
Abstract
Many cancers of neuroendocrine origin overexpress cholecystokinin-2 receptors (CCK-2R) including medullary thyroid cancer, small cell lung cancer and other lung carcinoids. Fluorine-18 labelled peptides targeting CCK-2R enable direct visualization and quantification of this receptor in vivo using positron emission tomography imaging. CP04 1 and MG11 2 are two previously described truncated peptides derived from the native CCK-2R hormone ligand, gastrin. The N-terminus of the MG11 2 octopeptide was chemically modified with various fluorine containing aromatic (4-fluorobenzoate), heterocyclic (6-fluoronicotinate) and aliphatic (2-fluoropropionate) moieties. To assess the impact these modifications had on CCK-2R binding, ligand-binding assays were conducted using A431 cells overexpressing human CCK-2R. MG11 2 modified by 4-fluorobenzoate (FB-MG11 3) demonstrated the highest binding affinity (0.20 nM) followed by MG11 2 modified by 6-fluoronicotinate (FNic-MG11 4; 0.74 nM) and 2-fluoropropionate (FP-MG11 5; 1.80 nM), respectively. Whilst indirect labelling of MG11 2 using fluorine-18 labelled activated esters of fluorobenzoate and 6-fluoronicotinate was unsuccessful, direct fluorine-18 labelling at the N-terminus modified with 6-nitronicotinate afforded a 47.6% radiochemical yield of [18F]FNic-MG11. Unfortunately, [18F]FNic-MG11 4 was chemically unstable, decomposing slowly through defluorination, thereby impeding any further work with this radiotracer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Theranostic radiopharmaceuticals are key to the realization of personalized medicine in oncology, leading to high therapeutic efficacy with low toxicity in patients with cancer (Hicks et al. 2017; Hofman et al. 2018; Turner 2018). Theranostic radiopharmaceuticals use the same targeting agent, often a peptide, for both diagnosis and therapy, contingent upon the nature of the incorporated radionuclide. Diagnostic radiopharmaceuticals labelled with positron emitting radionuclides such as fluorine-18 (F-18) can be used for positron emission tomography (PET) imaging that enables quantification of receptor density and drug interactions in vivo with high sensitivity (pico- to nano-molar range) (Vallabhajosula et al. 2011). The diagnostic scan stratifies patients according to their likelihood of benefiting from targeted radionuclide therapy. Only patients demonstrating high tumour uptake and suitable biodistribution of the diagnostic radiopharmaceutical are advanced to receive a therapeutic dose that uses the same targeting agent radiolabeled with a cytotoxic radionuclide, such as the beta-emitting lutetium-177 (Lu-177) (Rodrigues et al. 2021).
Cholecystokinin receptor subtype-2 (CCK-2R) is an important G protein-coupled receptor (GPCR) overexpressed in many types of neuroendocrine cancers including medullary thyroid carcinoma (92% incidence), small cell lung cancer (57% incidence), astrocytoma (65% incidence) and stromal ovarian cancer (100% incidence). Accordingly, significant efforts have been made to develop theranostic radiopharmaceuticals to image and treat cancers that overexpress CCK-2R (Rangger et al. 2017; Reubi et al. 1997). Under normal physiological conditions, CCK-2R preferentially binds the peptide hormone gastrin in the stomach, but can also bind with high affinity to the related hormone, cholecystokinin. Both hormones share the same C-terminally amidated pharmacophore (Trp-Met-Asp-Phe-NH2) that binds to CCK-2R, but they differ in the N-terminal sequence, with the pentaglutamic acid sequence of gastrin absent in cholecystokinin. A variety of peptides derived from these two CCK-2R ligands have been labelled with radionuclides and evaluated as CCK-2R targeting radiopharmaceuticals (Tornesello et al. 2011). Radiolabeled peptides derived from gastrin demonstrated improved tumour targeting in comparison to those derived from cholecystokinin (Laverman et al. 2011).
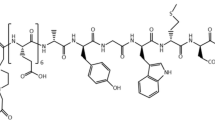
Radiolabelled derivatives of a CCK-2R targeting peptide derived from gastrin, CP04 1 (Fig. 1) have undergone successful clinical trials for imaging neuroendocrine tumours. These trials demonstrated that these radiolabeled peptides possessed high receptor affinity (IC50 in the sub-nM to low nM range), good cellular internalization and relative in vivo stability leading to high tumour to background ratios (Erba et al. 2018). We have recently developed ‘foldamer’ peptides based on CP04 1 that show significant improvements in tumour targeting and metabolic stability in comparison to CP04 1 (Corlett et al. 2021). Unfortunately, both CP04 1 and its analogues contain the highly ionic N-terminal hexa-glutamic acid sequence that results in undesired renal accumulation that when radiolabeled with a therapeutic radionuclide could lead to significant nephrotoxicity. Therefore, truncated analogues of CP04 1 have also been investigated, including the peptide MG11 2 that lacks all N-terminal glutamic acid residues. This shortened peptide displayed significantly reduced renal uptake and retention, spurring additional refinements of the core MG11 2 structure to further reduce kidney uptake and enhance tumor targeting (Erba et al. 2018; Grob et al. 2020; Hörmann et al. 2020; Klingler et al. 2019; Maximilian et al. 2020; Uprimny et al. 2020).

