attributed to citric acid, ethylenediaminetetraacetic acid, polyethyleneimine or polyethylene glycol (Qiu et al. 2011; Wang et al. 2011b). For the synthesis of TbF3@CeF3 NCs we have used the co-precipitation method in the presence of the nonionic surfactant Triton X-100. Prepared in this way NCs were covered by a SiO2 shell, and core/shell structures were formed.
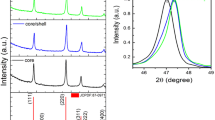
Cerium fluoride crystallizes in a trigonal crystal system with the space group \( P\bar{3}c1 \) 1(D 3d ) (Carnall et al. 1989; Cheetham et al. 1976). The Ce3+-ion in CeF3 crystals is coordinated by nine F− anions and has a C 2 site symmetry (Carnall et al. 1989). XRD patterns presented in Fig. 2, measured for the synthesized materials, fit well with standards from the ICSD database (Inorganic Crystal Structure Database) no 56773. Small nanoparticle sizes are responsible for the broadening of measured peaks. The broad band partially visible around 10° 2θ comes from amorphous silica. The characteristics of the pattern after surface covering is almost unchanged, which confirms that the applied process is safe for TbF3@CeF3 cores and does not influence their crystal structure.

XRD patterns of TbF3@CeF3 and core/shell TbF3@CeF3@SiO2 nanocrystals
The presence of a different phase is not visible in the XRD, which suggests that the TbF3 cores have the same structure as the CeF3 shell. The complex composition of the prepared product could also be responsible for the broadening of peaks in the XRD. Terbium fluoride usually crystallizes in the orthorhombic crystal structure with the space group Pnma. However, the displayed XRD patterns excluded the presence of phases other than trigonal CeF3. The explanation of this observation involves the manner in which these nanocrystals were synthesized. The precipitation of a product from the solution containing both lanthanide ions: Ce3+ and Tb3+, causes the formation of mixed fluorides containing both ions in the structure. The larger Ce3+ ions, present in the precipitated core, forced the formation of the trigonal structure.
Transmission electron microscopy was used for the imaging of the synthesized NCs and analysis of their morphology. Figure 3a shows TbF3@CeF3 fluoride NCs. Their sizes do not exceed 25 nm. Slight amounts of Triton X-100 molecules adsorbed on their surface suppress the formation of large agglomerates; however, they could still be observed. The formation of core/shell structures with a central core inside of the NCs, shown in Fig. 3d, confirms that occasionally the formed agglomerates are relatively weakly linked and could be broken due to the short ultrasonication. What is interesting, and remains unexpected, is the fact that in HR-TEM, the formation of the core/shell structure of these fluoride NCs results in a TbF3-rich core and a CeF3-dominated shell (Fig. 3b). Fourier transformation clearly indicates that both structures vary by differences in their lattice parameters. This situation is common in the obtained nanomaterial, which has been checked by HR-TEM. The reason for the crystallization mechanism leading to the presented structures is unknown. Most probable is that during the first stage of crystallization; when NH4F is added at concentrations lower than stoichiometric, Tb3+-rich nanoparticles are precipitated due to their lower solubility in the synthesis medium. Then, as additional reagent was being added, the Tb3+-rich nanoparticles acted as crystallization cores for the Ce3+-rich second phase. In fact, both phases crystallized in the \( P\bar{3}c1 \) crystal system, and the presence of more than a single phase in the XRD patterns was not observed. Also the luminescence analysis and the occurrence of ET confirmed that Ce3+ and Tb3+ must be present in the same phase.

TEM and HR-TEM images of TbF3@CeF3 (a,b) and TbF3@CeF3@SiO2 core/shell nanoparticles (c,d)
The subject of spontaneous organization of nanosystems can be found in the literature. Some examples are: the spontaneous self-organization of Cu2O/CuO core–shell nanowires from copper nanoparticles or the organization of single CdTe nanoparticles into luminescent nanowires (Ji et al. 2010; Tang et al. 2002). Also, individual nanocrystals can assemble into more complex structures (Grzyb et al. 2013).
