
Abstract
Following an acute myocardial infarction (AMI), early coronary artery reperfusion remains the most effective means of limiting the eventual infarct size. The resultant left ventricular systolic function is a critical determinant of the patient’s clinical outcome. Despite current myocardial reperfusion strategies and ancillary antithrombotic and antiplatelet therapies, the morbidity and mortality of an AMI remain significant, with the number of patients developing cardiac failure increasing, necessitating the development of novel strategies for cardioprotection which can be applied at the time of myocardial reperfusion to reduce myocardial infarct size. In this regard, the Reperfusion Injury Salvage Kinase (RISK) Pathway, the term given to a group of pro-survival protein kinases (including Akt and Erk1/2), which confer powerful cardioprotection, when activated specifically at the time of myocardial reperfusion, provides an amenable pharmacological target for cardioprotection. Preclinical studies have demonstrated that an increasing number of agents including insulin, erythropoietin, adipocytokines, adenosine, volatile anesthetics natriuretic peptides and ‘statins’, when administered specifically at the time of myocardial reperfusion, reduce myocardial infarct size through the activation of the RISK pathway. This recruits various survival pathways that include the inhibition of mitochondrial permeability transition pore opening. Interestingly, the RISK pathway is also recruited by the cardioprotective phenomena of ischemic preconditioning (IPC) and postconditioning (IPost), enabling the use of pharmacological agents which target the RISK pathway, to be used at the time of myocardial reperfusion, as pharmacological mimetics of IPC and IPost. This article reviews the origins and evolution of the RISK pathway, as part of a potential common cardioprotective pathway, which can be activated by an ever-expanding list of agents administered at the time of myocardial reperfusion, as well as by IPC and IPost. Preliminary clinical studies have demonstrated myocardial protection with several of these pharmacological activators of the RISK pathway in AMI patients undergoing PCI. Through the use of appropriately designed clinical trials, guided by the wealth of existing preclinical data, the administration of pharmacological agents which are known to activate the RISK pathway, when applied as adjuvant therapy to current myocardial reperfusion strategies for patients presenting with an AMI, should lead to improved clinical outcomes in this patient group.
Similar content being viewed by others
Introduction
For patients presenting with an acute myocardial infarction (AMI), it is well-established that early, effective myocardial reperfusion using either thrombolysis or primary percutaneous coronary intervention (PCI), remains the most powerful intervention for limiting myocardial infarct size. However, the morbidity and mortality from an AMI remains significant, necessitating the development of new strategies for cardioprotection, which can further reduce myocardial infarct size and improve clinical outcomes in this patient group. Ideally, any such novel cardioprotective strategy, would be available to be applied in conjunction with the current myocardial reperfusion therapy, and be demonstrated to confer a cardioprotective effect, in terms of myocardial infarct size reduction and improved clinical outcomes, over and above that elicited by coronary artery reperfusion and ancillary therapies (such as antiplatelet and antithrombotic treatments).
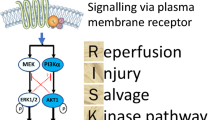
In this regard, the Reperfusion Injury Salvage Kinase (RISK) pathway, a term given to describe a group of survival protein kinases which include Akt and Erk1/2 that confer powerful cardioprotection, when specifically activated at the time of myocardial reperfusion, represents a novel target for cardioprotection in AMI patients [1, 2]. However, despite the abundance of preclinical data demonstrating effective cardioprotection with a variety of different agents given at the time of myocardial reperfusion to activate the RISK pathway, clinical studies are limited, a situation which should change with the revelation that both ischemic preconditioning and postconditioning also recruit the RISK pathway [3, 4], thereby regenerating interest in the myocardial reperfusion phase as a viable target for cardioprotection in AMI patients.
Ischemic preconditioning and postconditioning: ‘United’ by the RISK pathway
The requirement for intervening at myocardial reperfusion in AMI patients, renders ischemic preconditioning (IPC) ineffective as a cardioprotective intervention, given that its protective effect results from the application of one or more short-lived episodes of ischemia and reperfusion, applied before the index ischemic event [5], which is naturally unpredictable in AMI patients. This restricts its utility to scenarios in which the index myocardial ischemic episode can be reliably anticipated such as in those undergoing coronary artery bypass graft (CABG) surgery [6] or in unstable angina patients presenting with a threatening myocardial infarct [7]. However, emerging studies suggest that the signal transduction pathways underlying the cardioprotection elicited by IPC converge at the myocardial reperfusion phase, with the RISK pathway identified as a key component [8–10], thereby enabling the use of pharmacological agents that target components of the RISK pathway to harness the protective benefits of IPC for AMI patients.
As an interventional strategy which can be applied at the time of myocardial reperfusion, the recently introduced phenomenon of ischemic postconditioning (IPost), that describes the cardioprotective effect elicited by interrupting myocardial reperfusion with short-lived episodes of myocardial ischemia interspersed with reperfusion following the index ischemic event [11], offers an amenable strategy for cardioprotection in AMI patients, and its introduction has succeeded in renewing interest in the myocardial reperfusion phase as a target for cardioprotection [12, 13]. The clinical efficacy of IPost as a cardioprotective strategy has already been demonstrated in several small clinical studies of patients undergoing primary PCI, using an invasive IPost protocol comprising serial low-pressure coronary angioplasty inflations and deflations immediately following the deployment of the stent in the infarct-related coronary artery [14–17]. However, the widespread use of IPost in the clinical arena is likely to be limited by both its invasive nature and the fact that it is restricted to AMI patients undergoing PCI. A more amenable approach will be to mimic IPost, using pharmacological agents that target the RISK-pathway that has also been identified as underlying IPost-induced cardioprotection, thereby obviating the need for such an invasive IPost protocol.
This article will review the origins and evolution of the RISK pathway as a potential common cardioprotective pathway which can be activated by administering agents given either at the time of reperfusion or prior to the index ischemic event. The non-pharmacological activation of the RISK pathway as part of a common cardioprotective pathway which ‘unites’ IPC and IPost at the time of myocardial reperfusion, and the clinical implications of the RISK pathway, as a novel cardioprotective target will also be covered. This article will focus only on those protein kinases specifically modulated at the time of myocardial reperfusion in the setting of cardioprotection, and the reader is directed to several other reviews for a more comprehensive account detailing the contribution of protein kinases to cardioprotection [18–22].
