
Abstract
Purpose
Chronic granulomatous disease (CGD) is characterized by an inability of phagocytes to produce reactive oxygen species (ROS), which are required to kill some microorganisms. CGD patients are known to suffer from recurrent bacterial and/or fungal infections from the first year of life onwards. From 2009 to 2013, 12 cases of CGD were diagnosed in Morocco. We describe here these Moroccan cases of CGD.
Methods
We investigated the genetic, immunological and clinical features of 12 Moroccan patients with CGD from 10 unrelated kindreds.
Results
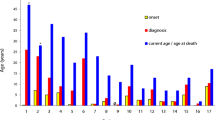
All patients were children suffering from recurrent bacterial and/or fungal infections. All cases displayed impaired NADPH oxidase activity in nitroblue tetrazolium (NBT), dihydrorhodamine (DHR) or 2′,7′ dichlorofluorescein diacetate (DCFH-DA) assays. Mutation analysis revealed the presence of four different mutations of CYBB in four kindreds, a recurrent mutation of NCF1 in three kindreds, and a new mutation of NCF2 in three patients from a single kindred. A large deletion of CYBB gene has detected in a patient. The causal mutation in the remaining one kindred was not identified.
Conclusion
The clinical features and infectious agents found in these patients were similar to those in CGD patients from elsewhere. The results of mutation analysis differed between kindreds, revealing a high level of genetic and allelic heterogeneity among Moroccan CGD patients. The small number of patients in our cohort probably reflects a lack of awareness of physicians. Further studies on a large cohort are required to determine the incidence and prevalence of the disease, and to improve the description of the genetic and clinical features of CGD patients in Morocco.


Similar content being viewed by others


References
Winkelstein JA, Marino MC, Johnston Jr RB, Boyle J, Curnutte J, Gallin JI, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore). 2000;79(3):155–69.
de Oliveira-Junior EB, Bustamante J, Peter PE, Condino-Neto A. The human NADPH oxidase: primary and secondary defects impairing the respiratory burst function and the microbicidal ability of phagocytes. Scand J Immunol. 2011;73(5):420–7.
Holland SM. Chronic granulomatous disease. Hematol Oncol Clin N Am. 2013;27(1):89–99. viii.
Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine (Baltimore). 2000;79(3):170–200.
Roos D, Kuhns DB, Maddalena A, Roesler J, Lopez JA, Ariga T, et al. Hematologically important mutations: X-linked chronic granulomatous disease (third update). Blood Cells Mol Dis. 2010;45(3):246–65.
Van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, Corbeel L, et al. Chronic granulomatous disease: the European experience. PLoS One. 2009;4(4):e5234.
Di Matteo G, Giordani L, Finocchi A, Ventura A, Chiriaco M, Blancato J, et al. Molecular characterization of a large cohort of patients with chronic granulomatous disease and identification of novel CYBB mutations: an Italian multicenter study. Mol Immunol. 2009;46(10):1935–41.
Jones LBKR, McGrogan P, Flood TJ, Gennery AR, Morton L, Thrasher A, et al. Special article: chronic granulomatous disease in the United Kingdom and Ireland: a comprehensive national patient-based registry. Clin Exp Immunol. 2008;152(2):211–8.
Roos D, Kuhns DB, Maddalena A, Bustamante J, Kannengiesser C, de Boer M, et al. Hematologically important mutations: the autosomal recessive forms of chronic granulomatous disease (second update). Blood Cells Mol Dis. 2010;44(4):291–9.
Francke U, Ochs HD, de Martinville B, Giacalone J, Lindgren V, Disteche C, et al. Minor Xp21 chromosome deletion in a male associated with expression of Duchenne muscular dystrophy, chronic granulomatous disease, retinitis pigmentosa, and McLeod syndrome. Am J Hum Genet. 1985;37(2):250–67.
Watkins CE, Litchfield J, Song E, Jaishankar GB, Misra N, Holla N, et al. Chronic granulomatous disease, the McLeod phenotype and the contiguous gene deletion syndrome-a review. Clin Mol Allergy. 2011;9(1):13.
Bousfiha AA, Jeddane L, Ailal F, Benhsaien I, Mahlaoui N, Casanova J-L, et al. Primary immunodeficiency diseases worldwide: more common than generally thought. J Clin Immunol. 2013;33(1):1–7.
Fattahi F, Badalzadeh M, Sedighipour L, Movahedi M, Fazlollahi MR, Mansouri SD, et al. Inheritance pattern and clinical aspects of 93 Iranian patients with chronic granulomatous disease. J Clin Immunol. 2011;31(5):792–801.
El Kares R, Barbouche MR, Elloumi-Zghal H, Bejaoui M, Chemli J, Mellouli F, et al. Genetic and mutational heterogeneity of autosomal recessive chronic granulomatous disease in Tunisia. J Hum Genet. 2006;51(10):887–95.
Köker MY, Camcıoğlu Y, van Leeuwen K, Kılıç SŞ, Barlan I, Yılmaz M, et al. Clinical, functional, and genetic characterization of chronic granulomatous disease in 89 Turkish patients. J Allergy Clin Immunol. 2013;132(5):1156–63.e5.
Abrar M. Granulomatose septique chronique (A propos d’un cas). Thèse de médecine, Casablanca, Faculté de Médecine et Pharmacie de Casablanca, 2003:123p. http://toubkal.imist.ma/handle/123456789/5355.
Jaouad IC, Elalaoui SC, Sbiti A, Elkerh F, Belmahi L, Sefiani A. Consanguineous marriages in Morocco and the consequence for the incidence of autosomal recessive disorders. J Biosoc Sci. 2009;41(5):575–81.
Barbouche M-R, Galal N, Ben-Mustapha I, Jeddane L, Mellouli F, Ailal F, et al. Primary immunodeficiencies in highly consanguineous North African populations. Ann N Y Acad Sci. 2011;1238:42–52.
Acknowledgments
We would particularly like to thank the patients and their families, whose trust, support, and cooperation were essential for the collection of the data used in this study. We thank Naim Btissam, Abdellah Mouden, Khattane Rachida and Labied Driss for technical support. We also thank the Hospital Biology Center (le Centre de Biologie des Hôpitaux; CBH) in Morocco for technical support for the NBT test. We thank the members of the Laboratory of Human Genetics of Infectious Diseases in Paris for secretarial and technical assistance. We thank the Moroccan PID Society and Hajar, the Association to Help Moroccan Children with Primary Immunodeficiencies (www.hajar-maroc.org) for financial support. The Laboratory of Human Genetics of Infectious Diseases is supported by institutional grants to INSERM and The Rockefeller University, and grants from the Agence Nationale de la Recherche (ANR13-ISV3-0001-01), the March of Dimes (6-FY08-278), HOMITUB E08153KK and NEOTIM (018736). Laila Ait Baba was supported by a PhD grant from the National Center for Technical Scientific Research (CNRST) in Morocco.
Conflict of Interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Fatima Ailal and Naima Hafidi contributed equally to the study.
Jacinta Bustamante, Hanane Salih Alj and Ahmed Aziz Bousfiha share senior coauthorship.
Rights and permissions
About this article
Cite this article
Baba, L.A., Ailal, F., El Hafidi, N. et al. Chronic Granulomatous Disease in Morocco: Genetic, Immunological, and Clinical Features of 12 Patients from 10 Kindreds. J Clin Immunol 34, 452–458 (2014). https://doi.org/10.1007/s10875-014-9997-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-014-9997-3