Figure 3c, d shows silica-covered-TbF3@CeF3@SiO2 nanoparticles. Their structure is well presented in HR-TEM and consists of small fluoride cores and relatively large silica shells with thicknesses of around 100 nm. The cores of the nanoparticles shown are composed of one to several TbF3@CeF3 nanocrystals.
The average size distribution of the synthesized core and core/shell nanoparticles and their zeta potential (ζ-potential) were measured using the DLS technique. The measured ζ-potential of the TbF3@CeF3 was +22.7 mV, and −29.8 mV for TbF3@CeF3@SiO2 nanoparticles. The measured values are in good agreement with our predictions and literature data for similar compounds. According to the literature, the isoelectric point (point of zero charge) for LnF3 nanoparticles is around pH = 6, and for silica, (SiO2) n particles, is ~pH = 3 (Philipse et al. 1994; Bogdan et al. 2011). The measured ζ-potential values for our samples (at pH = 5) indicate that after silica coating, there is a dramatic change in the ζ-potential, from a positive value +22.7 mV (protonated surface, e.g. LnOH2 +) to a negative value −29.8 mV (deprotonated silica surface, –SiO−). The average size distribution of the obtained nanostructures (Fig. 4) was approximately 49 nm for TbF3@CeF3 (a), and approx. 1,411 nm for TbF3@CeF3@SiO2 (b). These values are larger than the average size calculated from TEM images, because the hydrodynamic diameter of the analysed particles is larger than their real size (size of the individual nanoparticles), which is common in such DLS measurements (Bogdan et al. 2011). Additionally, the TbF3@CeF3@SiO2 nanostructures tend to form agglomerates, whose hydrodynamic diameter registered during the experiment is obviously much larger compared to individual core/shell type nanoparticles.

Particles size distribution obtained from DLS measurements; TbF3@CeF3 (a), TbF3@CeF3@SiO2 (b)
The synthesized NCs were examined by FT-IR spectroscopy in order to reveal the presence of Triton X-100 molecules on the NCs surface and silica in the core/shell type structure (Fig. 5). In the presented region of the infrared spectra there are no absorption peaks originating from Ln-F vibrations because they exhibit absorption at smaller wavenumbers, which is typical for inorganic fluorides. Both spectra show the most intense and broad absorption peak at around 3,394 cm−1, corresponding to O–H stretching vibrations of H2O (hydrogen-bonded water molecules) and O–H stretching vibrations of Triton X-100 molecules. In the case of the core/shell spectrum, this band is even more intense in comparison to the core, because of the overlapping of the O–H stretching vibrations from the previous molecules and silica (silanol groups, Si–OH). At 1,643, 1,633 and 1,595 cm−1 absorption peaks from the same molecules were registered and labelled as deformation vibrations of the O–H groups. The weak absorption bands at 2,980 cm−1 and 2,925 cm−1 were assigned to C–H stretching vibrations corresponding to the –CH3 and –CH2 groups of Triton X-100. Other peaks were labelled and assigned to the stretching (ν) and deformation (σ) vibrations corresponding to molecules of the surfactant, namely: νC=C(arom) (1,509 cm−1), σC–H (1,400 cm−1), σO–H (1,259 cm−1), νC–O (1,090, 1,048 cm−1) and σC–H (arom) (876, 832 cm−1). Only in the spectrum of the core/shell NCs, three intense and dominant peaks originating from silica were observed and assigned to: νSi–O-Si(asym) (1,180, 1,094 cm−1), νSi–O-(947 cm−1) and νSi–O-Si(sym) (798 cm−1). The presence of silica in the case of a core/shell type compound was confirmed. We also assumed that Triton X-100 molecules were adsorbed on the surface of the obtained NCs.