The origins and evolution of the RISK pathway
The Reperfusion Injury Salvage Kinase (RISK) pathway emerged as a concept in the late 1990s with the recognition that apoptotic cell death contributed to lethal reperfusion injury [23–25], and the knowledge that there existed certain pro-survival anti-apoptotic protein kinases, the original members of which were Akt and Erk1/2, which when specifically activated at the time of myocardial reperfusion conferred powerful cardioprotection [1, 2]. Studies had previously demonstrated activation of these protein kinases Akt [26, 27], Erk1/2 [28] and JNK [28, 29] at the time of myocardial reperfusion in control hearts, but clearly the activation of the RISK pathway in these settings was not sufficient to confer cardioprotection, and an additional pharmacological stimulus was required to enhance the activation of the RISK pathway. Other studies had confirmed the cardioprotective potential of both Akt [30, 31] and Erk1/2 [32] using transgenic activation of these kinases.
Alongside an ever expanding list of diverse pharmacological agents, demonstrated to confer cardioprotection when administered at the time of myocardial reperfusion through the activation of the RISK pathway (see Table 1), the concept of the RISK pathway has evolved to encompass several novel features: (a) the RISK pathway can be activated by interventions instituted prior to the index ischemic event, which includes pharmacological preconditioning agents, such as isoflurane [88] and opioids [89], as well as by IPC itself [4]; (b) the RISK pathway now includes other cardioprotective reperfusion salvage kinases such as PKC (primarily the PKC-ε isoform), PKG, p70s6K, and GSK-3β; (c) there are protein kinases such as PKC-δ and rho-kinase which when activated at the time of myocardial reperfusion are pro-injurious and counteract the cardioprotection elicited by the RISK pathway. The roles played by p38 and JNK MAPK are controversial, a term frequently used when discussing these protein kinases in the context of cardioprotection [19, 20, 94], with studies reporting both cardioprotective and pro-injurious roles of these kinases at the time of myocardial reperfusion [80, 88, 90]; and (d) the RISK pathway appears to be a core component of a common cardioprotective pathway which converges on the mitochondrial permeability transition pore, that appears to ‘unite’ both IPC and IPost at the time of myocardial reperfusion [95].
The critical time ‘Window’ for RISK pathway activation
Preclinical studies clearly demonstrate that cardioprotection at the time of myocardial reperfusion through the activation of the RISK pathway whether that be by administering insulin [35] or by applying an IPost protocol [96], must be instituted at the immediate onset of reperfusion to be effective, suggesting the existence of a critical time ‘window’ for cardioprotection. During the first few minutes of myocardial reperfusion, in response to the generation of ROS, an increase in mitochondrial Ca2+, the restoration of normal pH, the mitochondrial permeability transition pore (mPTP) opens [97–99], mediating cell death by uncoupling oxidative phosphorylation and inducing mitochondrial swelling [100, 101]. Pharmacologically inhibiting mPTP opening after the first few minutes of myocardial reperfusion have elapsed is ineffective, confirming the existence of this critical time ‘window’ of cardioprotection [102]. The implications of these findings for the patient presenting with an AMI are that, to be effective as a cardioprotective intervention, any pharmacological agent used to activate the RISK pathway, needs to be administered either prior to or at the immediate onset of myocardial reperfusion, and such a requirement should be a critical feature in the design of a clinical trial.
Activation of the RISK pathway by intervening at the time of reperfusion
The clinical requirement to demonstrate that a potential cardioprotective agent can attenuate myocardial injury, when given specifically at the time of myocardial reperfusion, has resulted in an increasing number of pharmacological agents being linked to the activation of the RISK pathway (see Table 1). Growth factors were the first group of agents demonstrated to exert a cardioprotective effect at the time of myocardial reperfusion through the activation of the RISK pathway, but this group has now grown to include various other receptor ligands as well as non-pharmacological activators of the RISK pathway such as IPost.
Growth factors as activators of the RISK pathway
Among the first growth factors demonstrated to confer cardioprotection when administered specifically at the time of myocardial reperfusion through the activation of one or more components of the RISK pathway are outlined below (and see Table 1). Ligand binding at the growth factor receptor, results in the activation of its receptor tyrosine kinase, which then activates the PI3K-Akt and Ras-MEK1-2-Erk1/2 signalling cascades.
Transforming Growth Factor-β1 (TGF-β1)
A cytokine that regulates cell growth and differentiation and modulates apoptosis in many cell types, has previously been demonstrated to confer cardioprotection [103]. Baxter et al. [33] demonstrated that TGF-β1 administered at the time of myocardial reperfusion or reoxygenation reduced myocardial infarct size and attenuated apoptotic cardiomyocyte death, respectively, effects which were abolished by the MEK1/2 inhibitor PD98059;
Insulin
Insulin has been reported to exert cardioprotection when administered at the time of myocardial reperfusion through the activation of the PI3K-Akt [34–36] component of the RISK pathway and the recruitment of downstream targets including the phosphorylation of p70S6K [35], BAD [35], and eNOS [36].
Insulin-like growth factor-1 (IGF-1)
A polypeptide which regulates cell proliferation and differentiation in different cell types in response to a diverse array of stimuli, has been extensively demonstrated to cardioprotect through the activation of Akt [27, 104–108] and Erk1/2 [105] and their downstream signalling elements including the inhibition of BAX [107, 109, 110], enhanced Bcl-2 expression [109, 110], and inhibition of mPTP opening [110]. Studies have demonstrated improved recovery of LV systolic function and reduced myocardial injury with IGF-1 given specifically at the time of myocardial reperfusion in the perfused rat heart, an effect which was abrogated by the PI3K inhibitor [37]. Yamashita et al. [27] demonstrated that mice heterozygously over-expressing IGF-1 displayed enhanced cardioprotective Akt activation in response to myocardial reperfusion when compared to wild type controls.
