
Abstract
The pandemic outbreak of the Corona viral infection has become a critical global health issue. Biophysical and structural evidence shows that spike protein possesses a high binding affinity towards host angiotensin-converting enzyme 2 and viral hemagglutinin-acetylesterase (HE) glycoprotein receptor. We selected HE as a target in this study to identify potential inhibitors using a combination of various computational approaches such as molecular docking, ADMET analysis, dynamics simulations and binding free energy calculations. Virtual screening of NPACT compounds identified 3,4,5-Trihydroxy-1,8-bis[(2R,3R)-3,5,7-trihydroxy-3,4-dihydro-2H-chromen-2-yl]benzo[7]annulen-6-one, Silymarin, Withanolide D, Spirosolane and Oridonin as potential HE inhibitors with better binding energy. Furthermore, molecular dynamics simulations for 100 ns time scale revealed that most of the key HE contacts were retained throughout the simulations trajectories. Binding free energy calculations using MM/PBSA approach ranked the top-five potential NPACT compounds which can act as effective HE inhibitors.
Graphic abstract

Similar content being viewed by others


Introduction
Coronaviruses (CoV) are a single (+)-stranded RNA containing a virus that appears as an oval-shaped envelop with spike-like protrusions [1, 2]. The first form of CoV has emerged in the Middle East which caused respiratory tract disease called the Middle East Respiratory Syndrome Coronavirus (MERS‐CoV) [3, 4]. The recent outbreak which created the global health threat is caused by a new form of CoV called CoV-2. The initial spread is traced to Hubei province, Wuhan, Republic of China. CoV infection develops asymptomatically as fever, cough, and severe shortness of breathing, nausea, vomiting and diarrhea symptoms [5]. The envelope of SARS-CoV composed of various types of proteins including envelope, membrane, nucleocapsid, replicase, spike glycoprotein, and other accuracy proteins 3a, 6, 7a and 7b [6,7,8,9,10,11,12].
Recent reports by Vankadari and Wilce, 2020 [13] and Wrapp et al., 2020 [14] showed that glycosylation occurs between the spike protein of coronaviruses and human host cells [15]. This spike protein recruits S1 (N-terminal half) and S2 (C-terminal half) fusion peptides [16] along with hemagglutinin-acetylesterase (HE) glycoprotein to interact with angiotensin-converting enzyme 2 (ACE2) [2], a surface receptor protein expressed in lungs, heart, kidneys, and intestine [17,18,19,20,21,22] cell adhesion and virulence [23, 24]. Studies also showed that the ectodomain possesses ~ 15 nM affinity towards the ACE2 receptor [23]. Various reports deduced that CoV-2 utilizes ACE2 for inserting viral particles into human cells [25,26,27,28]. After establishing a connection for sugar elements on cell membranes, the hemagglutinin–acetylesterase (HE) inserts messenger RNA and performs the replication process [29]. The World Health Organization (WHO) bulletin on COVID-19 pandemic reports 20,405,695 confirmed cases and 743,487 deaths in more than 215 countries, areas, or territories with COVID-19 cases as of 13 Aug 2020, 1.53 p.m. [30].
Plants are the main source of medication since Ancient times worldwide due to its medicinal values with less toxicity. Various scaffolds of phytochemicals including alkaloids, flavonoids, phenols, chalcones, coumarins, lignans, polyketides, alkanes, alkenes, alkynes, simple aromatics, saponins, peptides, terpenes, and steroids, are shown to exhibit a variety of medicinal properties such as antiviral, antibacterial, anticancer among others. Various diseases are being treated by plants and demonstrated to have no harmful side effects in the Ayurveda system of practice. In the drug discovery and development process, the immense therapeutic properties of plants enable researchers to utilize as starting lead molecules for developing drug-like natural molecules. Nowadays, lots of plant data repositories are available for researchers to exploit [31, 32]. For example, Kumar et al., 2018 [31] developed an indigenous plant database of Uttarakhand State, India depositing taxonomy, common names, location, medicinal uses, metabolites, interactions, targets, traditional knowledge, etc. Various studies already showed that the plant-based study could be an effective place for finding novel leads for COVID-19 drug discovery. Kumar et al. [32] screened natural metabolites against the main protease (Mpro) of COVID-19. Qamar et al. [33] performed the molecular docking and molecular dynamics simulations study of SARS-CoV-2 3CLpro target using phytochemicals. Pandey et al., 2020 carried out in silico analysis on SARS-CoV-2 spike protein using naturally occurring phytochemicals [34].
To combat the first line of infection developed due to CoV spike and ACE2 complex formation, effective strategies are needed to prevent this complex formation by developing small molecules that can target at its complex interface site. In this study, we employed virtual screening approach to screen NPACT (Naturally occurring Plant-based Anti-cancer Compound activity-Target database) compounds against HE target [32, 35] and the best-scoring molecules were validated using molecular dynamics simulations analysis to better understand the interactions and conformational changes to inhibit HE target [36, 37].