FT-IR absorption spectra of TbF3@CeF3 and TbF3@CeF3@SiO2 NCs
Luminescence properties
The synthesized NCs showed intense green luminescence under UV irradiation (254 nm) corresponding to the electronic transitions of the Tb3+ ions. This luminescence can be used for typical optoelectronic applications such as various types of displays or lightening. However, the range of potential applications could be extended by the imaging of cells in fluorescence or confocal microscopy. The relatively small size of the particles allows them to penetrate cell membranes or to join to their surfaces. Silica covering displays a promising method for increasing the biocompatibility and gives the possibility of modifying the surface of inorganic NCs. However, according to the luminescence studies described below, the emission intensity of the core is highly influenced by the presence of a silica shell.
Figure 6 shows the excitation and emission spectra of the obtained nanoparticles. The excitation spectra of both materials show broad and intense bands with a maximum around 250–260 nm. These broad and intense bands could be observed as a result of the 4f 1 → 4f 05d 1 transition of the Ce3+-ion (Guo et al. 2011). The presence of this broad band is evidence of ET between the Ce3+ and Tb3+ ions. Narrow and less intense bands in the range of 320–400 nm are connected with direct excitation of Tb3+ ions and are hindered by selection rules of f–f transitions. The covering of the TbF3@CeF3 NCs by silica changed the excitation spectra due to the absorption of UV light by SiO2 in this region (inset in Fig. 6). This effect is important, and therefore TbF3@CeF3@SiO2 showed much less effective luminescence than TbF3@CeF3. The emission spectra of the obtained NCs are typical for Tb3+-doped materials and are composed of narrow bands related to the f–f transitions of this ion.

Excitation and emission spectra of TbF3@CeF3 and TbF3@CeF3@SiO2 nanoparticles, and absorption spectrum of pure SiO2 as an inset (λ ex TbF3@CeF3 = 252 nm, λ ex TbF3@CeF3@SiO2 = 248 nm, λ em = 543 nm)
Both obtained materials can easily form stable colloidal solutions, which are presented in Fig. 6. Despite the similar emission spectral characteristics, the colloidal solutions of the core and the core/shell NCs showed slightly different colours of emission, which is due to SiO2 light scattering and absorption at the UV wavelengths. The emission intensity is an important factor; however, the toxicity for cells can be much more critical for bioimaging applications and therefore should be taken as the priority in the design of nanolabels.
The lower luminescence intensity of TbF3@CeF3@SiO2 is caused by the absorption of silica in the UV region, which results in a decreased excitation intensity. However, the measured luminescence decays, shown in Fig. 7, also indicate the formation of quenching centres after covering with silica. The FT-IR spectra suggest the presence of water molecules and, which seems even more possible, –OH groups in the SiO2 shell, which quenched the exited states of Tb3+ and additionally lowered the luminescence intensity of the core/shell type NCs. The calculated luminescence lifetimes are typical for Tb3+ ions, and the presence of the biexponential character of decays, in the case of the SiO2 covered NCs, could be explained by the two different surrounding environments of Tb3+ ions. Luminescence of the ions placed on the surface of NCs is quenched by –OH groups present in the SiO2 structure. Also the presence of a larger number of TbF3@CeF3 NCs incorporated into SiO2 could cause changes in the luminescence decay kinetics.

Luminescence lifetimes of TbF3@CeF3 and TbF3@CeF3@SiO2 nanoparticles (λ ex TbF3@CeF3 = 252 nm, λ ex TbF3@CeF3@SiO2 = 248 nm, λ em = 543 nm)
The disadvantages of SiO2 coating are undeniable and visible in the spectroscopic analysis presented. However, the same shell allows for the modification of NCs surface in a greater manner than uncoated particles. The receiving and designing of such nanostructures requires a compromise between a reduction in their luminescent properties and an expansion of their application possibilities.
Potential effects of NCs on living cells were assessed in normal (HSkMEC) or cancer (B16F0) cell cultures in vitro. 72-h expositions of the cells to NCs had no effect on the amount of cells in comparison to the control, as revealed by SRB assays (Fig. 8). Our observation indicates that NCs have no toxic or inhibitory effects on the tested cells. Furthermore, cell morphology was also typical with extended protrusions, with no visible aberrations or defects, as shown by optical microscopy (Figs. 9, 10). Higher concentrations of NCs (0.5 mg/mL and higher) may cause cell damage and a reduced viability (data not shown). However, these concentrations are of low applicability due to lower stability of colloidal solutions.