Chemical structures of unmodified CP04 1 and MG11 2
MG11 2 has been radiolabeled with different gamma-emitting radionuclides (Tc-99m, Lu-177 and In-111) for single-photon emission computed tomography (SPECT) imaging (Aloj et al. 2011; Ocak et al. 2011; von Guggenberg et al. 2009), as well as with the short-lived positron-emitting radionuclide Ga-68 for PET imaging (Summer et al. 2018). Whilst the superior resolution of PET imaging makes this the modality of choice compared to SPECT, logistical issues with Ga-68 (half-life: 68 min) mean that a longer lived more readily available PET radionuclide is a more feasible option. Fluorine-18 (half-life: 110 min) is the workhorse radionuclide for PET imaging as it can be generated in large amounts using low energy cyclotrons and importantly, it exhibits ideal physical properties. To date, an F-18 labelled MG11 2 analogue has yet got to be evaluated for PET imaging. Herein, we aim to develop fluorine-18 labelled MG11-based radiopharmaceuticals for evaluation as PET imaging agents.
Materials and Methods
General Methods and Reagents
Reagents and solvents were purchased in their highest purified condition from Sigma–Aldrich, Merck (Germany), Macrocyclics, Fluka and NovaBiochem (Germany). Purified water was obtained from an in-house Millipore water purification system. Amino acids used were purchased from GL Biochem (China) and ChemPep (USA). Anhydrous solvents were used in peptide synthesis. Anhydrous ether, dichloromethane (DCM), and triethylamine (TEA) were obtained from Sigma Aldrich. The automated peptide synthesizer (CEM Liberty Blue, John Morris Group, Victoria, Australia) was used to synthesize all peptides described in this research using standard Fmoc-based solid-phase peptide synthesis techniques. N,N,N-trimethyl-5-((2,3,5,6-tetrafluorophenoxy)carbonyl)pyridin-2-aminium bromide 6 (TFP-NMe4) was prepared as previously reported (Haskali et al. 2020; Olberg et al. 2010).
Peptides were purified using the preparative HPLC system (Agilent 1200 series) and using the Column; C18 150 mm × 21.2 mm, 5 µm 100 Å Phenomenex kinetex AXIA. Quality control analysis of the isolated peptides was performed using an analytical HPLC system (Agilent 1100 series) using a C18 150 mm × 4.6 mm 5 µm 130A Hypersil BDS column. Mass spectra were recorded on an Orbitrap high-resolution electrospray ionization mass spectrometer (HR–ESI–MS) (MSPF orbiTRAP infusion).
Radioactivity measurements were carried out with a CRC-15PET dose calibrator (Capintec) that was calibrated daily using Cs-137 and Co-57 sources (Isotope Products Laboratories). Radiation was detected using a solid state photodiode scintillator crystal detector (Knauer). Preparative high performance liquid radiochemical chromatography (HPLC) was performed using a Knauer 1050 pump, 2500 UV detector, and 5050 manager. Radiation was detected using a Knauer solid state photodiode scintillation crystal detector in a TO-5 case. Analytical HPLC was performed using a Shimadzu HPLC system consisting of a SCL-10AVP system controller, SIL-0ADVP auto-injector, LC-10 ATVP solvent delivery unit, CV-10AL control valve, DGU-14A degasser, and SPD-10AVPV detector. This was coupled to a radiation detector consisting of an Ortec model 276 photomultiplier base with a 925-SCINTACE-mate preamplifier, amplifier, bias supply, and SCA and a Bicron 1M11/2 photomultiplier tube. Radiochemical LC–MS analysis was performed on a Shimdzu LCMS-8030 model.
Peptide Synthesis
All peptides were synthesized using Rink amide resin (ChemPep-Florida, loading capacity 0.8 mmol/g, bead size 100–200 mesh) as solid support. Peptide synthesis was performed on a 0.1 mmol scale with a five fold molar excess of amino acids (0.2 M) on a CEM Liberty Blue module. DIC (5 eq) and Oxyma pure (5 eq) were used for amino acid coupling. Coupling to the Nα-deprotected peptide-resin was performed under microwave irradiation for 3 min (30 W, 90 °C). Fmoc deprotection was performed using a 0.1 M Oxyma pure solution containing 20% (v/v) piperidine in N,N-dimethylformamide (DMF) under microwave irradiation for 30 s (40 W, 40 °C). The N-terminus of MG11 2 was capped with 2-fluoropropionic acid, 4-fluorobenzoic acid, 6-fluoronicotinic acid or 6-nitronicotinic acid to afford the respective non-radioactive fluorine containing peptides. Fully assembled peptides on resin were transferred into a syringe fitted with frit and washed three times with DMF and DCM respectively. A cleavage cocktail was prepared (8 mL/peptide) consisting of 5% water, 5% thioanisole, 2.5% DODT, and 87.5% TFA in a falcon tube and drained into the syringe vessel containing the peptide on dried resin beads. This mixture was mixed for 2–3 h on an orbital shaker. The solution was then drained into a falcon tube and TFA was removed by purging with nitrogen for ~ 20 min or until 1/3 of the total volume remained. The peptide was triturated using ~ 30 mL of diethyl ether before being centrifuged at 3500 rpm for ~ 5 min. This process was repeated three times. The precipitate was purged with nitrogen to remove the remaining diethyl ether, leaving a dried pellet of peptide. The crude peptide was then suspended in 50% MeCN:H2O solution and immediately analyzed by analytical HPLC and mass spectrometry to assess synthesis efficacy and confirm product formation. The sample was then lyophilized and subsequently purified on a preparative HPLC system. The pure fractions were combined and lyophilized.
