
Abstract
A methodology using stripping voltammetry has been elaborated to enable sensitive and reliable protein–chromium complexation measurements. Disturbing effects caused by adsorption of proteins on the mercury electrode were addressed. At low concentrations of proteins (<60–85 nM), chromium–protein complexation measurements were possible. Chromium(VI) complexation was quantitatively determined using differently sized, charged, and structured proteins: serum albumin (human and bovine), lysozyme, and mucin. Generated results showed a strong relation between complexation and protein size, concentration, and the number of amino acids per protein mass. Complexation increased nonlinearly with increasing protein concentrations. The nature of this complexation was based on weak interactions judged from combined results with MALDI-TOF–MS and adsorptive cathodic stripping voltammetry.
Similar content being viewed by others
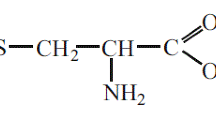

Avoid common mistakes on your manuscript.
1 Introduction
Chromium–protein complexation is an important process in the human body where, for example, trivalent chromium is known to maintain proper carbohydrate and lipid metabolism at a molecular level. Cr(III) is bound to an oligopeptide chromodulin which, when metal-saturated, can bind to an insulin-stimulated insulin receptor and activate the tyrosine kinase activity of the receptor [1]. Studies on chromium complexation and reported stability constants of different organic-chromium complexes are scarce in the literature. However, it has been shown that Cr(III) forms complexes with different organic species, including amino acids [2], bovine serum albumin (BSA) [3], other proteins, and DNA [4]. It has furthermore been shown that mainly trivalent chromium binds to proteins and DNA [4] even if hexavalent chromium is added, with tyrosine and cysteine, followed by histidine, methionine, and threonine as major binding sites for chromium to proteins [4]. Recent findings have shown that adsorption of proteins on chromium metal and chromium-containing alloys strongly enhances the release of metals in phosphate buffered saline [5]. Complexing agent-induced metal release was explained by adsorption-controlled surface complexation and subsequent detachment of metal complexes, as shown in the case of citric acid as complexing agent compared to different non-complexing solutions of the same pH [6]. The importance of such complexation-induced processes, proven to enhance the extent of metal release from chromium metal, ferrochromium alloys and stainless steels [5–7], should be taken into consideration when assessing potential health hazards induced by these materials in contact with proteins, e.g., in the human body (implants or inhaled metal particles), or in food processing. As chromium is widely used in many industrial alloys and implant materials [8], there is a need for a reliable and robust methodology to assess chromium–protein complexation in different solutions.
While there is plenty of information in the literature on the adsorption and determination of different proteins on the mercury electrode (for reviews, see [9–12]), the information available on chromium–protein complexation studies using stripping voltammetry is very limited. One exception is an early polarography study performed on the complexation of chromium to insulin and mitochondria [13].
Advantages and encountered difficulties of using stripping voltammetry with a mercury drop electrode to assess metal–protein complexation have been reported in the literature, e.g., [9, 14–21]. The foremost limitation of stripping voltammetry for protein–metal complexation studies is the adsorption of proteins on the charged and hydrophobic surface of the mercury drop, as proteins inevitable adsorb on almost all surfaces [22]. Detailed studies on protein adsorption on the mercury drop are hence essential for individual proteins, concentrations, and deposition times before evaluating the protein–metal complexation. Most reported studies [9, 14–16, 18, 20, 23–26] have used alternating current voltammetry or cyclic voltammetry to explore protein adsorption on the mercury electrode [9, 10, 27]. Other studies have used scan rate-dependent measurements [26–28], a procedure difficult to employ in the differential pulse mode [28]. Scheller et al. [25] investigated the limiting current of the reduction of disulfide bridges at different concentrations of proteins in solution. The investigation showed a linear increase with protein concentration reaching a constant value (full coverage of the mercury electrode). Other studies have reported [20, 24] that the extent of adsorption of proteins on the mercury electrode depends on potential, protein charge, and ionic strength of the solution, while the protein charge depends on protein isoelectric point (iep, point of zero surface charge), solution pH and solution composition. Very promising for protein–metal binding studies are also constant current chronopotentiometric stripping (CPS) analysis, where the “peak H” is evaluated as it is sensitive to changes in structure and binding of the protein [29–32].
The objectives of this study were to accurately assess the extent and nature of chromium–protein complexation in solution by using stripping voltammetry complemented with matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF–MS), and to elucidate its dependence on size, charge, and structure of the proteins [serum albumin (human and bovine), lysozyme (LYS), and mucin]. A robust methodology had to be elaborated for stripping voltammetry to enable these objectives, as the adsorption of proteins on the mercury drop interferes with the measurements.
Possibilities and limitations of using stripping voltammetry and MALDI-TOF–MS for chromium–protein complexation measurements are discussed.