Cell viability in TbF3@CeF3 or TbF3@CeF3@SiO2 nanoparticle treated cell cultures (SRB assay). a B16F0 cells, b HSkMEC cells. Control-cells treated with PBS, A-cells treated with 0.05 mg/mL TbF3@CeF3@SiO2, B-cells treated with 0.005 mg/mL TbF3@CeF3@SiO2, C-cells treated with 0.01 mg/mL TbF3@CeF3 (mass equivalent for 0.05 mg TbF3@CeF3@SiO2) D-cells treated with 0.001 mg/mL TbF3@CeF3 (mass equivalent for 0.05 mg TbF3@CeF3@SiO2)

Microscopy images of B16F0 cells treated with TbF3@CeF3 or TbF3@CeF3@SiO2 nanoparticles (scale bars at a, c and e: 40 μm, scale bars at b, d and f: 10 μm). a and b: control cells. c and d: cells treated with TbF3@CeF3 for 72 h, washed before imaging. e and f: cells treated with TbF3@CeF3@SiO2 for 72 h, washed before imaging

Microscopy images of HSkMEC cells treated with TbF3@CeF3 or TbF3@CeF3@SiO2 nanoparticles (scale bars at a, c and e: 40 μm, scale bars at b, d and f: 10 μm). a and b: control cells. c and d: cells treated with TbF3@CeF3 for 72 h, washed before imaging. e and f: cells treated with TbF3@CeF3@SiO2 for 72 h, washed before imaging
Conclusions
Luminescent nanoparticles based on TbF3@CeF3 can be synthesized by the co-precipitation method in Triton X-100 solution. During the nucleation and growth of nanocrystals, self-organization occurred resulting in the formation of the core/shell structure. These simultaneously formed core/shell nanocrystals were analysed by HR-TEM and EDX techniques what confirmed a complex character of the structures. The co-precipitation method allowed for obtaining small disks with an average diameter around 10 nm. The surface modification by SiO2 covering could be carried out by the described method. HR-TEM images showed that the precipitation process of discussed fluorides is complex and results. In the formation of NCs with a Tb3+-rich core and Ce3+-rich shell. The prepared fluoride NCs were used as the cores in the TbF3@CeF3@SiO2 structures. TEM images presented spherical shapes of NCs with a centrally placed fluoride core. Synthesized nanocrystals showed an intense green luminescence under UV irradiation what makes them good candidates as luminescent markers. Long luminescence lifetimes (2.52 and 3.53 ms) are other advantages of prepared NCs opening a possibility of time-resolved fluorescence lifetime imaging.
TbF3@CeF3@SiO2 showed lower luminescence intensity than the cores due to the absorption of UV exciting radiation by the SiO2 shell and its quenching properties. The cell viability and proliferation test indicated that both types of the NCs were neutral for the investigated cells with no toxic or antiproliferative effects in the cell cultures, both for normal and for cancer cells. Furthermore, NCs had no visible effect on the cell morphology, as was shown by microscopy imaging. These results suggest safety and adequacy of NCs for future use in medical or biological applications. Covering NCs by SiO2 shell opens up new possibilities like modification of the surface of core/shell structures or chemical connection with important organic molecules and drugs.