Materials and methods
Molecular data set and its preparation
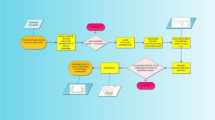
Different molecular modeling techniques were employed in this study viz, molecular docking, Absorption, Distribution, Metabolism, Excretion and Toxicity (ADMET) prediction, molecular dynamics simulations, and binding free energy calculations to obtain new leads from NPACT compounds [38]. NPACT compounds have diverse pharmacological effects with natural drug-like characteristics. These compounds possess desirable ADMET properties which leads to determining the potential drug candidates for the drug discovery process [35]. Figure 1 illustrates the workflow of this study.

Workflow of the proposed study
We selected CoV-2 hemagglutinin-acetylesterase (HE) glycoprotein as the target responsible for connecting sugar moieties (4, 9-O′-diacetyl sialic acid) on cell membranes of the receptor [39,40,41,42]. The HE acquires flexibility to bind with O-acetylated sialic acids which demolish the receptor and its membrane by working as HE fusion protein [43]. The X-ray crystal structure of Bovine coronavirus HE in complex with 4,9-O′-diacetyl sialic acid (PDB ID:3CL5; 1.80 Å resolution) was chosen as the receptor for virtual screening [2, 32]. To identify natural product-based drug leads, 1574 natural compounds were retrieved from the NPACT database [44]. These structure files were prepared using YASARA Structure (academic license) clean module such as removing crystallographic waters, adding polar hydrogens and assigning charges to titratable amino acids [45] followed by atom typing using Amber03 force field, and geometry optimization using the steepest gradient approach (100 iterations) [46, 47].
Virtual screening upon HE target
Virtual screening was performed on CoV-2 HE target receptor with 1574 NPACT compounds as ligand set using YASARA Structure package (version 19.12.14; academic license). YASARA Structure implements AutoDock Vina as the dock pose search algorithm and AMBER03 force field for scoring receptor-ligand interactions [48]. Initially, the docking procedure was validated by re-docking the co-crystal ligand of HE target 4,9-O′-diacetyl sialic acid with the 20 × 20 × 20 Å grid box size (dock site) The re-docked ligand was evaluated using root mean square deviation (RMSD) measure. The NPACT compounds were sorted based on the highest docking score to select top-scoring best compounds for further study. YASARA Structure employs its scoring function to identify best poses and high positive values indicate a better affinity of the ligand to receptor according to YASARA scoring conventions [49]. The binding energy was calculated using the following empirical equation:
where ΔGvdW = van der Waals term for docking energy; ΔGHbond = H bonding term for docking energy; ΔGelec = electrostatic term for docking energy; ΔGtor= torsional free energy term for the compound when the compound transits from unbounded to bounded state; ΔGdesolv = desolvation term for docking energy [49].
Lipinski’s rule and ADMET prediction
SwissADME webserver was used for computing ADMET properties of top-scoring compounds such as Lipinski rule of 5 [50], gastro-intestinal (GI) absorption, blood–brain barrier (BBB) permeant, cytochrome inhibition [51, 52]. The SMILES format of these compounds was submitted to SwissADME webserver.
Molecular dynamics simulations of top-scoring molecules with HE target
Molecular dynamics simulations were carried out in Desmond (Schrödinger Release 2019-3) package [48] with the best-scoring, top-five NPACT compounds of HE target as starting structures. We also performed simulations on the HE crystal structure in complex with 4,9-O′-diacetyl sialic acid (cognate ligand) to utilize it as control. The simulations were performed until 100 ns and plotted various metrics to verify whether structures were stable. For structural compliance under the Schrodinger interface, we again prepared the starting structures using Protein Preparation Wizard. The following tasks were carried out: addition of hydrogens, bond orders assignment and fill-in missing amino acid side chains and loops with optimization of hydrogen-bond assignment, and sampling of water orientations (pH 7.0). The simulation periodic box was generated using the System Builder module and solvated using a single point charge (SPC) water model [53] with Optimized Potentials for Liquid Simulations (OPLS) all-atom force field [54]. The system was minimized using the steepest descent technique for 1000 iterations. After equilibration, the unrestrained production phase was running under NPT (number of atoms, pressure and temperature were kept constant) ensemble for 100 ns at 300 K temperature and 1.01325 bar pressure. Nosé–Hoover thermostat (relaxation time = 1 ps) and the isotropic Martyna–Tobias–Klein barostat (relaxation time = 2 ps) was applied. Short-range interactions (cutoff = 9 Å) and long-range Coulombic interactions were evaluated using the smooth particle mesh Ewald (PME) method (PME) with RESPA integrator. The conformations captured in the simulation trajectories were exported at every 5 ps. After the completion of simulations, the stability of the system was assessed using RMSD, root mean square fluctuations (RMSF), Hydrogen bond analysis, radius of gyration (Rg), histogram for torsional bonds [52, 55,56,57,58].
Binding free energy calculations of top-scoring molecules with HE target
The single trajectory approach was used for calculating the binding free energy using the Molecular Mechanics/Poisson–Boltzmann Surface Area (MM/PBSA) using YASARA Structure. AMBER14 force field with APBS (Adaptive Poisson–Boltzmann Solver) was used to calculate solvation energy and treat electrostatics. 100 ns long simulation trajectory of top-five rank NPACT compounds and co-crystal ligand was supplied as input. The binding free energy was calculated using the following equations:
and
where ΔTDS is the conformation entropic contribution, and ΔGMM is the molecular mechanics interaction energy (electrostatic + van der Waals interaction) between protein and ligand. ΔGPB and ΔGSA depict the polar solvation energy and the nonpolar solvation energy, respectively [49].