Peptide Preparative HPLC Conditions
The peptides were purified using the preparative column described above, eluted at 8 mL/min with a gradient: 0.1% (v/v) TFA, starting at 0% MeCN for 1 min, increased to 85% over 30 min and then increased and maintained at 100% MeCN for another 10 min. Peptides were detected at 220 nm wavelength.
Peptide Analytical HPLC Conditions
The peptides were analyzed using the analytical column described above, eluted at 1 mL/min with a gradient: 0.1% (v/v) TFA, starting at 0% MeCN, increased to 100% over 30 min and then increased and maintained at 100% MeCN for another 10 min. Peptides were detected at 220 nm wavelength.
Radiofluorination
No-carrier-added (NCA) [18F]fluoride was obtained from a PETtrace 16.5 MeV cyclotron (Cyclotek Pty. Ltd.) incorporating a high pressure niobium target via the 18O(p,n)18F nuclear reaction (98% 18O isotopic enrichment). Radiochemical synthesis was performed on an iPHASE Flexlab radiochemistry module purchased from iPHASE Technologies Pty. Ltd. Both 4-nitrophenyl 4-[18F]fluorobenzoate 7 ([18F]7) and 4-nitrophenyl 6-[18F]fluoronicotinate 8 ([18F]8) were prepared as previously reported (Haskali et al. 2020) and described in brief below. [18F]Fluoride was recovered and processed using Sep-Pak Accell Plus QMA Plus Light Cartridge, 130 mg Sorbent per Cartridge, 37–55 µm.
4-Nitrophenyl 4-[ 18 F]Fluorobenzoate [ 18 F]7
[18F]Fluoride was recovered on a QMA solid phase extraction cartridge and released using a solution of potassium hydrogen carbonate (2–3 mg, 20–30 µmol) and kryptofix 222 (8–10 mg, 21–27 µmol) in milli-Q H2O:MeCN (1 mL, 30:70) into the reaction vessel. K222.K[18F]F complex was then azeotropically dried on the iPHASE Flexlab radiochemistry module using a stream of nitrogen and vacuum at 80 °C for 5 min followed by vacuum only at 80 °C for another 2 min. To the dried K222.K[18F]F complex, 4-(methoxy)phenyl(4-(4-nitrophenoxycarbonyl)phenyliodonium tosylate 9 (20 mg, 42 μmol) in DMSO:t-amyl alcohol (0.4:0.6, 1 mL) was added and the mixture was heated to 100 °C for 5 min. The reaction mixture was then diluted in 0.05% TFA H2O: MeCN (1.3:0.2, 3.0 mL) and purified by preparative HPLC to afford the title compound (564–762 MBq, isolated yield > 40% decay corrected to start of synthesis (SOS)).
4-Nitrophenyl 6-[ 18 F]Fluoronicotinate [ 18 F]8
[18F]Fluoride was recovered on a QMA solid phase extraction cartridge and released into the reaction vessel using a solution of potassium hydrogen carbonate (2–3 mg, 20–30 µmol) and kryptofix 222 (8–10 mg, 21–27 µmol) in milli-Q H2O:MeCN (1 mL, 30:70). The K222.K[18F]F complex was then azeotropically dried on the iPHASE Flexlab radiochemistry module. To the dried K222.K[18F]F complex, N,N,N-trimethyl-5-((4-nitrophenoxy)carbonyl)pyridin-2-aminium bromide 10 (10 mg, 33 μmol) in DMSO:t-amyl alcohol (0.4:0.6, 1 mL) was added and the mixture was heated to 100 °C for 5 min. The reaction mixture was then diluted in 0.05% TFA H2O:MeCN (1.3:0.2, 3.0 mL) and purified by preparative HPLC to afford the title compound (555–792 MBq, isolated yield > 50% decay corrected to start of synthesis (SOS)).
Attempted Radiosynthesis of [ 18 F]FB-MG11 3 and [ 18 F]FNic-MG11 4 from Synthons [ 18 F]7 and [ 18 F]8 Respectively
[18F]7 or [18F]8 (0.37–3.70 GBq) in DMSO (100–300 µL) containing triethylamine (10 µL, 0.73 mmol) was added to MG11 (1 mg, 0.98 µmol). The resulting reaction mixture was left incubating at room temperature or heated to 50 °C and 70 °C respectively. The crude reaction mixture was sampled every 5 min, diluted in water containing 0.1% TFA (100 µL) and analysed by HPLC to monitor reaction progression.