References
Abel K, Boyer JC, Van Veggel FCJM (2009) Hard proof of the NaYF4/NaGdF4 nanocrystal core/shell structure. J Am Chem Soc 131:14644–14655. doi:10.1021/ja906971y
Bao H, Yang J, Huang Y, Xu ZP, Hao N, Wu Z, Lu GQM, Zhao D (2011) Synthesis of well-dispersed layered double hydroxide core@ordered mesoporous silica shell nanostructure (LDH@mSiO2) and its application in drug delivery. Nanoscale 3:4069–4073. doi:10.1039/c1nr10718f
Bogdan N, Vetrone F, Ozin Ga, Capobianco Ja (2011) Synthesis of ligand-free colloidally stable water dispersible brightly luminescent lanthanide-doped upconverting nanoparticles. Nano Lett 11:835–840. doi:10.1021/nl1041929
Boulon G (2012) Fifty years of advances in solid-state laser materials. Opt Mater 34:499–512. doi:10.1016/j.optmat.2011.04.018
Carnall WT, Goodman GL, Rajnak K, Rana RS (1989) A systematic analysis of the spectra of the lanthanides doped into single crystal LaF3. J Chem Phys 90:3443–3457. doi:10.1063/1.455853
Chai RT, Lian HZ, Li CX, Cheng ZY, Hou ZY, Huang SS, Lin J (2009) In situ preparation and luminescent properties of CeF3 and CeF3:Tb3+ nanoparticles and transparent CeF3:Tb3+/PMMA Nanocomposites in the visible spectral range. J Phys Chem C 113:8070–8076. doi:10.1021/Jp900269b
Cheetham AK, Fender BEF, Fuess H, Wright AF (1976) A powder neutron diffraction study of lanthanum and cerium trifluorides. Acta Crystallogr B 32:94–97. doi:10.1107/S0567740876002380
Chen DQ, Yu YL, Huang P, Lin H, Shan ZF, Wang YS (2010a) Color-tunable luminescence of Eu3+ in LaF3 embedded nanocomposite for light emitting diode. Acta Mater 58:3035–3041. doi:10.1016/j.actamat.2010.01.035
Chen SY, Lu YH, Huang TW, Yan DC, Dong C-L (2010b) Oxygen vacancy dependent magnetism of CeO2 nanoparticles prepared by thermal decomposition method. J Phys Chem C 114:19576–19581. doi:10.1021/jp1045172
Crola Da Silva C, Lamerant-Fayel N, Paprocka M, Mitterrand M, Gosset D, Dus D, Kieda C (2009) Selective human endothelial cell activation by chemokines as a guide to cell homing. Immunology 126:394–404. doi:10.1111/j.1365-2567.2008.02906.x
Cubeddu R, Comelli D, D’Andrea C, Taroni P, Valentini G (2002) Time-resolved fluorescence imaging in biology and medicine. J Phys D Appl Phys 35:61–76. doi:10.1088/0022-3727/35/9/201
Di W, Li J, Shirahata N, Sakka Y, Willinger MG, Pinna N (2011a) Photoluminescence, cytotoxicity and in vitro imaging of hexagonal terbium phosphate nanoparticles doped with europium. Nanoscale 3:1263–1269. doi:10.1039/c0nr00673d
Di W, Ren X, Zhao H, Shirahata N, Sakka Y, Qin W (2011b) Single-phased luminescent mesoporous nanoparticles for simultaneous cell imaging and anticancer drug delivery. Biomaterials 32:7226–7233. doi:10.1016/j.biomaterials.2011.06.019
Diamente PR, Burke RD, Van Veggel FC (2006) Bioconjugation of Ln3+-doped LaF3 nanoparticles to avidin. Langmuir 22:1782–1788. doi:10.1021/la052589r
Evanics F, Diamente PR, Van Veggel FCJM, Stanisz GJ, Prosser RS (2006) Water-soluble GdF3 and GdF3/LaF3 nanoparticles physical characterization and NMR relaxation properties. Chem Mater 18:2499–2505. doi:10.1021/cm052299w
Fu YX, Sun YH (2009) Comparative study of synthesis and characterization of monodispersed SiO2@Y2O3:Eu3+ and SiO2 @ Y2O3:Eu3+@SiO2 core–shell structure phosphor particles. J Alloys Compd 471:190–196. doi:10.1016/j.jallcom.2008.03.055