Corticotrophin-1 (CT-1)
A member of the IL-6 group of cytokines had been originally isolated as a cardiomyocyte hypertrophic growth factor in 1995 [111], with subsequent studies implicating a cardioprotective role for this cytokine [112], through anti-apoptotic signalling pathways mediated by the activation of Erk1/2 [113]. Importantly, Brar et al. [38] found that both the pharmacological and genetic inhibition of PI3K, Akt and Erk1/2 abrogated the cardioprotective effect elicited by CT-1 when administered at the time of reoxygenation to neonatal rat cardiomyocytes, a finding confirmed using adult-rat cardiomyocytes and the perfused rat heart [39, 40].
Subsequent studies have linked growth factors such as fibroblast growth factor-2 (FGF-2) [41, 114], erythropoietin (EPO) [42, 43], and most recently the adipocytokines, leptin [45, 115], apelin (Simpkin et al. unpublished) and visfatin (Hausenloy et al. unpublished) with cardioprotection elicited at the time of myocardial reperfusion through the activation of one or more components of the RISK pathway (see Table 1). Most recently, Granulocyte Colony-Stimulating Factor (G-CSF), a cytokine that mediates the proliferation and differentiation of neutrophil progenitors, which has been found to prevent post-myocardial infarction cardiac remodelling through the JAK-STAT pathway [116], has been found to mediate cardioprotection when administered at the time of myocardial reperfusion through the activation of JAK2, STAT3, ERK1/2, Akt, and eNOS, with the suggestion that JAK2 which was reported to be upstream of the RISK pathway [44] may act as the intermediary between the cytokine receptor and the RISK pathway.
G-protein coupled receptor ligands as activators of the RISK pathway
Several agents have now been demonstrated to confer cardioprotection at the time of myocardial reperfusion by binding to their specific G-protein coupled receptor (GPCR) and activating the RISK pathway (see Table 1). Ligand binding at the GPCR, results in the transactivation of receptor tyrosine kinases which in turn activate the PI3K-Akt and Ras-MEK1-2-Erk1/2 signalling cascades.
-
(a)
Urocortin, a peptide related to corticotrophin-releasing factor, has been reported to protect neonatal cardiomyocytes against hypoxia-reoxygenation through the activation of Erk1/2, when given specifically at the time of reoxygenation [48], and reduce myocardial infarct size when administered at the time of myocardial reperfusion in both the perfused and in situ rat hearts [49]. Furthermore, it has been demonstrated that the cardioprotection elicited by both urocortin and its analogue, stresscopin, when given at the time of reoxygenation to neonatal rat cardiomyocytes was antagonized by the pharmacological and genetic inhibition of PI3K, Akt and Erk1/2 [46, 47].
-
(b)
Adenosine (through the use of various adenosine receptor agonists [51–53]), bradykinin [51, 54] and opioids [55] have all been demonstrated to reduce myocardial infarct size when administered at the time of myocardial reperfusion through the activation of the RISK pathway.
-
(c)
Adrenomedullin, a vasodilating peptide, which was first isolated from human phaeochromocytoma tissue in 1993 [117], binds to the calcitonin gene-related peptide like receptor. Animal studies have demonstrated that adrenomedullin given via adenoviral transfer [118, 119], acutely to the in situ rat heart [120], or specifically at the time of myocardial reperfusion [56], reduces myocardial infarct size and attenuates apoptotic cardiomyocyte death through the activation of the RISK pathway and downstream targets including BAX suppression, the phosphorylation of BAD, Bcl2 [118] and GSK-3β [119] and NO release [56].
-
(d)
Glucagon-Like Peptide-1 (GLP-1) is a gut incretin hormone which stimulates insulin secretion has emerged as a potential novel anti-diabetic agent [121]. Interestingly, our laboratory has demonstrated that GLP-1 administered at the time of myocardial reperfusion reduces myocardial infarct size through the activation of the RISK pathway [57].
Other receptor mediated activation of the RISK pathway
Natriuretic peptides
Both atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP), which bind to the natriuretic peptide receptor A, a membrane-bound guanylyl cyclase receptor, have been demonstrated to confer cardioprotection when given at the time of myocardial reperfusion [122, 123], although only ANP has been demonstrated to confer its protective effect through the activation of the RISK pathway. The mechanism through which ANP activates the RISK pathway is currently unclear.
Estrogens
Although the cardioprotective role of hormone replacement therapy is not clear, preclinical studies have demonstrated cardioprotection with estrogen replacement using a chronic myocardial infarction model in ovariectomized female mice through the activation of the PI3K-Akt pathway [124], and other studies have reported the cardioprotective effect of estrogens administered at the time of myocardial reperfusion [125]. More recently, the RISK pathway has been reported to mediate the infarct-limiting effects of 17-β estradiol and the phytoestrogen, genistein [60], a drug usually used to inhibit tyrosine kinase. Recent studies suggest that there exists a membrane bound estrogen receptor which has been demonstrated to be linked to activation of PI3K and MAPK’s [126, 127].
CGX-1051
A synthetic version of a peptide from Conus snail venom, has been reported by Zhang et al. [61] to reduce myocardial infarct size when administered at the time of myocardial reperfusion through MEK1/2 using the in situ rabbit heart.
Non-receptor mediated activation of the RISK pathway
Emerging studies suggest that components of the RISK pathway can be activated by pharmacological agents which exert their intracellular effect through non-receptor mediated mechanisms, the most extensively investigated being the volatile anesthetics.
Following the introduction of ischemic postconditioning as a cardioprotective strategy [11], and on the background of studies demonstrating the ability of volatile anesthetics to mimic the effects of ischemic preconditioning [128, 129] and exert cardioprotection when administered at the time of myocardial reperfusion [130, 131], a growing body of studies suggest that volatile anesthetics, when administered at the time of myocardial reperfusion are able to reduce myocardial injury through the recruitment of the RISK pathway [62], and its downstream targets, p70s6K, GSK-3β, eNOS, Bcl-2 and the mPTP [63, 68] both in the normal [63, 64, 67] and infarct-remodelled myocardium [65], thereby introducing the concept of ‘pharmacological’ or ‘anesthetic’ postconditioning.