2 Materials and methods
2.1 Chemicals and cleaning
The same chemicals were used as in an adsorption and metal release study on chromium surfaces [5]. Ultra-pure water (18.2 MΩ cm) and ultra-pure chemicals (Sigma-Aldrich) of grade puriss. p.a. including sodium acetate, diethylenetriaminepentaacetate (DTPA), sodium nitrate, and 30 % NaOH were used for the voltammetric studies. Puriss. p.a. mercury (Sigma-Aldrich) was used for the hanging mercury drop electrode (HMDE). All equipment and plastic vessels were acid cleaned using 10 % nitric acid (Sigma-Aldrich) to avoid any trace metal contamination. PTFE vessels were used for the Cr(VI) standards to minimize the risk of adsorption on the vessel walls. The Cr(VI) standard of 20 μg L−1 was prepared daily from a 10 mg L−1 Cr(VI) standard, in turn prepared from a stock solution of 1 g L−1 Cr(VI) in water (pH of ~7.4), based on K2Cr2O7 (from Sigma-Aldrich).
For the MALDI-TOF–MS measurements, 2,5-dihydroxybenzoic acid (DHB) and sinapinic acid (SA) and MALDI matrices were supplied by Bruker Daltonics (Germany). Trifluoroacetic acid (TFA), 2,6-dihydroxyacetophenone (DHAP), and MS calibration kit (ProteoMass™) containing cytochrome C (m/z 12,362 Da), apomyoglobin (m/z 16,952 Da), aldolase (m/z 39211 Da), and albumin from bovine serum (m/z 66,429 Da) were purchased from Sigma-Aldrich (Sweden).
Acetonitrile (ACN) was obtained from VWR International (Sweden). The water used for the MS experiment was purified using a Synergy 185 system from Millipore (USA).
2.2 Investigated proteins
Proteins were purchased from Sigma-Aldrich and used as received in all stripping voltammetry measurements. LYS from hen egg white (L6876) is a globular “hard” (high internal conformation stability) protein present in saliva and tear fluid with the dimensions 4.5 × 3 × 3 nm and a molecular weight of 14.1 kDa [33]. Serum albumin from human (HSA) (A8763) and bovine (BSA) (A3912) sources are “soft” globular proteins present in serum at high concentrations (ranging from 30 to 50 g L−1), which corresponds to ~60 % of the total amount of proteins present in serum [34]. Serum albumin has an estimated triangular structure with the dimensions 8 × 8 × 3 nm (determined by means of X-ray crystallography) and a molecular weight of ~66 kDa [35]. The large flexible glycoprotein bovine submaxillary gland mucin (BSM) (M3895) with a molecular weight of ~7 MDa [36] has important functions in mucous and in saliva where it binds water and thereby lubricates the epithelial membrane. BSM exists more or less as a random coil in solutions with a radius of gyration of around 130 nm [37].
2.3 Solutions
Phosphate buffered saline, PBS, (8.77 g L−1 NaCl, 1.28 g L−1 Na2HPO4, and 1.36 g L−1 KH2PO4) was adjusted to pH 7.4 using 50 % NaOH. The proteins were dispersed in this solution for at least 3 h before the measurements to equilibrate the samples. By considering the iep of the proteins used, LYS (iep = 11) [33] was net positively charged at pH 6.2 (the pH of the voltammetric measurements), BSM (iep = 3) [38] was net negatively charged due to its high content of sialic acid, and serum albumin, HSA and BSA (iep = 4.7) [34], also was net negatively charged at the same experimental conditions. Comparative chromium–protein complexation measurements were done at constant protein mass concentration (4 mg L−1) and constant molar concentration (59 nM), respectively. They were in addition conducted for different concentrations for BSM, i.e., 0.2, 0.3, 0.4, and 1 nM.
2.4 Metal–protein complexation studies by stripping voltammetry
A Metrohm 797 VA Computrace instrument [hanging mercury drop working electrode, Ag/AgCl (saturated KCl) reference electrode, platinum auxiliary electrode] was used for complexation measurements using differential pulse adsorptive cathodic stripping voltammetry (DPAdCSV). Measurements were conducted with freshly prepared protein solutions, not stored more than 2 days before use, Milli-Q water to dilute the protein solution (total sample volume 10 mL), and supporting electrolyte (2.5 mL) containing 0.2 M sodium acetate, 0.05 M DTPA, and 2.5 M sodium nitrate. The pH of the measured sample containing the supporting electrolyte was adjusted to 6.2 ± 0.1 (using 30 % ultrapure NaOH).
The limit of determination for Cr(VI) was 0.02 μg L−1 and the limit of detection (detected but not determined) was ~0.01 μg L−1. Complexation measurements were conducted by adding a known amount of standard solution of Cr(VI) to the protein containing solution (of known protein concentration) and measuring the free Cr(VI) content in the solution, i.e., indirect detection of the chromium–protein complexation.