Ghosh P, De la Rosa E, Oliva J, Solis D, Kar A, Patra A, Rosa ED, Haldar KK (2008) Enhancement of upconversion Emission of LaPO4:Er@Yb Core–Shell nanoparticles/nanorods. J Phys Chem C 112:9650–9658. doi:10.1021/jp801978b
Grzyb T, Lis S (2011) Structural and spectroscopic properties of LaOF:Eu3+ nanocrystals prepared by the sol–gel Pechini method. Inorg Chem 50:8112–8120. doi:10.1021/ic2005453
Grzyb T, Runowski M, Szczeszak A, Lis S (2012a) Influence of matrix on the luminescent and structural properties of glycerine-capped, Tb3+-doped fluoride nanocrystals. J Phys Chem C 116:17188–17196. doi:10.1021/jp3010579
Grzyb T, Szczeszak A, Rozowska J, Legendziewicz J, Lis S (2012b) Tunable luminescence of Sr2CeO4:M2+ (M=Ca, Mg, Ba, Zn) and Sr2CeO4:Ln3+ (Ln=Eu, Dy, Tm) nanophosphors. J Phys Chem C 116:3219–3226. doi:10.1021/jp208015z
Grzyb T, Gruszeczka A, Wiglusz RJ, Śniadecki Z, Idzikowski B, Lis S (2012c) Multifunctionality of GdPO4:Yb3+, Tb3+ nanocrystals–luminescence and magnetic behaviour. J Mater Chem 22:22989–22997. doi:10.1039/c2jm34863b
Grzyb T, Runowski M, Szczeszak A, Lis S (2013) Structural, morphological and spectroscopic properties of Eu3+-doped rare earth fluorides synthesized by hydrothermal method. J Solid State Chem 200:76–83. doi:10.1016/j.jssc.2013.01.012
Guo H (2006) Photoluminescent properties of CeF3:Tb3+ nanodiskettes prepared by hydrothermal microemulsion. Appl Phys B 84:365–369. doi:10.1007/s00340-006-2326-7
Guo C, Ding X, Seo HJ, Ren Z, Bai J (2011) Luminescent properties of UV excitable blue emitting phosphors MSr4(BO3)3:Ce3+ (M=Li and Na). J Alloys Compd 509:4871–4874. doi:10.1016/j.jallcom.2011.01.194
Henglein A, Giersig M (1999) Formation of colloidal silver nanoparticles: capping action of citrate. J Phys Chem B 103:9533–9539. doi:10.1021/jp9925334
Hu D, Chen M, Gao Y, Li F, Wu L (2011) A facile method to synthesize superparamagnetic and up-conversion luminescent NaYF4:Yb, Er/Tm@SiO2@Fe3O4 nanocomposite particles and their bioapplication. J Mater Chem 21:11276–11282. doi:10.1039/c1jm11172h
Janowski M, Bulte JWM, Walczak P (2012) Personalized nanomedicine advancements for stem cell tracking. Adv Drug Deliv Rev 64:1488–1507. doi:10.1016/j.addr.2012.07.008
Ji JY, Shih PH, Yang CC, Chan TS, Ma YR, Wu SY (2010) Spontaneous self-organization of Cu2O/CuO core–shell nanowires from copper nanoparticles. Nanotechnology 21:45603. doi:10.1088/0957-4484/21/4/045603
Johnson NJJ, Oakden W, Stanisz GJ, Prosser RS, Van Veggel FCJM (2011) Size-tunable, ultrasmall NaGdF4 nanoparticles: insights into their T1 MRI contrast enhancement. Chem Mater 23:3714–3722. doi:10.1021/cm201297x
Kang X, Cheng Z, Li C, Yang D, Shang M, Ma P, Li G, Liu N, Lin J (2011) Core–shell structured up-conversion luminescent and mesoporous NaYF4:Yb3+/Er3+@nSiO2@mSiO2 nanospheres as carriers for drug delivery. J Phys Chem C 115:15801–15811. doi:10.1021/jp203039t
Karbowiak M, Mech A, Bednarkiewicz A, Strek W, Kepinski L (2005) Comparison of different NaGdF4: Eu3+ synthesis routes and their influence on its structural and luminescent properties. J Phys Chem Solids 66:1008–1019. doi:10.1016/j.jpcs.2005.01.002