Studies have reported that morphine was able to potentiate the cardioprotective effect of isoflurane administered at the time of reperfusion [66]. Furthermore, isoflurane [68, 132] and pharmacological inhibition of the pro-apoptotic protein p53 [133], have been found to potentiate the cardioprotective effects of IPost, suggesting the existence of a threshold level of stimulation, required to mediate cardioprotection at the time of myocardial reperfusion.
Other non-receptor mediated agents demonstrated to confer cardioprotection at the time of myocardial reperfusion through the activation of the RISK pathway include: (a) the 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor (‘statins’), atorvastatin [69, 70]. Pravastatin, Pitavastatin, and Cerivastatin given pre-ischemically have been demonstrated to activate Akt at myocardial reperfusion [91]. Simvastatin has been shown in separate studies to confer cardioprotection when given at the time of myocardial reperfusion [134] and activate Akt [135]; (b) the antidiabetic biguanide, metformin, administered at the time of myocardial reperfusion, have been reported to mediate a reduction in myocardial infarct size through the activation of Akt in perfused non-diabetic and diabetic rat hearts, and have been demonstrated to delay mPTP opening in rat cardiomyocytes subjected to oxidative stress through PI3K (Bhamra et al. Unpublished) However, the mechanism through which these agents mediate the activation of the RISK pathway at the time of myocardial reperfusion is currently unclear.
Mechanical activation of the RISK pathway: Ischemic postconditioning
Our laboratory was the first to demonstrate the link between the cardioprotection elicited by IPost and the recruitment of the RISK pathway, in a study in which we reported that the activation of Akt and the downstream p70s6K, contributed to the infarct-size reduction in postconditioned perfused rat hearts [3]. Several subsequent studies have confirmed the contribution of Akt activation to IPost-induced cardioprotection (see Table 1) [62, 71–73, 76, 77], including in diseased MI and LVH-remodelled rat hearts [74, 75], although a single study using perfused rabbit hearts failed to demonstrate Akt activation in postconditioned hearts [78].
The other component of the RISK pathway, Erk1/2, has also been linked to IPost-induced protection with activation of Erk1/2 in postconditioned hearts [77, 78, 78, 79, 136]. Interestingly, Schwartz et al. [136] demonstrated both Akt and Erk1/2 activation using in situ porcine hearts subjected to a non-cardioprotective IPost protocol, suggesting perhaps that before protection is observed, a threshold level of activation of the RISK pathway may be required.
Subsequent studies have demonstrated the activation of PKC as a critical mediator of IPost-induced cardioprotection implicating the PKC-ε isoform as the survival kinase in this setting (see Table 1) [81, 82, 84]. Where PKC activation is situated in relation to the activation of Akt and Erk1/2 is unclear, although pharmacological activation of PKC at the time of myocardial reperfusion was suggested to be upstream of both the adenosine receptor and Akt, although this study did not examine events occurring in IPost directly [81]. Preliminary studies have implicated PKG activation in the cardioprotection elicited by IPost [73, 87]. Finally, using neonatal rat cardiomyocytes, Sun et al. [80] found that hypoxic postconditioning mediated cardioprotection through the reduction in both p38 and JNK activity, finding which do not agree with those demonstrating no effect [93] or their activation [88, 90] at the time of myocardial reperfusion, but the role of these kinases in the setting of cardioprotection has frequently courted controversy.
Intriguingly, Bopassa and colleagues [71] have reported that the infarct-limiting effect observed with low-pressure reperfusion of isolated perfused rat hearts could be abolished by reperfusing with pharmacological inhibitors of PI3K, suggesting the potential involvement of the RISK pathway in low-pressure reperfusion, although Akt activity was not measured in this study. This form of controlled reperfusion may, in common with IPost, be simply a form of modified reperfusion which cardioprotects through the RISK pathway [137].
The mechanism through which the RISK pathway is activated in the setting of IPost, is unclear although it may be due to the ligand binding of either adenosine [138] or opioids [139] with their cell surface receptor, although this has not been directly demonstrated. A more recent proposal is that a transient period of acidosis at the time of myocardial reperfusion, perhaps mediating a delayed restoration of neutral pH from reduced lactate wash-out, may contribute to both the activation of the RISK pathway and cardioprotection observed in postconditioned hearts [77], a finding that may be expected to attenuate mPTP opening given that the restoration of neutral pH at the time of myocardial reperfusion is a critical determinant of mPTP opening [99].
Activation of the RISK pathway by intervening prior to Ischemia
Interestingly, several studies suggest that components of the RISK pathway (including Akt, Erk1/2 p38 MAPK, JNK MAPK) can be activated by an intervention applied prior to the index ischemic period, whether that be by pharmacological agents such as ‘statins’ [91] (using pravastatin, pitavastatin and cerivastatin), a δ1-opioid agonist [89], isoflurane [88], pioglitazone [92] or Ag II [93], administered as preconditioning mimetics, or the cardioprotective phenomenon of IPC [4, 9, 10]. IPC or pre-treatment with ‘statins’ resulted in the activation of Akt and/or Erk1/2 at the time of myocardial reperfusion and crucially, the administration of a pharmacological inhibitor of the RISK pathway at the time of reperfusion, abolished cardioprotection [4, 91], suggesting that the activation of the RISK pathway was essential for protection.