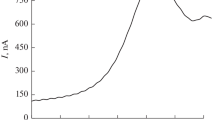
Standard analysis (described in [39] and developed in [40]) was performed with a pre-purging (argon) time of 300 s at the HMDE, followed by deposition (60 s, at −1.0 V) in the differential pulse mode (pulse amplitude 50 mV), starting and ending at −1.0 and −1.45 V, respectively, (sweep rate 33.3 mV s−1) and an equilibrium time after each addition of Cr(VI) of 300 s. Chromium determination using DTPA and nitrate is well established and reviewed in [41]. The equilibrium time period after addition of Cr(VI) to the protein solution may be important for complexation as metal–protein complexation is time dependent [42]. A relatively long, but still practically feasible time (300 s), was therefore introduced between every addition. The solutions were also stirred during this time period and at the pre-purging time to facilitate mixing. According to [39, 40], the chromium peak height is linearly dependent on the free chromium concentration up to 40–90 nM Cr(VI) and reveals a relative standard deviation of 5 % above 4 nM Cr(VI). The analyzed peak position for the chromium signal was at a potential of −1.25 ± 0.1 V. A typical voltammogram for chromium determination using this methodology is shown in Fig. 1. It reflects the reaction of the Cr3+/DTPA 1:1 complex ([Cr(H2O)HY]−), formed after deposition at −1 V by chromate and DTPA at the mercury drop, to [CrH2Y]− at −1.22 V, where “Y” symbolizes DTPA [43]. The presence of nitrate catalyses the reverse reaction ([CrH2Y]− → [Cr(H2O)HY]−) and increases therefore the peak height [43].

Typical voltammogram of chromium determination in a non-complexing solution [here: 10 mL deionized water, 2.5 mL supporting electrolyte, and 0.1 mL 20 μg L−1 Cr(VI)], showing the sample current–potential curves (solid lines) and measured current–potential curves after standard addition (dashed lines). Two standard additions and two replicate readings are shown
Mercury of the grade puriss. p.a. was used throughout the investigation. To ensure accurate measurements, cleaning and/or an exchange of the mercury and the capillary followed by an adjustment of the mercury drop size were conducted before analysis. All measurements were repeated at least once to prove the reproducibility.
Chromium–protein complexation studies were performed by adding Cr(VI) to a protein solution of as low protein concentration as possible. As the DPAdCSV method for chromium determination is very sensitive to electro-active (detectable) chromium, it was possible to use low (nM range or lower) protein concentrations to minimize the influence of protein adsorption on the mercury drop. An alternative method for metal complexation studies, i.e., to add protein to a metal containing solution, would enable better calculations of complexation since a complexation model can be used [17]. However, as this method cannot account for changing protein adsorption conditions during the measurement, this approach was not selected.
Cr(VI) was primarily investigated in this study due to the high sensitivity of the DPAdCSV technique for this species in solutions of low concentrations of proteins.
2.5 MALDI-TOF–MS measurements
As the MALDI-TOF–MS technique is regarded as a soft ionization technique with low fragmentation, often producing singly charged ions, it is suitable for studies of proteins and protein complexation. A Bruker Reflex III MALDI-TOF–MS (instrument from Bruker Daltonics with a SCOUT 384 ion source and Flex control software) was utilized. The methods used were optimized for 17 and 66 kDa in a linear mode.
A Bruker concentration MTP AnchorChip target plate with 400 nm anchors was used.
Three different matrices were used: DHB, SA, and DHAP, all at a concentration of 20 mg mL−1 dissolved in ACN:0.1 % TFA (1:1). For all studies, the dried droplet method was used with 0.5 μL of matrix applied to the MALDI target followed by 0.5 μL sample solution. Mass spectra were obtained by sweet spot searching (20 shots on each position summed up to 200), with a laser intensity of 75 %. Peak picking was obtained with the analyzing program XTOF using the centroid peak picking algorithm (Bruker Daltonics).
Cytochrome C, apomyoglobin, aldolase, and albumin from bovine serum at a concentration of 1 μM each were used for calibration.
Investigated samples included BSA and LYS (at concentrations of 1 μM without any chromium added) and each protein mixed with Cr(VI) to predetermined molar ratios (chromium to protein). The protein concentrations were adjusted to a molar ratio of 1:1 [19.2 μM Cr(VI):19.2 μM BSA] and 27:1 [173 μM Cr(VI):6.4 μM BSA], and of 1:3 [19.2 μM Cr(VI):57.6 μM LYS] and 9:1 [173 μM Cr(VI):19.2 μM LYS)].