Radiosynthesis of [ 18 F]FNic-MG11 4
[18F]Fluoride was recovered on a QMA solid phase extraction cartridge and released into the reaction vessel using a solution of potassium carbonate (2–3 mg, 14.5–22 µmol) and kryptofix 222 (8–10 mg, 21–27 µmol) in milli-Q H2O:MeCN (1 mL, 30:70). The K222.K[18F]F complex was then azeotropically dried on the iPHASE Flexlab radiochemistry module. A fraction (100–200 µL) of the dried K222.K[18F]F complex in DMSO (1 mL) was added to a 1.5 mL Eppendorf tube containing 6-nitronicotinoyl-MG11 peptide 11 (1 mg, 0.9 µmol) before being heated to 110 °C for 5 min. A small fraction (5–10 µL) of the mixture was diluted in 0.05% TFA in H2O:MeCN (1.3:0.2, 100 µL) and analysed by LC–MS using a Kinetex XB-C18 column (5 µm, 100 Å, 150 × 4.60 mm) eluted at 1.5 mL/min with a gradient of MeCN: 0.1% (w/v) formic acid, starting at 15% MeCN, increased to 90% B over 7 min and then maintained at 90% MeCN for 3 min. Radioactivity was detected using a scintillation detector.
Radiosynthesis of [ 177 Lu]Lu-DOTA-CP04 12 for in Vitro Ligand Binding Assays
[177Lu]Lu- DOTA-CP04 12 was prepared as reported previously with only slight modification. Briefly, DOTA-CP04 13 (30 µg, 14.6 nmol) was dissolved in aqueous sodium acetate (0.5 M, 100 µL (0.3 µg/µL peptide solution)) and then added to 0.4 M ammonium acetate/0.24 M 2,5-dihydroxybenzoic acid (200 µL, pH 4.5) containing ethanol (50 µL), L-methionine (50 µL of 10 mg/mL solution in milliQ water) and sodium ascorbate (50 µL, 0.05 M in milliQ water). 500–1000 MBq of non-carrier added [177Lu]LuCl3 in 0.04 M HCl (50–100 µL) was added to this mixture and the resulting solution was heated at 80 °C for 20 min. A fraction of this product (ca. 20 MBq) was diluted in Dulbecco's Modified Eagle's medium (DMEM) supplemented with 1% foetal calf serum (5 mL) to afford a 4 MBq/mL final concentration suitable for ligand binding assays.
In Vitro CCK-2 Receptor Ligand Binding Experiments
The human epidermoid carcinoma cell line A431, transfected with a plasmid encoding full length CCK-2R, was used for all competitive ligand binding assays, as previously described (Corlett et al. 2021). To prove specificity, negative control A431 cells transfected with an empty vector were also analysed. Briefly, 2 days before experimentation 650,000 cells were plated into each well of a 6-well plate and allowed to grow to confluence. On the day of experimentation, each novel peptide 3–5 was serially diluted in binding buffer (DMEM + 1% FCS) and mixed with [177Lu]Lu-DOTA-CP04 12 (~ 50,000 cpm). Each dilution was added to cells pre-washed with binding buffer and allowed to incubate at 37 °C for 1 h. Following incubation, peptide mixtures were removed and kept for analysis (unbound fraction). Cells were then washed twice in binding buffer, which was pooled with the unbound fraction. Cell lysates representing the bound fraction were created by incubating cells for 5 min with 1 M NaOH. All fractions were analysed using a gamma counter (Perkin Elmer 2480 Wizard2™; Perkin Elmer, Massachusetts, USA) and normalised according to protein concentration, which was determined using a Pierce BCA protein assay kit (ThermoFisher, Australia). Ligand binding curves were generated using GraphPad Prism software (GraphPad Software, California, USA) post non-linear regression analysis. Half maximal inhibitory concentration (IC50) values were approximated and expressed in nM of fluorinated peptides.
Results
Peptide Synthesis
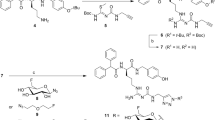
Peptides were synthesized on a microwave assisted CEM Liberty Blue module using a DIC/Oxyma coupling protocol. The N-terminus of the assembled MG11 peptide was capped with 4-fluorobenzoate, 6-fluoronicotinate, 2-fluoropropionate or 6-nitronicotinate to produce FB-MG11 3, FNic-MG11 4, FP-MG11 5 and Nitro-MG11 11, respectively (Fig. 2).

Synthesis of MG11-based peptides capped with different fluorine- and nitro-containing moieties
All peptides synthesized gave high yields and excellent chemical purity (Table 1). Samples of pure FB-MG11 3, FNic-MG11 4 and FP-MG11 5 were then used to prepare solutions for the subsequent competition ligand-binding assays.
Receptor Binding Study
To determine the relative binding affinity of FB-MG11 3, FNic-MG11 4 and FP-MG11 5 for the CCK-2 receptor, radioligand − receptor binding assays were performed using A431 cells stably expressing the CCK-2 receptor, with [177Lu]Lu-DOTA-CP04 12 employed as the competitive radioligand (Fig. 3). A total of seven concentrations of peptide were assayed, ranging from 0 to 40 nM. Unfortunately, due to solubility issues we were unable to assay at higher concentrations. This complicated the analysis of the CCK-2 receptor binding properties of these peptides with the IC50 values calculated to be used as a guide only for comparison. Nevertheless, the apparent binding affinity of all peptides tested were in the low nanomolar to sub-nanomolar range (Fig. 3). FB-MG11 3 demonstrated the highest binding affinity followed by FNic-MG11 4 whilst FP-MG11 5 clearly exhibited the lowest affinity. Accordingly, we chose to prepare fluorine-18 labelled versions of FB-MG11 3 and FNic-MG11 4.