The mechanism through which a protective stimulus applied pre-ischemically appears to recruit the RISK pathway at the time of myocardial reperfusion is unclear, although potential explanations include: (a) the activation of the RISK pathway observed at the time of myocardial reperfusion is in fact a continuation of that initiated pre-ischemically by the preconditioning stimulus. This would suggest that the kinase activation initiated by the preconditioning stimulus needs to be sustained into the myocardial reperfusion phase to confer cardioprotection. A recent study suggests that Akt activation is required for up to 50–60 min into reperfusion, whereas Erk1/2 activation is needed only the first 5–10 min of reperfusion to mediate the cardioprotection elicited by IPC [9]. Evidence in support of this explanation is provided by studies reporting that inhibiting either MEK1/2, PKC or the mKATP channel during the preconditioning phase attenuated the Erk1/2 and p38 activation observed in hearts preconditioned with either IPC, a δ1-opioid agonist or isoflurane [88, 89]; (b) the preconditioning stimulus primes the protein kinases by inducing their intracellular translocation to their sites of action, such that at the time of myocardial reperfusion, kinase activation is enhanced; (c) the preconditioning stimulus may induce the release of certain growth factors within the myocardium which then augment RISK pathway activation at the time of myocardial reperfusion in conjunction with ROS, as observed in the study demonstrating enhanced Akt activation at the time of myocardial reperfusion in mice heterozygously over-expressing IGF-1, which exhibited raised endogenous levels of IGF-1 and Akt activation [27].The enhanced Akt activation observed at the time of reperfusion was abolished in the presence of the antioxidant N-acetylcysteine [27], suggesting a potential signalling role for ROS in this setting; (d) adenosine A1/A2B receptor ligand binding at the time of myocardial reperfusion in preconditioned hearts may mediate the activation of the RISK pathway [9]. Solenkova et al. [9] reported that the administration of a non-specific adenosine receptor blocker at the time of myocardial reperfusion abrogated both the infarct-limiting effect of IPC as well as the Akt activation observed at the time of reperfusion in preconditioned hearts. Whether endogenous adenosine is generated in greater quantities in preconditioned hearts, or whether specific adenosine receptors are more sensitive to adenosine, an effect perhaps mediated by PKC, a known mediator of IPC, is currently unknown.
Interestingly, when comparing the recruitment of the RISK pathway, there appear in some cases to be differences between pharmacological preconditioning mimetics and IPC. For example, IPC was found to activate both p38 and Erk1/2 at the time of myocardial reperfusion, whereas only Erk1/2 was activated in hearts preconditioned with isoflurane [88]. Furthermore, Lecour et al. [10] reported that although the preconditioning-mimetic TNF-α, in common with IPC, exerted its cardioprotective effect through the activation of STAT-3, it did not elicit its protective effect through the conventional components of the RISK pathway, Akt and Erk1/2.
An important study by Bell et al. [93] has extended the contribution of RISK pathway activation to the phenomenon of delayed preconditioning (in which a preconditioning stimulus has been demonstrated to elicit cardioprotection 12–24 h later [140]), and demonstrated dissociation between RISK pathway activation and cardioprotection. Using AgII as their preconditioning mimetic they demonstrated the activation of both Akt and Erk1/2 at the time of myocardial reperfusion in hearts at 1, 6 and 24 h following the preconditioning stimulus, yet cardioprotection, corresponding to the time-points of classical and delayed preconditioning, was only observed at 1 and 24 h, respectively [93]. Why RISK pathway activation and cardioprotection were dissociated at 6 h is unclear, but it may suggest the requirement for another factor such as PKC to be present to mediate cardioprotection, or that the intracellular localization of components of the RISK pathway and its downstream effectors might not be optimized at the 6-h time-point.
Effector mechanisms of RISK pathway activation
When the concept of the RISK pathway was originally conceived, the antiapoptotic signalling pro-survival pathways recruited by Akt and Erk1/2, were proposed as the mechanism through which the RISK pathway conferred cardioprotection at the time of myocardial reperfusion [2, 1]. This notion has been subsequently supported by the many studies reporting, the infarct-limiting effect of pharmacologically activating the RISK pathway, to be associated with the recruitment of antiapoptotic signalling systems such as the phosphorylation and inhibition of the proapoptotic proteins BAX and BAD, the inhibition of caspase 3 activation, and the phosphorylation and activation of p70s6K (which acts to inhibit BAD [141]) and the phosphorylation and activation of the antiapoptotic protein Bcl-2 [2](see Table 1 and Fig. 1).

Scheme demonstrating the diverse variety of agents which activate the Reperfusion Injury Salvage Kinase (RISK) pathway in both a receptor and non-receptor mediated manner. Interestingly, in addition to being activated by intervening at the time of myocardial reperfusion, the RISK pathway can also be activated by interventions applied pre-ischemically such as ischemic preconditioning and volatile anesthetics, opioids, and ‘Statins’. The activation of the RISK pathway mediates cell survival through various pathways including various anti-apoptotic mechanisms, by inhibiting the opening of the mitochondrial permeability transition pore (mPTP) and by possibly inhibiting autophagy. The activation of the RISK pathway and the subsequent inhibition of mPTP opening provides a common cardioprotective pathway recruited at the time of myocardial reperfusion, ‘uniting’ the cardioprotective phenomena of ischemic preconditioning and postconditioning, which can be targeted by pharmacological agents at the time of myocardial reperfusion as a novel cardioprotective strategy in patients presenting with an AMI
Clearly, given the size of the myocardial infarct reduction elicited by the RISK pathway activation in the majority of studies, there must be other anti-necrotic protective mechanisms contributing to the cardioprotective effects of the RISK pathway. In this regard, the inhibition of the mitochondrial permeability transition pore (mPTP), a mitochondrial channel which mediates cell death at the time of myocardial reperfusion by uncoupling oxidative phosphorylation and inducing mitochondrial swelling [101, 100], have been identified as a down stream target of the RISK pathway [45, 71, 142, 143]. However, the mechanism through which the RISK pathway inhibits the opening of the mPTP is unclear, although there are several hypotheses (see Fig. 1): (a) GSK-3β, a downstream target of the RISK pathway has been linked to the inhibition of mPTP opening in the context of cardioprotection [142]; (b) eNOS, another downstream target of the RISK pathway has the potential for inhibiting mPTP opening either through the PKG-PKC-ε-mKATP channel signalling pathway [144–147] or it may suppress mPTP opening through the generation of nitric oxide [148]; (c) the inhibition of BAX translocation to mitochondria [149] and/or the activation of mitochondrial hexokinase II [150, 151] may act in concert to inhibit mPTP opening; (d) finally, Abdallah et al. [152] have demonstrated that activating PI3K using insulin can reduce the uptake of calcium by the sarcoplasmic reticulum, which may in turn act to inhibit mPTP opening at the time of myocardial reperfusion.