Kunath W, Zemlin F, Weiss K (1987) Refinement procedures for high-resolution electron microscopy. Optik 76:122–131
Lin W, Huang YW, Zhou XD, Ma Y (2006) In vitro toxicity of silica nanoparticles in human lung cancer cells. Toxicol Appl Pharmacol 217:252–259. doi:10.1016/j.taap.2006.10.004
Park MVDZ, Neigh AM, Vermeulen JP, De la Fonteyne LJJ, Verharen HW, Briedé JJ, Van Loveren H, De Jong WH (2011) The effect of particle size on the cytotoxicity, inflammation, developmental toxicity and genotoxicity of silver nanoparticles. Biomaterials 32:9810–9817. doi:10.1016/j.biomaterials.2011.08.085
Park Y, Kim HM, Kim JH, Moon KC, Yoo B, Lee KT, Lee N, Choi Y, Park W, Ling D, Na K, Moon WK, Choi SH, Park HS, Yoon S-Y, Suh YD, Lee SH, Hyeon T (2012) Theranostic probe based on lanthanide-doped nanoparticles for simultaneous in vivo dual-modal imaging and photodynamic therapy. Adv Mater 24:5755–5761. doi:10.1002/adma.201202433
Philipse AP, Van Bruggen MPB, Pathmamanoharan C (1994) Magnetic silica dispersions: preparation and stability of surface-modified silica particles with a magnetic core. Langmuir 10:92–99. doi:10.1021/la00013a014
Qiu S (2000) Synthesis of CeF3 nanoparticles from water-in-oil microemulsions. Powder Technol 113:9–13. doi:10.1016/S0032-5910(99)00249-1
Qiu H, Chen G, Sun L, Hao S, Han G, Yang C (2011) Ethylenediaminetetraacetic acid (EDTA)-controlled synthesis of multicolor lanthanide doped BaYF5 upconversion nanocrystals. J Mater Chem 21:17202–17208. doi:10.1039/c1jm12950c
Ren W, Tian G, Zhou L, Yin W, Yan L, Jin S, Zu Y, Li S, Gu Z, Zhao Y (2012) Lanthanide ion-doped GdPO4 nanorods with dual-modal bio-optical and magnetic resonance imaging properties. Nanoscale 4:3754–3760. doi:10.1039/c2nr30683b
Ricci PC, Salis M, Corpino R, Carbonaro CM, Fortin E, Anedda A (2010) A kinetics model for Tb3+ recombinations in low doped Tb:Lu1.8Y0.2SiO5 crystals. J Appl Phys 108:43512–43517. doi:10.1063/1.3467788
Runowski M, Grzyb T, Lis S (2012) Magnetic and luminescent hybrid nanomaterial based on Fe3O4 nanocrystals and GdPO4:Eu3+ nanoneedles. J Nanopart Res 14:1185–1188. doi:10.1007/s11051-012-1188-7
Selvan ST, Tan TTY, Yi DK, Jana NR (2009) Functional and multifunctional nanoparticles for bioimaging and biosensing. Langmuir 26:11631–11641. doi:10.1021/la903512m
Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR (1990) New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 82:1107–1112. doi:10.1093/jnci/82.13.1107
Tang Z, Kotov N, Giersig M (2002) Spontaneous organization of single CdTe nanoparticles into luminescent nanowires. Science 297:237–240. doi:10.1126/science.1072086
Bedekar V, Dutta DPD, Mohapatra M, Godbole SV, Ghildiyal R, Tyagi AK, SV (2009) Rare-earth doped gadolinia based phosphors for potential multicolor and white light emitting deep UV LEDs. Nanotechnology 20: 125707-125717. doi:10.1088/0957-4484/20/12/125707
Väisänen V, Härmä H, Lilja H, Bjartell A (2000) Time-resolved fluorescence imaging for quantitative histochemistry using lanthanide chelates in nanoparticles and conjugated to monoclonal antibodies. Luminescence 15:389–397. doi:10.1002/1522-7243(200011/12)15:6<389:AID-BIO626>3.0.CO;2-7
Wang F, Zhang Y, Fan X, Wang M (2006a) Facile synthesis of water-soluble LaF3:Ln3 + nanocrystals. J Mater Chem 16:1031–1034. doi:10.1039/b518262j