Results and discussion
Virtual screening of HE target
Virtual screening exercise was carried out to identify best NPACT compounds with the potential to inhibit the CoV-2 HE glycoprotein. The docking procedure was first validated by re-docking the co-crystal ligand, 4,9-O′-diacetyl sialic acid into its binding site and calculated the RMSD between the bound and docked conformations. The best dock pose (binding energy = 5.23 kcal/mol) with RMSD of 1.84 Å was obtained which depicted the robustness of the docking procedure to develop native-like poses as close as possible. The re-docked pose h ad captured 3 H-bonds (Leu212, Ser213 and Asn214) and 4 hydrophobic (Tyr184, Phe211, Leu266 and Leu267) crystal contacts. An exhaustive search to find poses less than 1 Å RMSD was unsuccessful since most of the crystal contacts were not retained and did not secure better binding energy. This unsuccessful attempt may be attributed to 13 torsional angles with symmetrical acetyl moieties of the co-crystal ligand. This re-docking validation with acceptable dock pose-ability promoted the task to dock 1574 lead-like NPACT compounds within the binding site of the CoV-2 HE target.
We sought NPACT compounds better than co-crystal ligand in terms of binding energy and key receptor contacts (Fig. 2). The threshold for binding energy was set to 6 kcal/mol to select NPACT compounds better than co-crystal ligand (5.23 kcal/mol). The top-five compounds were chosen whose binding energy ranged between 7.22 and 7.66 kcal/mol (Table 1). The ligand efficiency of top-five compounds was also in a similar range 0.18 to 0.27 compared to co-crystal ligand 0.19 which illustrated that the binding energy contributed by per atom of the compounds is nearly similar with the need to develop key contacts with CoV-2 HE target. Table 1 indicates that compound 3,4,5-Trihydroxy-1,8-bis[(2R,3R)-3,5,7-trihydroxy-3,4-dihydro-2H-chromen-2-yl]benzo[7]annulen-6-one showed highest binding energy of 7.66 kcal/mol, followed by Silymarin, Withanolide D, Spirosolane and Oridonin. The most shared key residues which developed Hydrogen bonds, hydrophobic contacts, pi-stacks, pi-alkyl and alkyl contacts in all the top-five compounds were The 114, Thr 159, Leu 161, Ala 176, Arg 177, Tyr 184, Phe 211, Leu 212, Ser 213, Asn 214 and Leu 267. The importance of each key contact in docking and molecular dynamics simulations of top-five compounds is separately discussed in the later section.

Docked poses of a 4,9-O′-diacetyl sialic acid, b 3,4,5-Trihydroxy-1,8-bis[(2R,3R)-3,5,7-trihydroxy-3,4-dihydro-2H-chromen-2-yl]benzo[7]annulen-6-one, c Silymarin, d Withanolide D, e Spirosolane and f Oridonin
Lipinsky’s rule and ADMET prediction of top-five NPACT compounds
The ADMET properties of top-five NPACT compounds as well as the co-crystal ligand (4,9-O′-diacetyl sialic acid) were calculated using SwissADME web-service. The pharmacokinetic properties of top-five compounds were close to co-crystal ligand (Table 2). We discussed earlier that the co-crystal ligand contains 13 torsional angles in its structure, 11 rotatable bonds were noted in co-crystal ligand in comparison to 2 to 4 rotatable bonds present in top-ranking NPACT compounds representing the rigidity of the molecular structure. The Hydrogen bond acceptor (HBA) and donor (HBD) profiles of co-crystal ligand were close to only two NPACT hits, 3,4,5-Trihydroxy-1,8-bis[(2R,3R)-3,5,7-trihydroxy-3,4-dihydro-2H-chromen-2-yl]benzo[7]annulen-6-one and Silymarin. Noteworthy, the counts of HBA and HBD highlighted the ability to establish key Hydrogen bonds. It appears that the other three top-scoring NPACT compounds which achieved the top places in the rank list will be attributed to its shape and hydrophobic contacts.
The entire compound set passed the ADMET prediction (ESOL Solubility (mg/ml, GI absorption, BBB permeant, Pgp substrate, a CYP1A2 inhibitor, CYP2C19 inhibitor, CYP2C9 inhibitor, CYP2D6 inhibitor and CYP3A4 inhibitor; Table 3) with few instances of low GI absorption and low BBB permeability. This observation highlights that there is a potential need to optimize molecules by simultaneously improving target interactions and ADMET properties.
Molecular dynamics simulations of top-five ranked compounds with HE target
To investigate the dynamic properties of the HE glycoprotein with top-ranked NPACT compounds necessary for structural changes related to the inhibition mechanism, molecular dynamics simulations of HE protein target in complex with top-ranked NPACT compounds were carried out for the 100 ns time scale using Schrodinger Desmond package. The simulations of co-crystal ligand served as the control for comparative study. The stability of these six docked complexes was evaluated using protein–ligand RMSD, protein–ligand contacts, secondary structural changes, and ligand RMSF among others.