3 Results and discussion
To enable studies of chromium–protein complexation using differently sized and charged proteins by means of stripping voltammetry, the known interference of protein adsorption on the HMDE had to be investigated for the specific proteins investigated. In the following, the extent of protein adsorption of the HMDE was explored for BSM, HSA/BSA and LYS, and the extent of protein–chromium complexation determined at low protein concentrations by means of stripping voltammetry and MALDI-TOF–MS.
3.1 Protein adsorption on mercury
The extent of protein adsorption onto the HMDE during the chromium–protein complexation measurement depends not only on protein concentration but may also depend on the chromium concentration in solution, as for instance Cr–DTPA complexes (via the supporting electrolyte) may competitively adsorb on the HMDE surface. This possibility was not taken into account during the adsorption studies. However, due to the larger size of the proteins, we expect the proteins to have a higher affinity for the HMDE surface compared to the small Cr–DTPA complexes as a consequence of gained entropy due to an exchange from counter-ions of the electrolyte to adsorbed proteins. The extent and influence of protein adsorption were evaluated by analyzing a reduction peak induced by the proteins at approximately −1.1 V (vs. Ag/AgCl), c.f. Fig. 2a, b. In Fig. 2a, b, the current–potential curve corresponding to the maximum peak of the concentration range (c.f. Figs. 3, 4) is shown, i.e., 0.016 and 0.018 g−L−1 for LYS, 0.014 g L−1 for HSA, and 0.6 g L−1 for BSM. The reduction process used to assess the amount of protein adsorption is unclear, however, similar data has been reported elsewhere [6, 9, 10, 44]. Several polarographic studies have observed a similar peak at −0.9 to −1.2 V (vs. SCE) at high (μM) protein concentrations [10] attributed to irreversibly [44] accepted electrons at the –S–S– bonds [9]. The reduction peak of this study was observed at significantly lower (nM) protein concentrations but of lower height at high (μM) concentrations compared to the literature findings. A similar peak has previously been observed for a sodium pyruvate solution in which –S–S– bonds were not present (but a C=O group) [6], c.f. Fig. 2c. These observations imply another possible reason for the reduction peak, namely the reduction of any accessible C=O group. Similar interpretations have been reported in Cosma et al. [45] reporting a weak peak at −1.0 to −1.1 V versus SCE in addition to a strong peak at −0.66 V versus SCE attributed to chlorophyll A. The presence of the reduction peak in this study originating from the protein solution (c.f. Fig. 2a, b) was not as pronounced as in the case of the sodium pyruvate solution (Fig. 2c). At present, this is not fully understood. The peak height was for all proteins concentration-dependent. As LYS was the only net positively charged protein at pH 6.2, it is furthermore expected to be adsorbed to a larger extent, and possibly to form stronger bonds at the negatively charged mercury electrode compared with the other proteins.

Current–potential curves of solutions (the same conditions as for chromium determination, see “Materials and methods”) containing a two independent replicate measurements of LYS, b HSA and BSM, and c 0.7 g L−1 sodium pyruvate (C3H3O3Na). Inset graph The same current scale as for sodium pyruvate (c). The protein concentrations that resulted in the maximum peaks which are shown here were 0.016 and 0.018 g L−1 for LYS, 0.014 g L−1 for HSA, and 0.6 g L−1 for BSM

Peak height corresponding to LYS over the accumulated LYS molar concentration obtained at a potential of −1.10 V versus the Ag/AgCl/KCl sat. reference electrode. The first (LYS_1) and second (LYS_2) sets of data were generated using different LYS solutions of the same batch

Peak height (−1.05 to −1.10 V) corresponding to mucin (BSM) and human serum albumin (HSA) proteins over the accumulated protein concentrations. The same protein solutions as used in the comparative chromium complexation capacity studies are used for the first measurement of BSM (BSM_1) and HSA (HSA_1)
The adsorption of proteins on the HMDE was studied by continuously adding proteins to an initially protein-free electrolyte solution and analyzing changes in the protein peak height (proportional to changes in peak area, as the peak width is constant). Changes in peak height versus the total protein concentration are illustrated for LYS in Fig. 3. An increased concentration initially resulted in an increased peak height that reached a maximum before it decreased and finally approached an almost constant value. This initial increase reaching a maximum is in concordance with previous studies on protein adsorption on mercury [25]. However, to the best of the knowledge of the authors, no other studies have reported decreasing peak heights and finally almost constant values at higher protein concentrations. A hypothesis is proposed to explain this observation. At low adsorption of proteins at the mercury drop (no peaks or linear increase of peak height), both adsorbed proteins and proteins in solution contribute to the peak height. The contribution from proteins in solution is reduced as the protein adsorption on the mercury drop increases (as visible as a slower increase of peak height, followed by decreasing peak height). Finally, at constant peak heights, only proteins adsorbed at the mercury drop surface are proposed to contribute to the observed peak as proteins in solution are shielded from the mercury surface. According to measurements of Stankovich and Bard [46] investigating the effect of scan rate and concentration of insulin solutions, the reduction wave at −1.2 V versus SCE was shown to be mostly diffusion-controlled (solution species) rather than adsorption-controlled. Findings of this study suggest that both diffusion and adsorption control are possible but largely related to the protein concentration. Adsorption control was evident in the case of high LYS concentrations (>1,000 nM, Fig. 3), an effect clearly illustrated by two independent measurements, Fig. 3. As the mercury drop size was slightly different between the measurements, the absolute values are different but the trends are similar. The first set of data was compiled over a more narrow concentration range (1,100–13,000 nM) compared to the second set, covering a much wider range (4–26,000 nM). Generated results show a minor influence of protein adsorption on the mercury drop at LYS concentrations <265 nM (no peak height or linear increase), however, as it was difficult to see any linearity between 60 and 265 nM, it is more safe to take a threshold value of 60 nM (0.0008 g L−1), below which no peak was observed.