Sanada et al. [91] have previously demonstrated that the administration of ‘statins’ pre-ischemically results in the phosphorylation of PI3K and the subsequent activation of ecto-5′-nucleotidase, an effect which would act to promote adenosine release at the time of myocardial reperfusion, which in itself could confer a cardioprotective effect.
Finally, an interesting recent study by Valentim et al. [50] suggests that the activation of the Akt but not the Erk1/2 component of the RISK pathway using urocortin may attenuate autophagy (a lysosomal degradative pathway that has emerged as a form of programmed cell death distinct from apoptosis), an effect which is in part due to the inhibition of Beclin-1, a critical mediator of autophagy. Furthermore, in support of a detrimental role for autophagy in the setting of myocardial ischemia-reperfusion injury, autophagocytosis has been reported to be inhibited in both preconditioned and postconditioned cardiomyocytes [153].
Pro-injurious protein kinases which counteract the RISK pathway
Somewhat intriguingly, it appears that the pro-survival RISK pathway may have its pro-injurious antithesis in the form of a collection of protein kinases that are detrimental, when activated at the time of myocardial reperfusion. This role is exemplified by the protein kinase, PKC-δ, the activation of which at the time of myocardial reperfusion increases myocardial infarct size [21]. Studies have reported that the selective inhibition of PKC-δ specifically at the time of myocardial reperfusion decreases myocardial infarct size [84, 154, 155]. Furthermore, Zatta et al. [84] have reported the inhibition of PKC-δ translocation to the mitochondria using in situ post-conditioned rat hearts.
Abnormal activation of Rho-kinase (ROCK), the downstream target of the small GTPase, Rho-A, mediates cardiovascular damage including myocardial ischemia-reperfusion injury and hypertension, and its inhibition underlies some of the pleiotropic effects of ‘statins’ [156, 157]. Studies have revealed that myocardial ischemia and reperfusion activate ROCK in ischemic myocardium and pharmacologically inhibiting its activation is cardioprotective [158] and this protective effect appears to be mediated through the activation of the PI3K-Akt-eNOS pathway [159], suggesting that ROCK exerts its pro-injurious effect by counteracting the RISK pathway. Importantly, data from Hamid et al. [160] have demonstrated that administering ROCK inhibitors specifically at the time of myocardial reperfusion reduces myocardial infarct size through the PI3K-Akt-NO pathway.
Sun et al. [80] demonstrated that the treatment of neonatal cardiomyocytes with hypoxic postconditioning reduced the activation of both JNK and p38 MAPK’s, and the pharmacological activation of these MAPK’s at the time of reoxygenation abolished the cardioprotective effect elicited by postconditioning, suggesting a detrimental role for these kinases at the time of reoxygenation. In contrast however, IPC has been reported to activate both p38 [88] and JNK [90] MAPK’s at the time of myocardial reperfusion, suggesting that the modulation of these particular kinase members of the RISK pathway in the setting of cardioprotection is more complex.
The RISK pathway as a target for cardioprotection: clinical application
As an interventional strategy which can be applied at the time of myocardial reperfusion for patients presenting with an AMI, the use of pharmacological agents to target the RISK pathway is certainly a viable proposition. The major clinical application of this cardioprotective strategy would be as adjuvant therapy to myocardial reperfusion for AMI patients undergoing either thrombolysis or primary PCI, although it could also be used to extend the ‘time window’ for intervention in those AMI patients in which a delay in myocardial reperfusion is anticipated. Patients receiving PCI for unstable angina/NSTEMI, and patients undergoing CABG surgery or cardiac transplantation as well as those patients surviving a cardiac arrest, also might experience acute myocardial ischemia-reperfusion injury and therefore might accrue benefit from such a cardioprotective strategy.
Many of the pharmacological agents linked to RISK pathway activation in basic science studies are already in clinical use today (see Tables 1 and 2), facilitating their investigation as potential cardioprotective agents. Importantly, the design of any clinical study investigating this form of cardioprotective strategy in AMI patients should ensure that the pharmacological agent is administered either prior to or at the immediate onset of myocardial reperfusion to ensure effective delivery of the agent to the reperfused myocardium and RISK pathway activation within the time-frame of the ‘window’ for cardioprotection. Several of the clinical studies reviewed below administered their cardioprotective agent to AMI patients after the onset of myocardial reperfusion, which may explain in part the lack of benefit of their treatment strategy (see Table 2).
Several pharmacological agents now known from pre-clinical studies to confer cardioprotection when given at the time of myocardial reperfusion through the activation of the RISK pathway have been previously investigated in clinical trials and are reviewed below (see Table 2). Clearly, whether the activation of the RISK pathway actually underlies the cardioprotective effect of these agents when used in the clinical arena is not clear, although data from our laboratory suggest that the RISK pathway does operate in human myocardial tissue to confer cardioprotection, in a study in which we have demonstrated that both EPO and IPost cardioprotect human atrial trabeculae harvested from patients undergoing CABG surgery through the activation of the RISK pathway (Mudgliari et al. Unpublished).
Activators of the RISK pathway which have demonstrated clinical cardioprotection
The large multi-centred Japanese clinical study entitled J-WIND-ANP [161], which recently reported its main findings at the AHA Scientific Sessions 2006, found that a 72 h infusion of Carperitide, a recombinant form of human ANP, conferred cardioprotection in over 600 patients presenting with an AMI undergoing PCI as evidenced by a 14.7% reduction in myocardial infarct size (measured by CK and troponin T) and a 5.1% increase in ejection fraction. In addition, the secondary endpoint of reperfusion injury (assessed by the presence of malignant ventricular arrhythmias during reperfusion periods, re-elevation of ST segments, and worsening of chest pain) was decreased by 25.9% and Carperitide also reduced the incidence of cardiac death and re-hospitalization for heart failure by 73.3% compared with placebo. Although these data are promising, further large-scale clinical studies using primary clinical outcome endpoints are required to determine whether the cardioprotective benefits of this treatment strategy extend to an improvement in clinical outcomes.