Wang ZL, Quan ZW, Jia PY, Lin CK, Luo Y, Chen Y, Fang J, Zhou W, O’Connor CJ, Lin J (2006b) Facile synthesis and photoluminescent properties of redispersible CeF3, CeF3: Tb3+, and CeF3:Tb3+/LaF3 (core/shell) nanoparticles. Chem Mater 18:2030–2037. doi:10.1021/Cm052360x
Wang Y, Qin W, Zhang J, Cao C, Lü S, Ren X (2009) Photoluminescence of colloidal YVO4:Eu/SiO2 core/shell nanocrystals. Opti Commun 282:1148–1153. doi:10.1016/j.optcom.2008.12.007
Wang F, Han Y, Lim CS, Lu Y, Wang J, Xu J, Chen H, Zhang C, Hong M, Liu X (2010) Simultaneous phase and size control of upconversion nanocrystals through lanthanide doping. Nature 463:1061–1065. doi:10.1038/nature08777
Wang G, Peng Q, Li Y (2011a) Lanthanide-doped nanocrystals: synthesis, optical-magnetic properties, and applications. Acc Chem Res 44:322–332. doi:10.1021/ar100129p
Wang M, Abbineni G, Clevenger A, Mao C, Xu S (2011b) Upconversion nanoparticles: synthesis, surface modification and biological applications. Nanomedicine 7:710–729. doi:10.1016/j.nano.2011.02.013
Wang Q, Bao Y, Zhang X, Coxon PR, Jayasooriya U, Chao Y (2012) Uptake and toxicity studies of poly-acrylic acid functionalized silicon nanoparticles in cultured mammalian cells. Adv Healthcare Mater 1:189–198. doi:10.1002/adhm.201100010
Wiglusz R, Grzyb T, Lis S, Strek W (2009) Preparation and spectroscopy characterization of Eu: MgAl2O4 nanopowder prepared by modified Pechini method. J Nanosci Nanotechnol 9:5803–5810. doi:10.1166/jnn.2009.1259
Yan ZG, Yan C (2008) Controlled synthesis of rare earth nanostructures. J Mater Chem 18:5046–5059. doi:10.1039/b810586c
Yang X, Liu J, He H, Zhou L, Gong C, Wang X, Yang L, Yuan J, Huang H, He L, Zhang B, Zhuang Z (2010) SiO2 nanoparticles induce cytotoxicity and protein expression alteration in HaCaT cells. Part Fibre Toxicol 7:1–12. doi:10.1186/1743-8977-7-1
Yang D, Kang X, Shang M, Li G, Peng C, Li C, Lin J (2011) Size and shape controllable synthesis and luminescent properties of BaGdF5:Ce3+/Ln3+ (Ln = Sm, Dy, Eu, Tb) nano/submicrocrystals by a facile hydrothermal process. Nanoscale 3:2589–2595. doi:10.1039/c1nr10203f
Zhang YW, Sun X, Si R, You LP, Yan C-H (2005) Single-crystalline and monodisperse LaF3 triangular nanoplates from a single-source precursor. J Am Chem Soc 127:3260–3271. doi:10.1021/ja042801y
Zhu L, Meng J, Cao XQ (2008) Synthesis and photoluminescent properties of silica-coated LaCeF3:Tb nanocrystals. J Nanopart Res 10:383–386. doi:10.1007/s11051-007-9262-2
Acknowledgments
Funding for this research was provided by the Polish Ministry of Science and Higher Education (Grant No N204 329736). MG thanks to Helmholtz Zentrum Berlin and to Foundation for Polish Science in Warsaw for financial support. T.G. holds a scholarship from the Foundation for Polish Science for Young Scientists (FNP). S.L. kindly acknowledges financial support from the National Science Centre (Grant 2012/06/M/ST5/00325).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Grzyb, T., Runowski, M., Dąbrowska, K. et al. Structural, spectroscopic and cytotoxicity studies of TbF3@CeF3 and TbF3@CeF3@SiO2 nanocrystals. J Nanopart Res 15, 1958 (2013). https://doi.org/10.1007/s11051-013-1958-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-013-1958-x