
Abstract
The anti-inflammatory drug, PMX205, is an antagonist of the C5a complement receptor and has been shown to be effective in rodent models of amyotrophic lateral sclerosis and Alzheimer’s disease. This cyclic hexapeptide (c[Arg-Trp-D-Cha-Pro-Orn]-Hca) has been reported to produce relatively low yields for both the linear peptide assembly and the cyclization reaction in solution and solid phase syntheses. During attempts to reproduce the solid phase methodology, a catastrophic loss of substitution was encountered which could be avoided or reduced by the use of 2-chlorotrityl resin. Likewise, the cyclization reaction could be significantly improved by the use of FDPP (pentafluorophenyl diphenylphosphinate) at high dilution (up to 80% purified yield). Both improvements are accomplished with commercially available products.
Similar content being viewed by others

Introduction
It has been observed that inflammation plays a major role in the progression of diseases like amyotrophic lateral sclerosis (Woodruff et al. 2008) and Alzheimer’s disease (Fonseca et al. 2009; Ager et al. 2010) and that inhibition of the major complement receptor C5aR by an antagonist results in a significant reduction of pathology in rodent models of these conditions. The patented (Abbenante et al. 2007) cyclic hexapeptide C5a antagonist PMX53 (c[Arg-Trp-D-Cha-Pro-Orn]-AcPhe), which has successfully passed phase 1 trials in humans (Köhl 2006), and its derivative PMX205 (c[Arg-Trp-D-Cha-Pro-Orn]-Hca) (Fig. 1) have been shown to be effective in causing a significant reduction in fibrillar amyloid deposits and activated glia in two mouse models of Alzheimer’s disease (Fonseca et al. 2009). The reduction in pathology was also accompanied by enhanced behavioral performance in passive avoidance tasks (Fonseca et al. 2009). In fact, PMX205 “may be more potent for treatment of brain disorders” (Lawhon 2009). Both compounds are capable of oral administration.

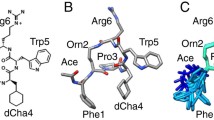
PMX205 structure
Initial repeated attempts (both manual and microwave) to synthesize PMX205 in this laboratory resulted in crude yields of 20% and less (as calculated from the crude molar yield and the initial substitution of the resin) for the linear hexapeptide following the method described in Fonseca et al. (2009). A loss of resin swelling was also observed after the Orn(Boc) coupling/deprotection step and Kaiser ninhydrin tests (Kaiser et al. 1970) confirmed a catastrophic loss of substitution between the D-cyclohexylalanine (D-Cha) deprotection and the ornithine deprotection (opaque blue to transparent pale blue ninhydrin reaction color). These observations led to the conclusion of a major side reaction in the solid phase synthesis of this short peptide and spurred efforts to attempt to elucidate and eliminate (or reduce) this side reaction so to improve yields in the optimization of this synthesis. Three approaches were attempted: changing the ornithine side chain protection from Boc to a more sterically hindered group (Dde: N-(1-(4,4-dimethyl-2,6-dioxocyclohexylidene)ethyl), changing the cyclization reaction from Orn Arg to D-Cha
Pro and replacing the resin linkage (Wang) to a more sterically hindered spacer (2-chlorotrityl chloride resin).
Arg to D-Cha
Pro and replacing the resin linkage (Wang) to a more sterically hindered spacer (2-chlorotrityl chloride resin).
Materials and Methods
Materials
Glass reaction vessels with sintered frits were obtained from Peptides International (11621 Electron Drive, Louisville, KY 40299, USA). All protected amino acids and aminoacyl resins as well as HATU (N-[(dimethylamino)-1H-1,2,2-triazolo[4,5-b]pyridine-1-ylmethylene]-N-methylmethanaminium hexafluorophosphate) and HOAt (1-hydroxy-7-azabenzotriazole) were supplied by aappTec (6309 Shepherdsville Rd., Louisville, KY 40228, USA). Anisole, ethanedithiol, DIPEA (N,N-diisopropylethylamine), piperidine, FDPP (pentafluorophenyl diphenylphosphinate) and Hca (hydrocinnaminic acid) were supplied by Sigma-Aldrich (3050 Spruce St., St. Louis, MO 63103, USA). Thioanisole and DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) were supplied by FLUKA (Sigma-Aldrich, 3050 Spruce St., St. Louis, MO 63103, USA). Redistilled TFA (trifluoroacetic acid) was from Advanced ChemTech (5609 Fern Valley Rd. Louisville, KY 40228, USA) and DIPC (diisopropylcarbodiimide) from AnaSpec (2149 O’Toole Ave., Suite F, San Jose, CA 95131, USA). DMF (dimethylformamide) over 4 Å sieve was obtained from Pharmco-Aaper (58 Vale Rd., Brookfield, CT 06804, USA). MTBE (methyl tert-butyl ether), hexane and Whatman DE52 were obtained from Fisher Scientific (Thermo Fisher Scientific Inc., 81 Wyman Street, Waltham, MA 02454). Some initial syntheses and all cleavages were performed with a CEM Discover microwave solid phase synthesis system (PO Box 200, Matthews, NC 25105, USA). Analytical RP-HPLC was accomplished on a Waters Model 510 system with a Waters 490E detector (Waters Corp., 34 Maple Street, Milford, MA 01757, USA) and a Vydac 214TP54 column (Grace, 2051 Waukegan Rd., Deerfield, IL 60015, USA) and semi-preparative RP-HPLC with a Waters Delta Prep 4000 with a Waters 486 detector and a Vydac 259VHP1522 polymer column. ESI–MS was performed with Waters Micromass LCT and LCT Premier mass spectrometers.