The protein–ligand RMSD plots for all too-scoring molecules showed the stability of the docked complexes attained only after 17 ns. This profile viewpoint is similar to co-crystal ligand which attained stability around 17 ns (Fig. 3). Notably, the RMSD fluctuations were ~ 3 Å for all compounds. Similar to RMSD plots, the protein RMSF fluctuations were higher in the residue index window of 120 to 170 residue positions since some of the amino acid residues present in this window were pocket residues facilitating ligand binding (Figs. S1 to S6). The secondary structure elements corresponding to pocket residues exhibited intactness in the β-sheets and loop regions (Figs. S7 to S12). Visual inspection of the fluctuating residues of RMSD plots in this window is enriched with loop elements which showed up peaks of around ~ 3 Å in its plots. The ligand RMSD plot (Fig. 4a) showed the co-crystal ligand and 3,4,5-Trihydroxy-1,8-bis[(2R,3R)-3,5,7-trihydroxy-3,4-dihydro-2H-chromen-2-yl]benzo[7]annulen-6-one experienced fluctuations of 0.3 Å to 1 Å. Other ligands did not display large deviations of RMSD values. The compactness deduced by radius of gyrations measure revealed similar notion for co-crystal ligand as well as 3,4,5-Trihydroxy-1,8-bis[(2R,3R)-3,5,7-trihydroxy-3,4-dihydro-2H-chromen-2-yl]benzo[7]annulen-6-one (Fig. 4b). Intramolecular hydrogen bonding was observed in Silymarin and Oridonin compounds (Fig. 4c) which accounted more than 30% preserved contacts in its respective simulation trajectories. The molecule surface area (MSA) of 3,4,5-Trihydroxy-1,8-bis[(2R,3R)-3,5,7-trihydroxy-3,4-dihydro-2H-chromen-2-yl]benzo[7]annulen-6-one was around 410 to 440 Å2 in most of the frames in the trajectory (Fig. 4d). The solvent accessible surface area (SASA) and polar surface area (PSA) for Silymarin was more than 500 Å2 in most of the intervals (Fig. 4e, f). The 3,4,5-Trihydroxy-1,8-bis[(2R,3R)-3,5,7-trihydroxy-3,4-dihydro-2H-chromen-2-yl]benzo[7]annulen-6-one was better-docked in the HE glycoprotein target whereas Silymarin did not developed strong contacts even its PSA did not contribute strong hydrogen bonding and often are solvent exposed in most of the frames.

RMSD plot of HE target with co-crystal ligand and top-five NPACT compounds as a function of simulation time. a 4,9-O′-diacetyl sialic acid, b 3,4,5-Trihydroxy-1,8-bis[(2R,3R)-3,5,7-trihydroxy-3,4-dihydro-2H chromen-2-yl]benzo[7]annulen-6-one, c Silymarin, d Withanolide D, e Spirosolane and f Oridonin. Color legends: Cα (blue color), side chains (green color), heavy atoms (yellow color), ligand with protein (dark pink color), ligand with ligand (pink color)

Various measures of the molecular dynamics simulations of co-crystal ligand and top-five NPACT compounds with HE target. a RMSD, b rGyr, c intra HB, d MolSA, e SASA and f PSA. Color legends: 4,9-O′-diacetyl sialic acid (yellow color), 3,4,5-Trihydroxy-1,8-bis[(2R,3R)-3,5,7 trihydroxy-3,4-dihydro-2H-chromen-2yl]benzo[7]annulen-6-one (magenta color), Silymarin (green color), Withanolide D (purple color), Spirosolane (orange color) and Oridonin (blue color)
Preservation of intermolecular contacts in molecular dynamics simulations
Crystal structure of HE with 4,9-O′-diacetyl sialic acid revealed that its ligand-binding site is composed of two adjacent hydrophobic pockets to accommodate 5-N-acetyl and 9-O′-acetyl moieties. The 5-N-acetyl group is held tightly by creating a hydrogen bond with Leu 212 residue whereas 9-O′-acetyl group is held close to Tyr 184 residue with strong hydrophobic contacts. It is well-established that the 9-O′-acetyl moiety is vital for receptor binding and acts as a switch for visual particle attachment. Two water-bridges and other two hydrogen bonding centers were also present. These include threonines at 114th and 215th position (water-mediated contacts), Ser 213 and Asn 214 (hydrogen bonds). Figure 5 plots the different types of intermolecular interactions (hydrogen bond, hydrophobic and water bridges) made by each pocket residue with its bound ligand. The 2D interaction maps of re-docked and top-five NPACT molecules depicting the preservation of contacts throughout the simulation trajectory is given in Fig. 6.