Calculated IC50 values from binding assays performed on human epidermoid carcinoma cells (A431), engineered to overexpress the full-length CCK-2 receptor. Competition binding curves were generated by incubating the peptides 3–5 with the known CCK-2 receptor binding agent, [177Lu]Lu-DOTA-CP04 12. IC50 experiments were performed in three technical replicates
Radiochemistry
Initial attempts at indirect fluorine-18 labelling of MG11 2 using the pre-activated fluorine-18 labelled acylation synthons, [18F]7 and [18F]8, were performed as described previously (Haskali et al. 2020). MG11 2 was treated with [18F]7 or [18F]8 in DMSO and triethylamine (TEA) at room temperature for 5–60 min (Fig. 4). Every 5 min the crude reaction mixture was sampled to monitor reaction progression. Unfortunately, the reaction of [18F]7 with MG11 2 did not generate [18F]FB-MG11 3. Similarly, the reaction of [18F]8 with MG11 2 did not yield [18F]FNic-MG11 4. Acylation of MG11 2 with [18F]7 and [18F]8 was then attempted at elevated temperatures (50 °C and 70 °C), but only trace amounts (˂1%) of [18F]FB-MG11 3 and [18F]FNic-MG11 4 were formed. Partial hydrolysis of [18F]7 and [18F]8 amongst other unidentified radioactive byproducts were observed in the reaction mixture.

Unsuccessful attempts of indirect fluorine-18 labelling of MG11 2 using [18F]7 or [18F]8 acylation synthons
A direct labelling approach was next investigated by preparing the N,N,N-trimethylpyridin-2-aminium-tethered MG11 analogue 14. Treatment of MG11 2 with N,N,N-trimethyl-5-((2,3,5,6-tetrafluorophenoxy)carbonyl)pyridin-2-aminium bromide 6 (TFP-NMe4) yielded a complex mixture. Disappointingly, the desired analogue, 14 was only a minor product that decomposed over long reaction times (Fig. 5). These results collectively highlight the unique difficulty in modifying the N-terminus of the unprotected MG11 2 peptide (vida infra).

Attempted synthesis of N,N,N-trimethylpyridin-2-aminium tethered MG11 analogue 14
Given the difficulties in radiolabeling the N-terminus of the unprotected MG11 peptide in solution, the attachment of 6-nitronicotinate on solid phase was investigated. Within aromatic systems the nitro group is a good leaving group that often effectively facilitates radiofluorination substitutions (Preshlock et al. 2016). We hypothesized that 6-nitronicotinoyl-MG11 11 (Nitro-MG11) could be an effective precursor to allow the synthesis of [18F]FNic-MG11 4 via direct labelling. Nitro-MG11 11 was successfully prepared using Fmoc-SPPS, with the N-terminus of the protected, resin bound MG11 peptide capped with 6-nitronicotinic acid. The peptide was then cleaved off the resin and fully deprotected and purified to generate Nitro-MG11 11 with good yields and purity. Direct radiofluorination of Nitro-MG11 11 via nucleophilic aromatic substitution of the nitro group with fluoride-18 to generate [18F]FNic-MG11 4 was then investigated (Fig. 6).

Direct radiofluorination of Nitro-MG11 11 yields [18F]FNic-MG11 4
Dried nucleophilic ‘naked’ [18F]fluoride was prepared using a mixture of Kryptofix 222 (K222) and K2CO3. The reactive K222/K2CO3/18F– complex was treated with Nitro-MG11 11 in DMSO at 110 °C. To monitor the progress of the reaction, a sample of the reaction mixture was taken every 5 min for up to 20 min and analysed using HPLC. Pleasingly, within 5 min this reaction generated [18F]FNic-MG11 4 with a 47.6% radiochemical yield (based on HPLC integration) (Fig. 7). Intriguingly, the yield decreased to 33.4%, 28.8% and 27.3% at 10, 15 and 20 min, respectively. Furthermore, the fraction of [18F]FNic-MG11 4 present in the reaction mixture heated for only 5 min decreased to 20.7% after three hours at room temperature, indicating product instability under basic conditions. The remaining radioactivity present was free [18F]fluoride ions, indicating that [18F]FNic-MG11 4 decomposes through a substitution process that releases fluoride-18.

Radiochromatogram of the [18F]FNic-MG11 4 crude reaction mixture at 5 min, detected using a scintillation detector. After 3 h at room temperature, the % area of [18F]FNic-MG11 4 decreased with only the % area of free [18F]fluoride increasing. HPLC Conditions: Kinetex XB-C18 column (5 µm, 100 Å, 150 × 4.60 mm) eluted at 1.5 mL/min with a gradient of MeCN: 0.1% (w/v) formic acid, starting at 15% MeCN, increased to 90% B over 7 min and then maintained at 90% MeCN for 3 min
Discussion
Over the last two decades, many CCK-2R targeting radiotracers with high affinity have been developed for SPECT and PET imaging. However, fluorine-18 labelled CCK-2R targeting radioligands have remained out of reach. Although the N-termini of MG11 2 and other CCK-2R targeting peptides based on the gastrin hormone do not interact directly with the receptor, they have been shown to induce interesting folding characteristics of the peptides that may augment binding affinity (Morley et al. 1968). Accordingly, we chose to investigate the CCK-2R binding affinity of MG11-based peptides modified with a range of fluorine-containing aliphatic, aromatic and heteroaromatic moieties that act as scaffolds to introduce fluorine-18 at the N-terminus.