Methods
Peptide Synthesis
Syntheses were performed manually in fritted glass reaction vessels so that each coupling and deprotection could be monitored by qualitative Kaiser ninhydrin (Kaiser et al. 1970) and isatin (proline) (Kaiser et al. 1980) tests. All syntheses were performed on a 1.5 mmol scale with 2.5 equivalents of the coupling reagents, COMU (4-[[[(1-cyano-2-ethoxy-2-oxoethylidene)amino]oxayl](dimethylamino)methylene]morpholinium hexafluorophosphate) (El-Faham and Albericio 2010), HATU and HOAt (Carpino 1993) with DIPEA for 2 h in DMF without preactivation. HOAt active esters were formed in situ with DIPC in DMF. Re-couplings were performed as required. Fmoc deprotections were accomplished by two (10 and 20 min) washes with 2% piperidine, 2% DBU (Fields 1995) in DMF (v/v). Cleavage and deprotection of the peptidoresins was accomplished with a cocktail of TFA, thioanisole, anisole and ethanedithiol (9.0:0.5:0.3:0.2, v/v) (Reagent R—Albericio et al. 1990) in a CEM Discover microwave reactor (30 min at 38°C and 20 W). Crude peptide reaction mixtures were precipitated with MTBE/hexane (1:1, v/v) overnight at −20°C and the precipitates collected by low speed centrifugation and dried under vacuum.
Hca-Orn(Dde)-Pro-D-Cha-Trp(Boc)-Arg(Pbf)-Wang Resin
The peptide was assembled using COMU/DIPEA couplings, except for Orn(Dde) and Hca which were coupled as the HOAt active esters (Hca required recoupling). The Dde protecting group was removed with three washes of fresh 2% hydrazine·H2O in DMF/CH2Cl2 (1:1, v/v) (Mellor et al. 2004) prior to cleavage.
Hca-Orn[Arg(Pbf)-Trp(Boc)-D-Cha-NH2]-Pro-2-chlorotrityl-Wang Resin
This branched peptide was assembled as the protected tripeptide (with HOAt active ester couplings) prior to the removal of the Orn-Dde with three washes of fresh 2% hydrazine·H2O in DMF/CH2Cl2 (1:1, v/v). The peptide assembly was then continued with HATU/HOAt/DIPEA couplings and a final amino terminal deprotection prior to cleavage.
Hca-Orn(Boc)-Pro-D-Cha-Trp(Boc)-Arg(Pbf)-2-chlorotrityl-Wang Resin
The peptide was assembled using HOAt active ester couplings. Trp(Boc) and Hca required recoupling, the latter with HATU/HOAt/DIPEA, to achieve a negative Kaiser test. Cleavage from the resin was performed as described.
RP-HPLC
Analytical RP-HPLC was performed on a Waters Model 510 system with a Waters 490E detector and a C4 Vydac 214TP54 column running a 0.1% TFA/CH3CN gradient (5–95% over 60 min at 1.0 ml/min). Peptide purification was achieved by repetitive chromatographic runs on a Vydac 259VHP1522 semi-preparative column using a Waters Delta Prep 4000 and a Waters 486 detector running a 0.1% TFA/CH3CN gradient (5–45% over 45 min at 8.0 ml/min). Fractions were collected, pooled and lyophilized as appropriate to achieve >95% homogeneity.