Analogous investigations conducted for HSA and BSM (net negatively charged at pH 6.2, same as the mercury drop) are illustrated in Fig. 4. Non-reproducible threshold values were obtained for both proteins. No significant effect of protein adsorption on the mercury drop was observed for concentrations up to 85 nM for BSM, and up to 211 nM for HSA, in a first measurement. For these proteins (net negatively charged at pH 6.2, same as the mercury drop), the peak corresponding to the protein (at −1.1 V) increased linearly with added concentration. However, in a second measurement using the same batch and preparation, but slightly different mercury drop size, a poorly resolved peak (at −1.1 V) was observed due to protein reduction. This peak could not be quantified in the case of BSM and was difficult to quantify for HSA. For BSM, the signal was <0.1 nA, and for HSA, adsorption was non-significant up to 85 nM (linear increase of peak height), Fig. 4, which is in contrast to the first measurement (>211 nM).
The poor reproducibility between the different measurements for HSA, Fig. 4, and BSM could be attributed to small differences of the mercury drop size that may result in slight differences in surface charge (still net negatively charged), effects not crucial for the adsorption of LYS being net positively charged, but notable for BSM and HSA being net negatively charged. From these adsorption studies, it was assumed that a protein concentration exceeding 60 nM of LYS, 85 nM of BSM, and 85 nM of HSA/BSA resulted in an adsorption of proteins on the mercury drop that hindered accurate studies of proteins in solutions and hence protein-complexation studies. In addition, the protein peak potential shifted to more negative potentials for all proteins investigated (LYS, HSA, and BSM) with increasing protein concentrations, i.e., up to 40 mV for LYS, 50 mV for BSM, and 55 mV for HSA. This shift was not pronounced at the low concentrations proposed for complexation studies. Similar shifts to more negative potentials due to adsorption processes have been previously reported for peptides and fulvic substances [23, 26].
3.2 Chromium–protein complexation—influence of protein type and protein concentration
To form a stable chromium complex, the protein must first reduce Cr(VI) (as negatively charged chromate) to Cr(III) (positively charged) [47]. As Cr(III) results in significantly lower signals using DPAdCSV, an electrochemical reduction of Cr(VI) to Cr(III) taking place that is not induced by the formation of protein–chromium complexes may erroneously be assigned as protein complexation. Investigations were therefore conducted with BSM (negatively charged) to assess if a Cr(III) addition, instead of a Cr(VI) addition, would result in different chromium–protein complexation. Differences in complexation between differently charged chromium species and charged proteins could be a consequence of electrostatic repulsion between similar charged species. No significant complexation was observed either with Cr(VI) or with Cr(III) [linearly increasing signal with addition of Cr(III) (R 2 = 0.993) and for Cr(VI) (R 2 = 0.995)] at a protein concentration of 0.6 nM (0.004 g L−1) (data not shown). The measurements were performed with a significantly shorter equilibrium time after the addition of Cr(III), only 5 s compared with 300 s [for Cr(VI)], due to a decreasing signal of Cr(III) with time [41]. Even though these findings imply similar results independent of chromium speciation in the case of BSM, the situation may be different for LYS, HSA, and/or BSA (not investigated in this study) as suggested by Tkaczyk et al. for BSA [3]. The approach of this study should at this stage hence be only applied for the methodology adding Cr(VI).
Chromium–protein complexation investigations by means of stripping voltammetry were conducted for proteins of varying size, charge, and structure (LYS, BSA, HSA, and BSM) in concentrations lower than the proposed threshold values to minimize the effect of adsorbed proteins on the HMDE surface, with the exception of LYS at 280 nM, Fig. 5. The results are presented both as a function of given mass concentration (4 mg L−1), Fig. 5, and given molar concentration (59 nM), Fig. 6.