The recently described phenomenon of ischemic postconditioning (IPost) had already been demonstrated to reduce myocardial injury in several small clinical studies of AMI patients undergoing PCI [14–17]. In these studies, a series of low-pressure inflations and deflations of the coronary angioplasty balloon administered upstream of the deployed stent in the infarct-related coronary artery were demonstrated to attenuate myocardial reperfusion injury and reduce myocardial infarct size as measured by cardiac enzymes and nuclear scanning [14–17]. Further large clinical studies are required to determine whether the cardioprotective benefits translate to improved clinical outcomes in this patient group.
The administration of high-dose ‘statins’ to patients with acute coronary syndromes more than 24 h following PCI has been demonstrated to improve clinical outcomes [162, 163], but whether high dose ‘statins’ given at the time of myocardial reperfusion, confers any further cardioprotective effect is unclear. A small preliminary study, has demonstrated that high-dose atorvastatin (80 mg) administered before PCI in patients with non-ST elevation MI or unstable angina conferred clinical benefit [173].
The administration of high-dose adenosine as an adjunct to myocardial reperfusion in AMI has demonstrated myocardial protection in several small clinical studies [174, 175]. Larger randomized controlled studies, in which intravenous adenosine was commenced after the onset of reperfusion therapy, reported an 11% reduction in myocardial infarct size but no improvement in clinical outcomes with this treatment strategy [164, 165]. Adequately powered larger clinical studies administering adenosine prior to the onset of myocardial reperfusion are required to provide evidence of improved clinical outcomes with this treatment strategy.
In a small clinical study comprising 10 AMI patients with impaired LV systolic function undergoing primary PCI, a 72 h infusion of glucagon-like peptide-1 (GLP-1), administered more than 3 h following reperfusion, was demonstrated to improve ejection fraction from 29 to 39% [166]. However, the effect of GLP-1 on subsequent myocardial injury was not investigated in this study. The use of GLP-1 as both an antidiabetic and potential cardioprotective agent is limited by the fact that it is rapidly broken down by endogenous dipeptidyl peptidase-IV (DPPIV). The longer acting GLP-1 analogues, which are resistant to DDPIV breakdown such as Exenatide, or the novel DPPIV inhibitors Sitagliptin and Vildagliptin, which act to augment endogenous GLP-1, may offer more promise, but their cardioprotective effect needs to be first determined in preclinical studies.
Activators of the RISK pathway which have failed to demonstrate clinical cardioprotection
The cardioprotective potential of the glucose-insulin-potassium (GIK) cocktail was comprehensively examined in the large multi-centred randomized clinical trial comprising 20,201 patients undergoing PCI or thrombolysis for an ST-elevation MI, and was found to confer no beneficial effect in terms of mortality, cardiac arrest, cardiogenic shock and re-infarction at 30 days [176]. Potential explanations for the lack of cardioprotection include: (a) the preclinical studies demonstrating a reduction in myocardial infarct size used insulin alone on the most part [35], with only one study demonstrating cardioprotection with GIK therapy at the time of myocardial reperfusion [177]; (b) a delay in the administration of GIK therapy in relation to the onset of myocardial reperfusion therapy and the prolonged myocardial ischemic time, with pre-clinical studies suggesting benefit with insulin therapy at the immediate onset of myocardial reperfusion [35, 178], and following a shorter myocardial ischemic time [178].
Several small clinical studies have demonstrated reduced myocardial injury [179] and preserved left ventricular function [180] with the use of volatile anesthetics as preconditioning agents in patients undergoing CABG surgery. However, a recent meta-analysis has reported no beneficial effect of volatile anesthetics on rates of myocardial infarction and mortality in patients undergoing CABG surgery [181]. Larger clinical trials are required to determine whether volatile anesthetics are beneficial in cardiac surgery.
A recent small clinical trial examining G-CSF as adjunctive therapy to PCI for an acute anterior MI, in which subcutaneous G-CSF was injected daily for 5 days commencing after myocardial reperfusion, but within 24 h, failed to show any clinical benefit [182], with other clinical studies reporting high restenosis rates [183] and serious side effects [184] with this treatment strategy.
Activators of the RISK pathway that have the potential to demonstrate clinical cardioprotection
Erythropoietin (EPO) which is already in clinical use for raising haematocrit in anemic patients with chronic renal and cardiac failure shows great promise as both a neuroprotective and cardioprotective agent. Ehreneich et al. [167] demonstrated that an intravenous infusion of 33,000 iu of recombinant EPO administered daily for 3 days, was safe and improved functional recovery and showed a trend for reducing cerebral infarct size in 20 patients presenting with an acute ischemic stroke. A preliminary study has demonstrated that the long-acting EPO analogue, darbopoeitin alfa is safe when administered as a single 300 μg intravenous bolus prior to primary PCI in patients presenting with an AMI [168]. Clinical studies examining the cardioprotective potential of EPO in AMI patients and in patients undergoing CABG surgery are now underway.
Whether hormone replacement therapy is cardioprotective in post-menopausal women is unclear. However, a small clinical study comprising both men and women has demonstrated that estrogen administration has the ability to reduce ST-segment shift and reduce chest pain in patients undergoing single vessel elective PCI [169], but whether this translates to any cardioprotective benefit in patients presenting with an AMI remains to be examined.
Clinical trials are underway examining the cardioprotective potential of pharmacological PKC-ε activators and pharmacological PKC-ε inhibitors in different clinical settings of acute myocardial ischemia-reperfusion injury.
Several small clinical studies have demonstrated that the pharmacological inhibition of rho-kinase (ROCK) using fasudil, to be beneficial as an anti-anginal agent [170], to be neuroprotective in acute ischemic stroke [171] and to improve endothelial function in heart failure patients [172]. Whether ROCK inhibitors administered at the time of myocardial reperfusion to AMI patients, are beneficial, remains to be determined.