Cyclization
Crude peptides were purified by semi-preparative RP-HPLC prior to cyclization with a 1.2 equivalent excess of FDPP and a 3.6 equivalent excess of DIPEA in DMF that had been thoroughly purged with argon (0.0005 M concentration) for 4 days. Cyclic product formation was monitored by ESI–MS (Fairweather et al. 2010). The DMF was removed by rotary evaporation and the residue triturated with petroleum ether overnight prior to vacuum drying and semi-preparative RP-HPLC.
Electrospray Ionization Mass Spectrometry
Samples were prepared and injected in GC–MS grade methanol onto Waters Micromass LCT and LCT Premier mass spectrometers.
[Purified cyclized peptide was converted into the acetate salt by passage through DEAE cellulose (Whatman DE52) with water/CH3CN (2:1, v/v) into acetic acid and lyophilized prior to animal testing.]
Results and Discussion
The first reaction to the observed loss of substitution during the manual synthesis of the linear hexapeptide was to attempt a microwave synthesis. When this approach continued to produce negative peptidoresin weight gains and atypically low crude yields (<20% before purification) a series of di- and tripeptides based on the Wang-Arg(Pbf) resin were manually synthesized to exclude diketopiperazine formation as the cause of the problem. Having established that the consistent loss of resin swelling and substitution was associated only with the Orn(Boc) coupling/deprotection step, the filtrates from each coupling and deprotection were collected, evaporated and analyzed by mass spectrometry, but no recognizable product ions could be found in the spectra of the relevant filtrates (presumably due to masking by the more abundant reactant ions). Lacking evidence of the nature of the observed side reaction, it was hypothesized from molecular modeling of the resin-bound peptide that the most likely mechanism was an intramolecular nucleophillic attack by the α-amino of ornithine on the carbonyl of the argine ester linkage to the resin—analogous to diketopiperazine formation (Pedroso et al. 1986; Carpenter et al. 1994) (Fig. 2).

Tentative mechanism to explain the premature cleavage side reaction (which results in the loss of substitution) and its prevention. a nucleophilic attack on the resin ester linkage carbonyl causes premature cleavage of the protected cyclic pentapeptide. b steric hindrance from the 2-chlorotrityl linker prevents nucleophilic attack by the Orn(Boc) α-amino group
The first approach to eliminating or, at least, limiting the loss of substitution during the synthesis of this peptide was to discontinue the use of base in the Orn and Hca couplings (carbodiimide rather than aminium/phosphonium activation) and replacing the Boc-Orn side chain protection with the more sterically hindered Dde group (Hca-Orn(Dde)-Pro-D-Cha-Trp(Boc)-Arg(Pbf)-Wang resin). After hydrazinolysis and cleavage from the resin, a yield of 25.53% [416.8 mg] (based on the formula weight of the TFA salt) was obtained for the HPLC purified linear peptide (mwcalc. 856) (Table 1). Although an improvement, this would not be considered an optimized linear synthesis.
The second approach was to reconstruct the linear sequence as a “branched” peptide, so reordering the amino acids that the cyclization reaction would now be between the -D-Cha-NH2 and the HO-Pro- (Hca-Orn[Arg(Pbf)-Trp(Boc)-D-Cha-NH2]-Pro-2-chlorotrityl-Wang resin). The anticipated advantage would be a more facile cyclization (not obtained—Table 1), but the potential for diketopiperazine formation with Pro as the first amino acid on the resin needed to be addressed. To this end, a 2-chlorotrityl resin linkage was employed (Chan and White 2004). Thus the tripeptide: Pro-Orn(Dde)-Hca was assembled followed by hydrazinolysis to remove the Dde group before continuing the synthesis from the δ-amino of the Orn side chain. The yield of the HPLC purified branched peptide after cleavage was 31.76% [323.8 mg] (TFA salt) (Table 1).
On the basis of the proposed mechanism for the loss of substitution observed in previous syntheses, it was speculated that if 2-chlorotrityl resin prevented diketopiperazine formation in prolyl peptides, it may also prevent the on-resin pentapeptide cyclization and loss of substitution plaguing this synthesis (Fig. 2). It was decided to return to the original protocol of Fonseca et al. (2009), but using Wang 2-chlorotrityl-Arg(Pbf) as the solid support (Hca-Orn(Boc)-Pro-D-Cha-Trp(Boc)-Arg(Pbf)-2-chlorotrityl-Wang resin). No loss of resin swelling or deprotected ninhydrin color was observed during the synthesis and the yield of the HPLC purified linear peptide was 62.92% [1.027 g] (TFA salt) (Table 1), a respectable, if not optimal, purified yield for a hexapeptide.