Various intermolecular interactions made by HE pocket residues with co-crystal ligand and top-five NPACT compounds, captured during molecular dynamics simulations. a 4,9-O′-diacetyl sialic acid, b 3,4,5-Trihydroxy-1,8-bis[(2R,3R)-3,5,7-trihydroxy-3,4-dihydro-2H-chromen-2-yl]benzo[7]annulen-6-one, c Silymarin, d Withanolide D, e Spirosolane and f Oridonin. Bar colors: hydrogen bond (green), hydrophobic contacts (purple) and water-bridge (blue)

Preserved contacts of co-crystal ligand and top-five NPACT compounds with HE target, captured during molecular dynamics simulations. a 4,9-O′-diacetyl sialic acid, b 3,4,5-Trihydroxy-1,8-bis[(2R,3R)-3,5,7-trihydroxy-3,4-dihydro-2H-chromen-2-yl]benzo[7] annulen-6-one, c Silymarin, d Withanolide D, e Spirosolane and f Oridonin
The co-crystal ligand, 4,9-O′-diacetyl sialic acid preserved almost all entire set of crystal contacts viz. Leu 212 (5-N-acetyl moiety, 98%), Asn 214 (Carboxylate group, 76%), Thr 215 (water-bridges, 56%). Ser 213 preferred to develop hydrogen bond with terminal carboxylate group (98%) instead of hydroxyl group attached at 6th carbon atom of sialic acid. Two new contacts were established which were noteworthy. Thr 215 developed water-mediated hydrogen bond with 4-N-acetyl moiety due to its proximity to carboxylate group (53%). Arg 177 also developed hydrogen bond directly or through water bridges in certain frames (Figs. 5a and 6a). All these contacts were indeed captured in the dock pose (Fig. 2a) boosting the robustness of the docking procedure. The best-scoring NPACT molecule, 3,4,5-Trihydroxy-1,8-bis[(2R,3R)-3,5,7-trihydroxy-3,4-dihydro-2H-chromen-2yl]benzo[7]annulen-6-one developed π-stack with Phe 211 residue (5-N-acetyl pocket) and π-alkyl contact with Leu 267 (9-O′-acetyl pocket) in its dock pose (Fig. 2b). The former crystal contact was preserbed in 37% of the time scale whereas the latter contact was traced below 30% (Figs. 5b, 6b). Surprisingly, the new hydrogen bond contact of Lys 163 generated by docking was lost in the dynamics which replaced by a new hydrogen bond by Ile 175 (39%).
Silymarin in its dock conformation created two crystal contacts (π-stack with Phe 211 and hydrogen bond with Thr 114) and the new contacts (π-stack by Phe 245 and hydrogen bond with Ser 116 and Arg 177) (Fig. 2c). As discussed above in the intramolecular hydrogen bonding measure, Silymarin developed intramolecular hydrogen bond between its hydroxyl and carbonyl group in 33% of the simulation time. Although absent in its dock pose, three crystal contacts by Leu 212, Asn 214 and Thr 215 residues mediating direct hydrogen bond and through water bridges were noted in the histogram (Figs. 5c and 6c). It should be accounted more than 30% of the frames captured in the simulation trajectory. Withanolide D made a new hydrogen bond contact with Arg 177 residues in its dock pose (Fig. 2d) which were lost during the dynamics simulation. The interaction histogram plot found Phe 211 residue forming hydrophobic contacts. Either of any crystal contacts or new contacts were captured in more than 30% of the frame. It is evident that Withanolide D surface is hydrophobically enclosed in the ligand-binding site. A new hydrophobic interaction by Phe 207 were also observed (< 30%) (Figs. 5d, 6d).
Spirosolane developed a sole π-alkyl crystal contact with Phe 211 (5-N-acetyl pocket) in its docking calculations along with two new alkyl contacts with Leu 161 and Ala 176 (Fig. 2e). Fortunately, a water-bridges was formed with Tyr 184 residue (9-O′-acetyl pocket) in 36% frames. Two hydrophobic crystal contacts (Tyr 184 and Phe 211) were plotted in the histogram in addition to two new contacts (Arg 177: hydrogen bond and hydrophobic; Phe 207-hydrophobic) (Figs. 5e and 6e). Similar to Spirosolane, Oridonin established only a π-alkyl crystal contact with Phe 211 residue (Fig. 2f). It also developed two intramolecular hydrogen bonds with its hydroxyl and carbonyl group (hydroxyl-carbonyl hydrogen bond: 89%; hydroxyl–hydroxyl hydrogen bond: 36%). A combination of new contacts with varied interaction types were observed viz hydrogen bond (Tyr 55), water-bridges (Arg 53, Tyr 55), and hydrophobic contacts (Tyr 55, Tyr 343). Crystal hydrogen bond with Leu 212 were also noted. Finally, the comparison of contacts developed in the dock pose and subsequent preservation in the dynamics simulation provides strong evidence about the optimal binding of the NPACT top-scoring compounds. Indeed, selected NPACT compounds retained certain crystal contacts in addition new contacts (Fig. S13).