Stripping voltammetry measurements of free non-complexed Cr(VI) (peak height) as a function of known concentration of Cr(VI) (by addition) in protein solutions (BSM—0.6 nM, BSA—59 nM, HSA—59 nM, LYS—281 nM) of constant mass concentration (3.97 mg L−1). A reference electrolyte containing no protein (single measurement) is included for comparison. Data is based on one measurement with two replicate readings (error bars) for BSM and LYS, and on two separate measurements, each with two replicate readings for BSA and HSA. The dotted lines are only added as visual guides

Stripping voltammetry measurements of free non-complexed Cr(VI) (peak height) as a function of known concentration of Cr(VI) (by addition) in protein solutions (BSM—0.4 g L−1, BSA—0.004 g L−1, HSA—0.004 g L−1, LYS—0.0008 g L−1) of constant molar concentration (59 nM). A reference electrolyte containing no protein (single measurement) is included for comparison. Data based on one measurement with two replicate readings (error bars) for BSM and LYS, and on two separate measurements, each with two replicate readings for BSA and HSA. The dotted lines are only added as visual guides
Generated results for the same solution without any proteins are included for comparison and show a relatively linear increase of the chromium peak height with added Cr(VI) (Figs. 5, 6). At 40 nM Cr(VI), the peak height was ~175 nA for the solution without proteins. At low mass concentrations of BSM (0.6 nM, Fig. 5) and at low molar concentration of LYS (59 nM, Fig. 6), the chromium signal was enhanced compared to the non-protein containing solution. This has also been previously observed for glycine [6]. This catalytic effect is known for nitrate which therefore is a component of the supporting electrolyte with the function to enhance the signal by oxidizing Cr(II)–DTPA back to Cr(III)–DTPA [41].
Observed differences in complexation capacity with chromium correlated with corresponding protein size (BSM > HSA/BSA > LYS). At constant mass concentration, Fig. 5, the molar concentration of BSM was the lowest (0.6 nM), followed by BSA/HSA (59 nM) and LYS of 281 nM. LYS, with the highest molar concentration, resulted in the strongest complexation to added chromium, which partially also could be an effect of protein adsorption on the HMDE, while BSM of very low molar concentration (0.6 nM) did not result in any complexation [an initially relatively linear increase in peak height with added Cr(VI)].
According to the findings of this study, the number of amino acid groups available for complexation plays a larger role in the metal complexation process compared with the structure of the protein or protein charge. For example, even though BSM has a large carbohydrate content that constitutes ~80 % of the total protein by mass [48], it has a larger number of amino acid groups compared with LYS and BSA due to its large molecular size (7,000 kDA) in comparison to LYS (14.1 kDa, 129 amino acids per protein [33]) and BSA (66 kDA, 582 amino acids per protein [49]) at the same molar concentration. As a consequence, BSM also has the highest degree of chromium complexation, Fig. 6. In comparison, LYS has the largest number of amino acids at constant mass concentration compared to BSM and BSA, Fig. 5, and therefore the highest chromium complexation.
The results clearly show that LYS, the smallest protein investigated, formed the significantly largest number of chromium complexes per mass unit of protein, Fig. 5, followed by a significantly lower complexation to chromium for HSA and BSA, and BSM. In contrast, at constant molar concentration, Fig. 6, the largest protein, BSM, formed the largest number of chromium complexes, followed by HSA/BSA [approximately one chromium every 20th protein, based on the amount of added Cr(VI) without giving any signal (3 nM) in a 59 nM protein solution], and LYS, which showed no measurable complexation at this concentration.
Detailed studies on the effect of protein concentration on chromium complexation were conducted with BSM. To avoid any significant adsorption of BSM on the mercury drop, low protein concentrations, 1 nM and less, were investigated, Fig. 7. From the results, it was evident that complexation [corresponding to the reduction signal of free non-complexed Cr(VI)] increased nonlinearly with increasing concentration. At the highest molar concentration investigated (1 nM BSM), ~18 chromium ions were complexed to one protein. At lower BSM concentrations, complexation could not be quantified but was significantly lower. The observed nonlinearity of BSM to chromium complexation in this concentration interval could be an effect of the sum of two processes, (i) enhancement of the chromium signal, similar to the effect of nitrate [41], as previously discussed and (ii) lowering of the signal by protein complexation to chromium.