Ligand binding assays with our novel MG11 peptides demonstrated that N-terminal modification with a 4-fluorobenzoyl moiety led to the highest increase in CCK-2R binding affinity, compared with 6-fluoronicotinate and 2-fluoropropanoate (Fig. 3). These results indicate that the introduction of lipophilic/aromatic groups at the N-terminus of MG11 2 improves binding affinity. It is proposed that the Ala-Tyr-Gly-Trp region of the gastrin hormone is involved in a β-turn, inducing a “U” shape conformation (Morley et al. 1968). We have also recently demonstrated that 13-residue peptide analogues of the gastrin hormone (based on CP04) adopt a β-hairpin conformation (Corlett et al. 2021). As such, we speculate that the aromatic fluorobenzoyl or fluoronicotinoyl moieties may facilitate intramolecular π–π interactions with the tyrosine and/or tryptophan residues stabilizing the biological active conformation.
Peptides are commonly labelled with fluorine-18 via indirect methods using pre-labelled fluorine-18 synthons. Thus, we also investigated the use of known [18F]fluoroacylation synthons [18F]7 and [18F]8 to prepare [18F]FB-MG11 3 and [18F]FNic-MG11 4, respectively, from MG11 2. Surprisingly, acylation of MG11 2 did not proceed, suggesting the N-terminus of this peptide is inaccessible (Fig. 4). This lack of reactivity is consistent with the proposed folded conformation of MG11, which may result in occlusion of the N-terminal amine.
While indirect labeling is the most common method routinely employed for the preparation of fluorine-18 labelled peptides, recent advancements have demonstrated that direct labeling of peptides can also be performed with clear advantages using highly reactive precursors. Nicotinate scaffolds with leaving groups at the 2-position undergo facile radiofluorination through fluoride-18 ion attack at the ipso-carbon atom. Thus, peptides incorporating N,N,N-trimethylpyridin-2-aminium groups encompass an important class of precursors for direct radiofluorination of peptides (Cardinale et al. 2017; Dornan et al. 2018; Naka et al. 2020). Using this knowledge, we investigated the preparation of an MG11 analogue capped at the N-terminus with an N,N,N-trimethylpyridin-2-aminium group to facilitate direct radiofluorination. Unfortunately, treatment of MG11 2 with activated ester TFP-NMe4 6 did not generate the N,N,N-trimethylpyridin-2-aminium tethered peptide 14 (Fig. 5), further confirming the inaccessibility of the α-amine of the N-terminal D-Glu residue.
The challenges of modifying the N-terminus of the MG11 peptide in solution led us to incorporate a reactive nicotinate moiety directly onto the fully protected peptide on solid phase. The nitro group is a well-established leaving group on aromatic systems that can facilitate fluorine-18 substitution (Preshlock et al. 2016). As such, we prepared 6-nitronicotinate capped MG11 (Nitro-MG11 11) by standard SPPS techniques. Treatment of Nitro-MG11 11 with fluoride-18 successfully generated the radiolabelled [18F]FNic-MG11 4 with a 47.6% radiochemical yield within 5 min at 110 °C. However, [18F]FNic-MG11 4 was not stable under basic conditions and decomposed with increased reaction time, and even at room temperature after 3 h, releasing free [18F]fluoride. Further, the precursor Nitro-MG11 11 demonstrated similar instability, decomposing to a compound with m/z 1120.4 Da, indicating loss of HNO2 from 11.
We hypothesise that the decomposition of Nitro-MG11 11 and [18F]FNic-MG11 4 progresses through an intramolecular nucleophilic aromatic substitution with an internal nucleophile, resulting in displacement of NO2– from 11 or [18F]F– from 4. The most likely nucleophile is the phenol of the tyrosine residue (Fig. 8), though other groups including the tryptophan residue cannot be ruled out. The carboxylate of the neighboring glutamate residue is unlikely to be a suitable nucleophile as this would lead to a highly strained paracyclophane ring. This cyclisation process accounts for both the observance of the product with m/z 1120.4 Da and the decomposition of [18F]FNic-MG11 4 with loss of [18F]fluoride.

Proposed decomposition mechanism of the Nitro-MG11 11 precursor and [18F]FNic-MG11 4
Conclusion
Three MG11 peptide analogues containing fluorinated acyl groups were prepared and shown to bind with nM affinity to the CCK-2R receptor. Fluorine-18 labelling of these peptides was explored, with a direct radiofluorination approach successfully generating [18F]FNic-MG11 4. [18F]FNic-MG11 4 demonstrated poor chemical stability, decomposing through intramolecular displacement of fluoride. Peptides with improved chemical stability may be formed in future work through the use of chemical spacers between the N-terminal residue and the fluorine-18 containing scaffold.