Binding free energy calculations of selected NPACT compounds with HE target
The free energy of binding of top-five NPACT compounds and HE cognate ligand was computed using MM/PBSA approach. The simulation trajectory with 100 ns time scale was supplied to YASARA structure binding energy macro. The cognate ligand, 4,9-O′-diacetyl sialic acid secured in − 38.56 kJ/mol whereas the 3,4,5-Trihydroxy-1,8-bis[(2R,3R)-3,5,7-trihydroxy-3,4-dihydro-2H-chromen-2-yl]benzo[7]annulen-6-one obtained the best binding for energy of − 61.088 kJ/mol (Fig. 7). This observation highlights that 3,4,5-Trihydroxy-1,8-bis[(2R,3R)-3,5,7-trihydroxy-3,4-dihydro-2H-chromen-2-yl]benzo[7]annulen-6-one developed better crystal contacts on par with cognate ligand along with new contacts resulting in securing the top rank energy among the selected NPACT compounds. If also 3,4,5-Trihydroxy-1,8-bis[(2R,3R)-3,5,7-trihydroxy-3,4-dihydro-2H-chromen-2-yl]benzo[7]annulen-6-one contributes for creating a new ligand binding hypothesis: π-stack with Phe 211 residues and π-alkyl or broadly hydrophobic contacts in 5-N-acetyl and 9-O′-diacetyl pockets, respectively. Hydrogen bonding with Leu 212 proved to be beneficial. Oridoin ranked second place with a binding energy of − 57.144 kJ/mol simply due to new contacts generated both during docking and dynamics simulations. Spirosolane and Silymarin occupied the next two rank positions. Withanolide D obtained a positive binding energy of 22.85 kJ/mol implying no affinity according to MM/PBSA scoring. It is related to the absence of any dominant intermolecular contacts with HE target compared to other selected NPACT compounds.

Binding free energy calculations of co-crystal ligand and top-five NPACT compounds with HE target. The co-crystal ligand of HE target, 4,9-O′-diacetyl sialic acid, is shown in orange color bar. The NPACT compounds are represented in blue colour bars
Conclusion
The absence of an effective therapeutic drug or vaccine had already aggrevated the situation of COVID-19 pandemic outbreak. Since the first line of viral invasion and infection is facilitated by prominent interaction between Human ACE2 and CoV-2 HE proteins, targeting the HE glycoprotein receptors forms the primary strategy to develop effective inhibitors guided by computer-aided drug design approaches. This study prioritizes the lead compounds against HE complex from NPACT natural compound repository using a hierarchical procedure of virtual screening, molecular dynamics simulations and binding free energy calculations. 3,4,5-Trihydroxy-1,8-bis[(2R,3R)-3,5,7-trihydroxy-3,4-dihydro-2H-chromen-2yl]benzo[7]annulen-6-one, Silymarin, Withanolide D, Spirosolane and Oridonin were the promising natural compounds with better binding affinity profile. Molecular dynamics simulations were performed for all the docked complexes for 100 ns time scale which indicated the structural stability of the protein–ligand complexes. Various assessment measures such as RMSD, RMSF, radius of gyration, surface area measures collectively supported the structural stability of the complexes. Moreover, crystal contacts of cognate ligand were also retained in few NPACT compounds in its simulations trajectories which illustrated that NPACT compounds have a similar binding pattern of 4,9-O′-diacetyl sialic acid. Furthermore, MM/PBSA calculations were employed to calculate the binding free energy of top-five scoring NPACT compounds revealing that a better interactions at two adjacent hydrophobic binding pockets of ligand-binding aid in enhancing the binding free energy while ensuring key receptor contacts. Although this study provides an important insights in the preliminary level of HE drug designing, the ligand-binding hypothesis deduced by molecular docking and molecular dynamics simulations can be utilized to search and optimize natural molecules which requires experimental validation through in vitro and in vivo studies.
References
Pene F, Merlat A, Vabret A, Rozenberg F, Buzyn A, Dreyfus F, Cariou A, Freymuth F, Lebon P (2003) Coronavirus 229E-related pneumonia in immunocompromised patients. Clin Infect Dis 37(7):929–932
Zeng Q, Langereis MA, van Vliet AL, Huizinga EG, de Groot RJ (2008) Structure of coronavirus hemagglutinin-esterase offers insight into corona and influenza virus evolution. Proc Natl Acad Sci 105(26):9065–9069
Coleman CM, Frieman MB (2014) Coronaviruses: important emerging human pathogens. J Virol 88(10):5209–5212
Zaki AM, Van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA (2012) Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367(19):1814–1820
Porcheddu R, Serra C, Kelvin D, Kelvin N, Rubino S (2020) Similarity in case fatality rates (CFR) of COVID-19/SARS-COV-2 in Italy and China. J Infect Dev Ctries 14(02):125–128
Huang C, Ito N, Tseng C-TK, Makino S (2006) Severe acute respiratory syndrome coronavirus 7a accessory protein is a viral structural protein. J Virol 80(15):7287–7294
Huang C, Peters C, Makino S (2007) Severe acute respiratory syndrome coronavirus accessory protein 6 is a virion-associated protein and is released from 6 protein-expressing cells. J Virol 81(10):5423–5426