Lacking the resources to duplicate the original cyclization protocol (an equiv. of DIPEA and dropwise addition of 1.1 equiv. of PyBOP {benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate} at −10°C, pH 7.5–8.0, for 5 h—Reid et al. (2003), Abbenante et al. (2007) and Fonseca et al. (2009)), it was decided to adopt the method of Fairweather et al. (2010) using the racemization free (Chen and Xu 1991; Fairweather et al. 2010) reagent, FDPP (pentafluorophenyl diphenylphosphinate), but at an 10× higher dilution (Fig. 3). Although a much longer procedure (4 days vs. 5 h), it has the advantages of a single addition of reagent and room temperature. The cyclization was monitored by ESI–MS until the linear peptide (M + H 857) was completely replaced by the cyclic product (M + H 839—mwcalc. 838). No evidence of dimer or trimer ions was observed. Surprisingly, the HPLC purified yields (calculated from: mmoles cyclic peptide TFA salt/mmoles linear peptide TFA salt) of the
Orn
Arg cyclization were significantly better than the D-Cha
Pro cyclization (78.23% [285.3 mg] and 81.8% [735.0 mg] vs. 66.17% [300.3 mg]), but much better than the reported 33% cyclization yield of the original protocol (Reid et al. 2003) (Figs. 4, 5).

Pentafluorophenol diphosphinate

Analytical C4 RP-HPLC of HPLC purified PMX205: 0.125 mg/50 μl formic acid. 5–95%B (60 min)—A: 0.1% TFA aq. B: 0.1% TFA/CH3CN. (a) Gray trace—214 nm. (b) Black trace—280 nm

ESI–MS spectrum (Waters Micromass LCT Premier) of HPLC purified PMX205
The overall yields (mmoles cyclic peptide TFA salt/starting resin substitution) for all three approaches described here were also an improvement on literature. Although Fonseca et al. (2009) do not give synthetic or cyclization yields, Reid et al. (2003) report a 13% overall yield for the solution phase synthesis and cyclization of PMX53 (Ac-Phe-[Orn-Pro-D-Cha-Trp-Arg]), a related compound, while the first approach described here (Hca-Orn(Dde)-Pro-D-Cha-Trp(Boc)-Arg(Pbf)-Wang resin) led to a purified yield of 19.97% [285.3 mg] and the second approach (Hca-Orn[Arg(Pbf)-Trp(Boc)-D-Cha-NH2]-Pro-2-chlorotrityl-Wang resin) gave a 21.02% [300.3 mg] purified yield. The greatest improvement was seen with the third approach (Hca-Orn(Boc)-Pro-D-Cha-Trp(Boc)-Arg(Pbf)-2-chlorotrityl-Wang resin) which produced an overall purified yield of 51.45% [735.0 mg]. (Table 1)
Conclusion
The adoption of 2-chlorotrityl resin to the linear solid phase synthesis and FDPP to the cyclization of PMX205 leads to greatly improved yields without the use of any non-commercial protected amino acids, resins or reagents.
Abbreviations
- Boc:
-
tert-Butyloxycarbonyl
- COMU:
-
4-[[[(1-cyano-2-ethoxy-2-oxoethylidene)amino] oxayl](dimethylamino)methylene]morpholinium hexafluorophosphate
- DBU:
-
1,8-diazabicyclo[5.4.0]undec-7-ene
- D-Cha:
-
D-cyclohexylalanine
- DEAE:
-
Diethylaminoethyl
- Dde:
-
N-(1-(4,4-dimethyl-2,6-dioxocyclohexylidene)ethyl)
- DIPC:
-
Diisopropylcarbodiimide
- DIPEA:
-
N,N-diisopropylethylamine
- DMF:
-
Dimethylformamide
- ESI–MS:
-
Electrospray ionization mass spectroscopy
- FDPP:
-
Pentafluorophenyl diphenylphosphinate
- Fmoc:
-
9-fluorenylmethyloxycarbonyl
- fw:
-
Formula weight
- GC-MS:
-
Gas chromatography mass spectrometry
- HATU:
-
N-[(dimethylamino)-1H-1,2,2-triazolo[4,5-b]pyridine-1-ylmethylene]-N-methylmethanaminium hexafluorophosphate
- Hca:
-
Hydrocinnaminic acid
- HOAt:
-
1-hydroxy-7-azabenzotriazole
- MTBE:
-
Methyl tert-butyl ether
- mw:
-
Molecular weight
- Pbf:
-
2,2,4,6,7-pentamethyl-2,3-dihydrobenzofuran-5-sulfonyl
- PyBOP:
-
Benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate
- RP-HPLC:
-
Reversed phase high performance liquid chromatography
- TFA:
-
Trifluoroacetic acid