Data Availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
Change history
07 January 2022
A Correction to this paper has been published: https://doi.org/10.1007/s10989-021-10351-4
References
Aloj L, Aurilio M, Rinaldi V, D’ambrosio L, Tesauro D, Peitl PK, Maina T, Mansi R, Von Guggenberg E, Joosten L (2011) Comparison of the binding and internalization properties of 12 DOTA-coupled and 111 In-labelled CCK2/gastrin receptor binding peptides: a collaborative project under COST Action BM0607. Eur J Nucl Med Mol Imaging 38(8):1417–1425
Cardinale J, Martin R, Remde Y, Schäfer M, Hienzsch A, Hübner S, Zerges A-M, Marx H, Hesse R, Weber K, Smits R, Hoepping A, Müller M, Neels OC, Kopka K (2017) Procedures for the GMP-compliant production and quality control of [18F]PSMA-1007: a next generation radiofluorinated tracer for the detection of prostate cancer. Pharmaceuticals 10(4):77–95
Corlett A, Sani M-A, Van Zuylekom J, Ang C-S, von Guggenberg E, Cullinane C, Blyth B, Hicks RJ, Roselt PD, Thompson PE, Hutton CA, Haskali MB (2021) A new turn in peptide-based imaging agents: foldamers afford improved theranostics targeting cholecystokinin-2 receptor-positive cancer. J Med Chem 64:4841–4856
Dornan MH, Simard J-M, Leblond A, Juneau D, Delouya G, Saad F, Ménard C, DaSilva JN (2018) Simplified and robust one-step radiosynthesis of [18F]DCFPyL via direct radiofluorination and cartridge-based purification. J Labelled Comp Radiopharm 61:757–763
Erba PA, Maecke H, Mikolajczak R, Decristoforo C, Zaletel K, Maina-Nock T, Peitl PK, Garnuszek P, Froberg A, Goebel G, de Jong M, Jabrocka-Hybel A, Konijnenberg M, Virgolini I, Nock B, Lenda-Tracz W, Pawlak D, Rangger C, Trofimiuk-Müldner M, Sowa-Staszczak A, Tomaszuk M, von Guggenberg E, Scarpa L, Hubalewska-Dydejczyk A (2018) A novel CCK2/gastrin receptor-localizing radiolabeled peptide probe for personalized diagnosis and therapy of patients with progressive or metastatic medullary thyroid carcinoma: a multicenter phase I GRAN-T-MTC study. Pol Arch Intern Med 128(12):791–795
Grob NM, Häussinger D, Deupi X, Schibli R, Behe M, Mindt TL (2020) Triazolo-peptidomimetics: novel radiolabeled minigastrin analogs for improved tumor targeting. J Med Chem 63:4484–4495
Haskali MB, Farnsworth AL, Roselt PD, Hutton CA (2020) 4-Nitrophenyl activated esters are superior synthons for indirect radiofluorination of biomolecules. RSC Med Chem 11(8):919–922
Hicks RJ, Kwekkeboom DJ, Krenning E, Bodei L, Grozinsky-Glasberg S, Arnold R, Borbath I, Cwikla J, Toumpanakis C, Kaltsas G, Davies P, Hörsch D, Tiensuu Janson E, Ramage J (2017) ENETS consensus guidelines for the standards of care in neuroendocrine neoplasia: peptide receptor radionuclide therapy with radiolabeled somatostatin analogues. Neuroendocrinology 105(3):295–309
Hofman MSP, Violet JM, Hicks RJP, Ferdinandus J, Thang SPM, Akhurst TM, Iravani AMD, Kong GM, Ravi Kumar AM, Murphy DGMBB, Eu PB, Jackson PP, Scalzo M, Williams SGM, Sandhu SM (2018) [177 Lu]-PSMA-617 radionuclide treatment in patients with metastatic castration-resistant prostate cancer (LuPSMA trial): a single-centre, single-arm, phase 2 study. Lancet Oncol 19(6):825–833
Hörmann AA, Klingler M, Rezaeianpour M, Hörmann N, Gust R, Shahhosseini S, Ev G (2020) Initial in vitro and in vivo evaluation of a novel CCK2R targeting peptide analog labeled with lutetium-177. Molecules 25(19):4585–4604
Klingler M, Rangger C, Summer D, Kaeopookum P, Decristoforo C, von Guggenberg E (2019) Cholecystokinin-2 receptor targeting with novel C-terminally stabilized HYNIC-minigastrin analogs radiolabeled with technetium-99m. Pharmaceuticals (basel) 12(1):13–28
Laverman P, Joosten L, Eek A, Roosenburg S, Peitl PK, Maina T, Mäcke H, Aloj L, von Guggenberg E, Sosabowski JK (2011) Comparative biodistribution of 12 111 In-labelled gastrin/CCK2 receptor-targeting peptides. Eur J Nucl Med Mol Imaging 38(8):1410–1416
Maximilian K, Anton Amadeus H, Von Elisabeth G (2020) Cholecystokinin-2 receptor targeting with radiolabeled peptides: current status and future directions. Curr Med Chem 27:1–21
Morley JS, Blaschko HKF, Gregory RA, Harris GW, Kenner GW (1968) Structure-function relationships in gastrin-like peptides. Proc Royal Soc B 170(1018):97–111
Naka S, Watabe T, Kurimoto K, Uemura M, Soeda F, Neels OC, Kopka K, Tatsumi M, Kato H, Nonomura N, Shimosegawa E, Cardinale J, Giesel FL and Hatazawa J (2020) Automated [18F]PSMA-1007 production by a single use cassette-type synthesizer for clinical examination. EJNMMI Radiopharm Chem 5(18).