Schaecher SR, Mackenzie JM, Pekosz A (2007) The ORF7b protein of severe acute respiratory syndrome coronavirus (SARS-CoV) is expressed in virus-infected cells and incorporated into SARS-CoV particles. J Virol 81(2):718–731
Shen S, Lin P-S, Chao Y-C, Zhang A, Yang X, Lim SG, Hong W, Tan Y-J (2005) The severe acute respiratory syndrome coronavirus 3a is a novel structural protein. Biochem Biophys Res Commun 330(1):286–292
Nieto-Torres JL, DeDiego ML, Álvarez E, Jiménez-Guardeño JM, Regla-Nava JA, Llorente M, Kremer L, Shuo S, Enjuanes L (2011) Subcellular location and topology of severe acute respiratory syndrome coronavirus envelope protein. Virology 415(2):69–82
Peiris J, Lai S, Poon L, Guan Y, Yam L, Lim W, Nicholls J, Yee W, Yan W, Cheung M (2003) Coronavirus as a possible cause of severe acute respiratory syndrome. The Lancet 361(9366):1319–1325
Chu D, Peiris J, Chen H, Guan Y, Poon LL (2008) Genomic characterizations of bat coronaviruses (1A, 1B and HKU8) and evidence for co-infections in Miniopterus bats. J Gen Virol 89(5):1282–1287
Vankadari N, Wilce JA (2020) Emerging COVID-19 coronavirus: glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerg Microbes Infect 9(1):601–604
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, Abiona O, Graham BS, McLellan JS (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367(6483):1260–1263
Xiong X, Tortorici MA, Snijder J, Yoshioka C, Walls AC, Li W, McGuire AT, Rey FA, Bosch B-J, Veesler D (2018) Glycan shield and fusion activation of a deltacoronavirus spike glycoprotein fine-tuned for enteric infections. J Virol 92(4):e01628-01617
Sainz B, Rausch JM, Gallaher WR, Garry RF, Wimley WC (2005) Identification and characterization of the putative fusion peptide of the severe acute respiratory syndrome-associated coronavirus spike protein. J Virol 79(11):7195–7206
Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N et al (2000) A novel angiotensin converting enzyme related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res 87:E1–E9
Zhang H, Kang Z, Gong H, Xu D, Wang J, Li Z, Cui X, Xiao J, Meng T, Zhou W (2020) The digestive system is a potential route of 2019-nCov infection: a bioinformatics analysis based on single-cell transcriptomes. BioRxiv
Wan Y, Shang J, Graham R, Baric RS, Li F (2020) Information: read & look at all below!
Hofmann H, Hattermann K, Marzi A, Gramberg T, Geier M, Krumbiegel M, Kuate S, Überla K, Niedrig M, Pöhlmann S (2004) S protein of severe acute respiratory syndrome-associated coronavirus mediates entry into hepatoma cell lines and is targeted by neutralizing antibodies in infected patients. J Virol 78(12):6134–6142
Hofmann H, Pyrc K, van der Hoek L, Geier M, Berkhout B, Pöhlmann S (2005) Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc Natl Acad Sci 102(22):7988–7993
Hofmann H, Simmons G, Rennekamp AJ, Chaipan C, Gramberg T, Heck E, Geier M, Wegele A, Marzi A, Bates P (2006) Highly conserved regions within the spike proteins of human coronaviruses 229E and NL63 determine recognition of their respective cellular receptors. J Virol 80(17):8639–8652
Song W, Gui M, Wang X, Xiang Y (2018) Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog 14(8):e1007236
Millet JK, Kien F, Cheung C-Y, Siu Y-L, Chan W-L, Li H, Leung H-L, Jaume M, Bruzzone R, Peiris JSM (2012) Ezrin interacts with the SARS coronavirus Spike protein and restrains infection at the entry stage. PLoS One 7(11):e49566
Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, Si H-R, Zhu Y, Li B, Huang C-L (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579(7798):270–273
Walls AC, Park Y-J, Tortorici MA, Wall A, McGuire AT, Veesler D (2020) Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181:281–292.e6
Letko M, Marzi A, Munster V (2020) Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol 5(4):562–569
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N-H, Nitsche A (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181:271–280
Holmes KV (2003) SARS-associated coronavirus. N Engl J Med 348(20):1948–1951
Homepage WC (2020) https://covid19.who.int/. Accessed 13 Aug 2020
Kumar A, Kumar R, Sharma M, Kumar U, Gajula M, Singh KP (2018) Uttarakhand medicinal plants database (UMPDB): a platform for exploring genomic, chemical, and traditional knowledge. Data 3(1):7
Kumar A, Choudhir G, Shukla SK, Sharma M, Tyagi P, Bhushan A, Rathore M (2020) Identification of phytochemical inhibitors against main protease of COVID-19 using molecular modeling approaches. J Biomol Struct Dyn (just-accepted):1–21
ul Qamar MT, Alqahtani SM, Alamri MA, Chen L-L (2020) Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J Pharm Anal
Pandey P, Rane JS, Chatterjee A, Kumar A, Khan R, Prakash A, Ray S (2020) Targeting SARS-CoV-2 spike protein of COVID-19 with naturally occurring phytochemicals: an in silico study for drug development. J Biomol Struct Dyn:1–11
Mangal M, Sagar P, Singh H, Raghava GP, Agarwal SM (2013) NPACT: naturally occurring plant-based anti-cancer compound-activity-target database. Nucl Acids Res 41(D1):D1124–D1129