Stripping voltammetry measurements of free non-complexed Cr(VI) (peak height) as a function of known concentration of Cr(VI) (by addition) in BSM solutions of different molar concentrations (0.2, 0.3, 0.4, and 1.0 nM). A reference electrolyte containing no protein (single measurement, 0 nM BSM) is included for comparison
MALDI-TOF–MS measurements were conducted for comparison and to possibly support the findings by means of stripping voltammetry measurements, Fig. 8. The best mass spectra to investigate complexation were obtained with DHAP, in comparison to DHB and SA, although all matrices showed the same results regarding complexation. The DHAP matrix is considered as a soft matrix, suitable for studies of protein quaternary structures and non-covalent protein complexes [50]. Any bonding of chromium to the protein investigated would result in a positive mass shift of 52 Da for each chromium entity when compared to the native mass of the protein. No strong bonds were evident between chromium and any of the proteins investigated (LYS and BSA), Fig. 8, as no significant mass shifts or additional peaks evident for chromium complexation were observed. A degraded MS-signal was observed for the highest chromium concentrations [27:1 and 9:1 Cr(VI):protein, for BSA and LYS, respectively], Fig. 8a (top), an effect possibly induced by the increased ion concentration, even though MALDI is generally regarded as a relatively high ionic strength tolerant MS-method. The correct mass of the peak was difficult to be determined from the broad peak, and no clear indication of a mass shift was observed despite the high chromium to BSA ratio (27:1) investigated. The results clearly show that there were no strong interactions such as covalent bonds between the proteins and chromium. Similar conclusions were drawn from stripping voltammetry measurements as no visible peak shifts [13] of either the chromium peak at −1.22 V or the protein peak (both at different protein concentrations) were observed (data not shown).

Mass spectra of a BSA [M+H]+ and b LYS [M+H]+ generated by means of MALDI-TOF–MS. Lower spectra Pure protein (1 μM), middle spectra Cr(VI) to protein ratio of 1:1 (BSA) and 3:1 (LYS), and upper spectra Cr(VI) to protein ratio of 27:1 (BSA) and 9:1 (LYS). The spectra are offset for clarity
However, weaker associations such as electrostatic interactions and interactions of less than one chromium per protein may still take place, but it is possible that these will not be detected with MALDI-TOF–MS.
Both methods used, stripping voltammetry and MALDI-TOF–MS, imply weak chromium complexation with BSA and LYS, at those conditions studied. Further studies should include evaluation of the “peak H”, shown to be sensitive to protein changes [11, 29–32] as well as electrochemical investigations to possibly assess different binding of trivalent and hexavalent chromium to proteins, as this study only investigates hexavalent chromium.
4 Conclusions
Stripping voltammetry and MALDI-TOF–MS measurements were conducted aiming for an in-depth understanding of chromium complexation with proteins of different concentration, structure and charge. The following main conclusions are drawn:
-
The effect of protein adsorption at the hanging mercury drop needs to be taken into account when using stripping voltammetry for studies of metal–protein complexation in solution. As the extent of adsorption is strongly related to protein concentration, reproducible complexation studies can only be conducted in solutions of low protein concentration [<60 nM (LYS) and <85 nM (HSA/BSA/BSM)].
-
Chromium complexation was favored by an increased protein size and by an increased molar concentration of proteins in solution. The highest complexation per mass was evident for LYS, the protein with the largest number of amino acid groups per mass (and net positively charged), compared with BSA/HSA and BSM. BSM showed in contrast the highest complexation to chromium at identical molar concentration compared with LYS and BSA/HSA due to its larger number of amino acid groups per mole protein.
-
Generated results by means of both stripping voltammetry and MALDI-TOF–MS measurements suggest weaker interactions such as electrostatic interactions between chromium and protein in solution rather than stronger interactions such as covalent bonding. This interpretation is justified from the absence of any peak shift of either the protein or the chromium peak in the stripping voltammogram and by the absence of peak shifts in the mass spectra of MALDI-TOF–MS.