References
Abbenante G, Fairlie DP, Reid RC (2007) Process for the preparation of cyclic peptides. US patent 2007/0245926 A1
Ager RR, Fonseca ML, Chu S-H et al (2010) Microglial C5aR (CD88) expression correlates with amyloid-β deposition in murine models of Alzheimer’s disease. J Neurochem 113:389–401
Albericio F, Kneib-Cordonier N, Biancalana S et al (1990) 5-(4-(9-fluorenylmethyloxycarbonyl)aminomethyl-3,5-dimethoxyphenoxy)-valeric acid (PAL) handle for the solid-phase synthesis of C-terminalpeptide amides under mild conditions. J Org Chem 55:3730–3743
Carpenter KA, Weltrowska G, Wilkes BC, Schmidt R, Schiller PW (1994) Spontaneous diketopiperazine formation via end-to-end cyclization of a nonactivated linear tripeptide: an unusual chemical reaction. J Am Chem Soc 116:8450–8458
Carpino LA (1993) 1-Hydroxy-7-azabenzotriazole. An efficient peptide coupling additive. J Am Chem Soc 115:4397–4398
Chan WC, White PD (2004) Basic procedures. In: Chan WC, White PD (eds) Fmoc solid phase peptide synthesis—a practical approach. Oxford University Press, Oxford, pp 41–76
Chen S, Xu J (1991) Pentafluorophenyl diphenylphosphinate a new efficient coupling reagent in peptide chemistry. Tetrahedron Lett 32:6711–6714
El-Faham A, Albericio F (2010) COMU: a third generation of uranium-type coupling reagents. Pept Sci 16:6–9
Fairweather KA, Sayyadi N, Luck IJ, Clegg JK, Joliffe KA (2010) Synthesis of all L-cyclic tetrapeptides using pseudoprolines as removable turn indicators. Org Lett 12:3136–3139
Fields GB (1995) Methods for removing the Fmoc group. In: Pennington MW, Dunn BM (eds) Methods in molecular biology, vol 35. Springer Protocols, Humana Press, New York, pp 17–27
Fonseca ML, Ager RR, Chu S-H et al (2009) Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. J Immunol 183:1375–1383
Kaiser E, Colescott RL, Bossinger CD, Cook PI (1970) Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal Biochem 34:595–598
Kaiser E, Bossinger CD, Colescott RL, Olsen DB (1980) Color test for terminal prolyl residues in the solid-phase synthesis of peptides. Anal Chim Acta 118:149–151
Köhl J (2006) Drug evaluation: the C5a receptor antagonist PMX53. Curr Opin Mol Ther 8:529–538
Lawhon C (2009) Drug rescues memory lost to Alzheimer’s disease. U.C. Irvine Communications: UCIrvine Today, July 14, 2009
Mellor SL, Wellings DA, Fehrentz J-A et al (2004) Synthesis of modified peptides. In: Chan WC, White PD (eds) Fmoc solid phase peptide synthesis—a practical approach. Oxford University Press, Oxford, pp 137–181
Pedroso E, Grandas A, de las Heras X, Eritja R, Giralt E (1986) Diketopiperazine formation in solid phase peptide synthesis using p-alkoxybenzyl ester resins and Fmoc amino acids. Tetrahedron Lett 27:743–746
Reid R, Abbenante G, Taylor SM, Fairlie DP (2003) A convergent solution phase synthesis of the macrcocycle Ac-Phe-[Orn-Pro-D-Cha-Trp-Arg], a potent new antiinflammatory drug. J Org Chem 68:4464–4471
Woodruff TM, Constantini KJ, Crane JW et al (2008) The complement factor C5a contributes to pathology in a rat model of amyotrophic lateral sclerosis. J Immunol 181:8727–8734
Acknowledgments
We are grateful to the National Institutes of Health (NINDS, NS35144, A.J. Tenner, PI) for partial support (to ARC) of this work. We thank J. Greaves and S. Sorooshian of the UCI Mass Spectrometer Facility for assistance and C.G. Glabe for his support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
deLisle Milton, R.C., Milton, S.C. & Chamberlin, A.R. Improving the Fmoc Solid Phase Synthesis of the Cyclic Hexapeptide Complement C5a Antagonist, PMX205. Int J Pept Res Ther 17, 337 (2011). https://doi.org/10.1007/s10989-011-9273-9
Accepted:
Published:
DOI: https://doi.org/10.1007/s10989-011-9273-9