Patel CN, Kumar SKP, Pandya HA, Modi KM, Patel DP, Gonzalez FJ (2017) Retrieval of promiscuous natural compounds using multiple targets docking strategy: a case study on kinase polypharmacology. In: 2017 IEEE international conference on bioinformatics and biomedicine (BIBM). IEEE, pp 288–291
Kumar SP, Patel CN, Jha PC, Pandya HA (2017) Molecular dynamics-assisted pharmacophore modeling of caspase-3-isatin sulfonamide complex: recognizing essential intermolecular contacts and features of sulfonamide inhibitor class for caspase-3 binding. Comput Biol Chem 71:117–128
Patel CN, Georrge JJ, Modi KM, Narechania MB, Patel DP, Gonzalez FJ, Pandya HA (2018) Pharmacophore-based virtual screening of catechol-o-methyltransferase (COMT) inhibitors to combat Alzheimer’s disease. J Biomol Struct Dyn 36(15):3938–3957
Organization WH (2020) Coronavirus disease (COVID-2019) situation reports (https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports/). Copyright© 2020 Massachusetts Medical Society. Accessed 13 Aug 2020
Wagner R, Matrosovich M, Klenk HD (2002) Functional balance between haemagglutinin and neuraminidase in influenza virus infections. Rev Med Virol 12(3):159–166
Yuan P, Thompson TB, Wurzburg BA, Paterson RG, Lamb RA, Jardetzky TS (2005) Structural studies of the parainfluenza virus 5 hemagglutinin-neuraminidase tetramer in complex with its receptor, sialyllactose. Structure 13(5):803–815
De Groot RJ (2006) Structure, function and evolution of the hemagglutinin-esterase proteins of corona-and toroviruses. Glycoconj J 23(1–2):59–72
Tortorici MA, Walls AC, Lang Y, Wang C, Li Z, Koerhuis D, Boons G-J, Bosch B-J, Rey FA, de Groot RJ (2019) Structural basis for human coronavirus attachment to sialic acid receptors. Nat Struct Mol Biol 26(6):481–489
Chen Y, de Bruyn Kops C, Kirchmair J (2017) Data resources for the computer-guided discovery of bioactive natural products. J Chem Inf Model 57(9):2099–2111
Kumar SP, Patel CN, Rawal RM, Pandya HA (2020) Energetic contributions of amino acid residues and its cross-talk to delineate ligand-binding mechanism. Proteins: Struct Funct Bioinform 88:1207–1225
Krieger E, Darden T, Nabuurs SB, Finkelstein A, Vriend G (2004) Making optimal use of empirical energy functions: force-field parameterization in crystal space. Proteins: Struct Funct Bioinform 57(4):678–683
Patel CN, Narechania MB (2018) Targeting epidermal growth factor receptors inhibition in non-small-cell lung cancer: a computational approach. Mol Simul 44(17):1478–1488
Patel CN, Kumar SP, Modi KM, Soni MN, Modi NR, Pandya HA (2019) Cardiotonic steroids as potential Na+/K+-ATPase inhibitors—a computational study. J Recept Signal Transduction 39(3):226–234
Krieger E, Vriend G (2015) New ways to boost molecular dynamics simulations. J Comput Chem 36(13):996–1007
Lipinski CA (2004) Lead-and drug-like compounds: the rule-of-five revolution. Drug Discov Today: Technol 1(4):337–341
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7:42717
Karim A, Mishra A, Newton MH, Sattar A (2019) Efficient toxicity prediction via simple features using shallow neural networks and decision trees. ACS Omega 4(1):1874–1888
Babu S, Nagarajan SK, Madhavan T (2019) Understanding the structural features of JAK2 inhibitors: a combined 3D-QSAR, DFT and molecular dynamics study. Mol Divers 23(4):845–874
Meshram RJ, Bagul KT, Aouti SU, Shirsath AM, Duggal H, Gacche RN (2020) Modeling and simulation study to identify threonine synthase as possible drug target in Leishmania major. Mol Divers:1–22
Ahmad S, Raza S, Uddin R, Azam SS (2017) Binding mode analysis, dynamic simulation and binding free energy calculations of the MurF ligase from Acinetobacter baumannii. J Mol Graph Model 77:72–85
Tietze D, Kaufmann D, Tietze AA, Voll A, Reher R, König G, Hausch F (2019) Structural and dynamical basis of G protein inhibition by YM-254890 and FR900359: an inhibitor in action. J Chem Inf Model 59(10):4361–4373
Bhoi MN, Borad MA, Jethava DJ, Acharya PT, Pithawala EA, Patel CN, Pandya HA, Patel HD (2019) Synthesis, biological evaluation and computational study of novel isoniazid containing 4H-Pyrimido [2, 1-b] benzothiazoles derivatives. Eur J Med Chem 177:12–31
Gmelch TJ, Sperl JM, Sieber V (2020) Molecular dynamics analysis of a rationally designed aldehyde dehydrogenase gives insights into improved activity for the non-native cofactor NAD+. ACS Synth Biol 9:920–929
Acknowledgements
This work is supported by the Financial Assistance Programme—Department of Science and Technology [Grant No. GSBTM/MD/JDR/1409/2017-18] and Gujarat Council on Science and Technology [GUJCOST/Supercomputer/2019-20/1359]. The authors gratefully acknowledge the Department of Botany, Bioinformatics and Climate Change Impacts Management, Gujarat University for providing an opportunity to access the bioinformatics research facilities.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Patel, C.N., Kumar, S.P., Pandya, H.A. et al. Identification of potential inhibitors of coronavirus hemagglutinin-esterase using molecular docking, molecular dynamics simulation and binding free energy calculation. Mol Divers 25, 421–433 (2021). https://doi.org/10.1007/s11030-020-10135-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-020-10135-w