References
Vincent JB (2000) J Nutr 130:715–718
Nakayama E, Kuwamoto T, Tsurubo S, Tokoro H, Fujinaga T (1981) Anal Chim Acta 130:289
Tkaczyk C, Huk OL, Mwale F, Antoniou J, Zukor DJ, Petit A, Tabrizian M (2010) J Biomed Mater Res 94A:214
Salnikow K, Zhitkovich A, Costa M (1992) Carcinogenesis 13:2341
Lundin M, Hedberg Y, Jiang T, Herting G, Wang X, Thormann E, Blomberg E, Odnevall Wallinder I (2012) J Colloid Interface Sci 366:155
Hedberg Y, Hedberg J, Liu Y, Odnevall Wallinder I (2011) Biometals 24:1099
Midander K, de Frutos A, Hedberg Y, Darrie G, Odnevall Wallinder I (2010) Integr Environ Assess Manag 6:441
Mikulewicz M, Chojnacka K (2010) Biol Trace Elem Res 137:127
Heyrovsky M (2004) Electroanalysis 16:1067
Honeychurch MJ (1997) Bioelectrochem Bioenergy 44:13
Paleček E, Ostatná V (2007) Electroanalysis 19:2383
Paleček E (2009) Electroanalysis 21:239–251
Christian GD, Knoblock EC, Purdy WC, Mertz W (1963) Biochim Biophys Acta 66:420
Buffle J, Cominoli A (1981) J Electroanal Chem 121:273
Buffle J, Greter FL (1979) J Electroanal Chem 101:231
Buffle J, Vuilleumier JJ, Tercier ML, Parthasarathy N (1987) Sci Total Environ 60:75
Chekmeneva E, Díaz-Cruz JM, Ariño C, Esteban M (2006) Anal Biochem 348:252
Cominoli A, Buffle J, Haerdi W (1980) J Electroanal Chem 110:259
Heyrovský M, Mader P, Veselá V, Fedurco M (1994) J Electroanal Chem 369:53
Mota AM, Pinheiro JP, Gonçalves ML (1994) Water Res 28:1285
Tanford C (1952) J Am Chem Soc 74:6036
Claesson PM, Blomberg E, Fröberg JC, Nylander T, Arnebrant T (1995) Adv Colloid Interface Sci 57:161
Greter FL, Buffle J, Haerdi W (1979) J Electroanal Chem Interfacial Electrochem 101:211
Raspor B (1991) J Electroanal Chem Interfacial Electrochem 316:223
Scheller F, Jänchen M, Prümke H-J (1975) Biopolymers 14:1553
Harlyk C, Bordin G, Nieto O, Rodriguez AR (1997) Electroanalysis 9:608
Guidelli R, Aloisi G, Becucci L, Dolfi A, Rosa Moncelli M, Tadini Buoninsegni F (2001) J Electroanal Chem 504:1
Forsman U (1981) J Electroanal Chem 122:215
Paleček E, Ostatná V, Černocká H, Joerger AC, Fersht AR (2011) J Am Chem Soc 133:7190
Doneux T, Ostatná V, Paleček E (2011) Electrochim Acta 56:9337
Ostatná V, Kuralay F, Trnková L, Paleček E (2008) Electroanalysis 20:1406
Ostatná V, Uslu B, Dogan B, Ozkan S, Palecek E (2006) J Electroanal Chem 593:172
Imoto T, Johnson LN, North ACT, Phillips DC, Rupley JA (1972) Vertebrate lysozymes. In: Boyer P (ed) The enzymes. Academic Press, New York
Roach P, Farrar D, Perry CC (2006) J Am Chem Soc 128:3939
He XM, Carter DC (1992) Nature 358:209
Bastardo L, Claesson P, Brown W (2002) Langmuir 18:3848
Bettelheim FA, Hashimoto Y, Pigman W (1962) Biochim Biophys Acta 63:235
Bettelheim FA (1963) Ann NY Acad Sci 106:247
Metrohm, Application Bulletin 116, http://products.metrohm.com/applications/ab-application-bulletins/ab_101-200/prod-AB-116.aspx. Accessed 25 March 2012
Golimowski J, Valenta P, Nürnberg HW (1985) Fresenius J Anal Chem 322:315
Bobrowski A, Królicka A, Zarebski J (2009) Electroanalysis 21:1449
Turner DR (ed) (1995) Metal speciation and bioavailability in aquatic systems. Wiley, Buffins Lane
Sander S, Navrátil T, Novotný L (2003) Electroanalysis 15:1513
Kuznetsov BA, Shumakovich GP, Mestechkina NM (1988) J Electroanal Chem Interfacial Electrochem 248:387
Cosma P, Longobardi F, Agostiano A (2004) J Electroanal Chem 564:35
Stankovich MT, Bard AJ (1977) J Electroanal Chem Interfacial Electrochem 85:173
Rollinson CL (1975) The chemistry of chromium, molybdenum and tungsten. Robert Maxwell MC, Oxford
Svensson O, Arnebrant T (2010) Curr Opin Colloid Interface Sci 15:395
Peters TJ (1985) Adv Protein Chem 37:161
Cohen L, Strupat K, Hillenkamp F (1997) J Am Soc Mass Spectrom 8:1046
Acknowledgments
Financial support from the Swedish Research Council, VR, is highly acknowledged. The authors are members of the Stockholm Particle Group, an operative network between three universities in Stockholm: Royal Institute of Technology, Karolinska Institutet, and Stockholm University, supported by the Swedish Research Council. Cusanuswerk, Germany, is acknowledged for the financial support of Yolanda Hedberg. The Swedish Steel Association and Carl Trygger Foundation for the voltammetric equipment are highly acknowledged. Dr. Troy Lowe and Dr. Jonas Hedberg, Division of Surface and Corrosion Science, KTH are both highly acknowledged for their scientific support.
Conflict of interest
The authors declare that they have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Hedberg, Y., Lundin, M., Jacksén, J. et al. Chromium–protein complexation studies by adsorptive cathodic stripping voltammetry and MALDI-TOF–MS. J Appl Electrochem 42, 349–358 (2012). https://doi.org/10.1007/s10800-012-0404-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-012-0404-6