Ocak M, Helbok A, Rangger C, Peitl PK, Nock BA, Morelli G, Eek A, Sosabowski JK, Breeman WA, Reubi JC (2011) Comparison of biological stability and metabolism of CCK2 receptor targeting peptides, a collaborative project under COST BM0607. Eur J Nucl Med Mol Imaging 38(8):1426–1435
Olberg DE, Arukwe JM, Grace D, Hjelstuen OK, Solbakken M, Kindberg GM, Cuthbertson A (2010) One step radiosynthesis of 6-[18F]fluoronicotinic acid 2,3,5,6-tetrafluorophenyl ester ([18F]F-Py-TFP): a new prosthetic group for efficient labeling of biomolecules with fluorine-18. J Med Chem 53:1732–1740
Preshlock S, Tredwell M, Gouverneur V (2016) 18F-labeling of arenes and heteroarenes for applications in positron emission tomography. Chem Rev 116(20):719–766
Rangger C, Klingler M, Balogh L, Postenyi Z, Polyak A, Pawlak D, Mikołajczak R, von Guggenberg E (2017) 177Lu labeled cyclic minigastrin analogues with therapeutic activity in CCK2R expressing tumors: preclinical evaluation of a kit formulation. Mol Pharm 14(9):3045–3058
Reubi JC, Schaer J-C, Waser B (1997) Cholecystokinin (CCK)-A and CCK-B/gastrin receptors in human tumors. Cancer Res 57(7):1377–1386
Rodrigues M, Winkler K-K, Svirydenka H, Nilica B, Uprimny C, Virgolini I (2021) Long-term survival and value of 18F-FDG PET/CT in patients with gastroenteropancreatic neuroendocrine tumors treated with second peptide receptor radionuclide therapy course with 177Lu-DOTATATE. Life 11:198
Summer D, Rangger C, Klingler M, Laverman P, Franssen GM, Lechner BE, Orasch T, Haas H, von Guggenberg E, Decristoforo C (2018) Exploiting the concept of multivalency with 68Ga-and 89Zr-labelled fusarinine C-minigastrin bioconjugates for targeting CCK2R expression. Contrast Media Mol Imaging 2018:1–12
Tornesello AL, Aurilio M, Accardo A, Tarallo L, Barbieri A, Arra C, Tesauro D, Morelli G, Aloj L (2011) Gastrin and cholecystokinin peptide-based radiopharmaceuticals: an in vivo and in vitro comparison. J Pept Sci 17(5):405–412
Turner JH (2018) An introduction to the clinical practice of theranostics in oncology. Br J Radiol 91(1091):20180440–20180440
Uprimny C, von Guggenberg E, Svirydenka A, Mikołajczak R, Hubalewska-Dydejczyk A, Virgolini IJ (2020) Comparison of PET/CT imaging with [18F]FDOPA and cholecystokinin-2 receptor targeting [68Ga]Ga-DOTA-MGS5 in a patient with advanced medullary thyroid carcinoma. Eur J Nucl Med Mol Imaging 48:935–936
Vallabhajosula S, Solnes L, Vallabhajosula B (2011) A broad overview of positron emission tomography radiopharmaceuticals and clinical applications: what is new?, in Semin Nucl Med pp 246–264, Elsevier.
von Guggenberg E, Sallegger W, Helbok A, Ocak M, King R, Mather SJ, Decristoforo C (2009) Cyclic minigastrin analogues for gastrin receptor scintigraphy with technetium-99m: preclinical evaluation. J Med Chem 52:4786–4793
Acknowledgements
The authors wish to acknowledge funding from the Peter MacCallum Cancer Center through Peter MacCallum Cancer Foundation Grant #1728 and the National Health and Medical Research Council (NHMRC) for New Investigator grant funding (APP1158863). The authors also wish to acknowledge the Higher Education Commission (HEC) of Pakistan for funding Dr. Naeem-Ul-Haq Khan’s PhD candidature and travel expenses.
Author information
Authors and Affiliations
Contributions
Dr. NK performed peptide synthesis to generate analogues 3–5. Dr. NK also assisted with the write up of this manuscript. Dr. AC performed the ligand binding assays and assisted with the write up of this manuscript. Prof. Hutton facilitated some of the work described in this manuscript in his lab and he contributed to the design and write-up of this work. Dr. MBH was primarily responsible for the study design, peptide synthesis and radiochemistry development. He also contributed to the write up of this manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: the misspelled word “Cholecystokinin” in Keywords and "assisted" in Author Contribution section has been corrected. Also, the supplementary file was updated with the correct article title.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khan, NUH., Corlett, A., Hutton, C.A. et al. Investigation of Fluorine-18 Labelled Peptides for Binding to Cholecystokinin-2 Receptors with High Affinity. Int J Pept Res Ther 28, 6 (2022). https://doi.org/10.1007/s10989-021-10310-z
Accepted:
Published:
DOI: https://doi.org/10.1007/s10989-021-10310-z