Conclusions
The Reperfusion Injury Salvage Kinase (RISK) pathway, when originally described, referred to the protein kinases, Akt and Erk1/2, which when specifically activated at the time of myocardial reperfusion conferred powerful cardioprotection against lethal reperfusion injury. The list of pharmacological agents identified as mediating their cardioprotective effect at the time of myocardial reperfusion through the activation of the RISK pathway is ever expanding and now includes growth factors, G-protein coupled receptor ligands, and other non-receptor acting agents. The RISK pathway has evolved to encompass other pro-survival kinases such as PKC-ε, p70S6K, PKG and GSK-3β, and constitutes the core component of a common cardioprotective pathway recruited at the time of myocardial reperfusion by both ischemic preconditioning and postconditioning which converges on the inhibition of the mitochondrial permeability transition pore, as one of the potential cardioprotective mechanisms of the RISK pathway.
The concept of the RISK pathway as a target for intervening at the time of myocardial reperfusion in patients presenting with an AMI, along with the effective clinical application of ischemic postconditioning in AMI patients, has succeeded in re-igniting interest in the myocardial reperfusion phase as a target for cardioprotection. The RISK pathway also provides a pharmacological target for intervention in other patient groups experiencing acute myocardial ischemia-reperfusion injury such as patients undergoing CABG surgery, patients having cardiac transplant surgery, and those patients surviving a cardiac arrest. Several small clinical studies have demonstrated myocardial protection with pharmacological agents known to activate the RISK pathway. Large placebo-controlled randomized clinical studies are now required to determine whether the administration of pharmacological agents, known to activate the RISK pathway, as adjunctive therapy to myocardial reperfusion confers any cardioprotective benefit, in terms of meaningful clinical out comes, for patients presenting with an AMI. Clearly, the valuable information obtained from the wealth of pre-clinical data that exist, should form the centrepiece in the design of such clinical trials.
References
Yellon DM, Baxter GF (1999) Reperfusion injury revisited: is there a role for growth factor signaling in limiting lethal reperfusion injury? Trends Cardiovasc Med 9:245–249
Hausenloy DJ, Yellon DM (2004) New directions for protecting the heart against ischaemia-reperfusion injury: targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway. Cardiovasc Res 61:448–460
Tsang A, Hausenloy DJ, Mocanu MM, Yellon DM (2004) Postconditioning: a form of “modified reperfusion” protects the myocardium by activating the phosphatidylinositol 3-kinase-Akt pathway. Circ Res 95:230–232
Hausenloy DJ, Tsang A, Mocanu M, Yellon DM (2005) Ischemic Preconditioning Protects by Activating Pro-Survival Kinases at Reperfusion. Am J Physiol Heart Circ Physiol 288:H971–H976
Murry CE, Jennings RB, Reimer KA (1986) Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 74:1124–1136
Yellon DM, Alkhulaifi AM, Pugsley WB (1993) Preconditioning the human myocardium. Lancet 342:276–277
Heusch G (2001) Nitroglycerin and delayed preconditioning in humans: yet another new mechanism for an old drug? Circulation 103:2876–2878
Hausenloy DJ, Tsang A, Mocanu MM, Yellon DM (2005) Ischemic preconditioning protects by activating prosurvival kinases at reperfusion. Am J Physiol Heart Circ Physiol 288:H971–H976
Solenkova NV, Solodushko V, Cohen MV, Downey JM (2006) Endogenous adenosine protects preconditioned heart during early minutes of reperfusion by activating Akt. Am J Physiol Heart Circ Physiol 290:H441–H449
Lecour S, Suleman N, Deuchar GA et al (2005) Pharmacological preconditioning with tumor necrosis factor-alpha activates signal transducer and activator of transcription-3 at reperfusion without involving classic prosurvival kinases (Akt and extracellular signal-regulated kinase). Circulation 112:3911–3918
Zhao ZQ, Corvera JS, Halkos ME et al (2003) Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol 285:H579–H588
Vinten-Johansen J, Yellon DM, Opie LH (2005) Postconditioning: a simple, clinically applicable procedure to improve revascularization in acute myocardial infarction. Circulation 112:2085–2088
Yellon DM, Opie LH (2006) Postconditioning for protection of the infarcting heart. Lancet 367:456–458
Laskey WK (2005) Brief repetitive balloon occlusions enhance reperfusion during percutaneous coronary intervention for acute myocardial infarction: a pilot study. Catheter Cardiovasc Interv 65:361–367
Staat P, Rioufol G, Piot C et al (2005) Postconditioning the human heart. Circulation 112:2143–2148
Ma X, Zhang X, Li C, Luo M (2006) Effect of postconditioning on coronary blood flow velocity and endothelial function and LV recovery after myocardial infarction. J Interv Cardiol 19:367–375
Yang X-C, Liu Y, Wang L-F, Cui L, Ge Y-G, Wang H-S, Li W-M, Xu Li, Ni Z-H, Liu H-S, Zhang L, Wang T, Jia H-M, Vinten-Johansen J., Zhao Z-Q (2006) Permanent reduction in myocardial infarct size by postconditioning in patients after primary coronary angioplasty. Circulation 114:II-812
Ravingerova T, Barancik M, Strniskova M (2003) Mitogen-activated protein kinases: a new therapeutic target in cardiac pathology. Mol Cell Biochem 247:127–138
Armstrong SC (2004) Protein kinase activation and myocardial ischemia/reperfusion injury. Cardiovasc Res 61:427–436
Hausenloy DJ, Yellon DM (2006) Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc Res 70:240–253
Inagaki K, Churchill E, Mochly-Rosen D (2006) Epsilon protein kinase C as a potential therapeutic target for the ischemic heart. Cardiovasc Res 70:222–230
Michel MC, Li Y, Heusch G (2001) Mitogen-activated protein kinases in the heart. Naunyn Schmiedebergs Arch Pharmacol 363:245–266
Gottlieb RA, Burleson KO, Kloner RA, Babior BM, Engler RL (1994) Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest 94:1621–1628
Freude B, Masters TN, Robicsek F et al (2000) Apoptosis is initiated by myocardial ischemia and executed during reperfusion. J Mol Cell Cardiol 32:197–208
Zhao ZQ, Morris CD, Budde JM et al (2003) Inhibition of myocardial apoptosis reduces infarct size and improves regional contractile dysfunction during reperfusion. Cardiovasc Res 59:132–142
Mockridge JW, Marber MS, Heads RJ (2000) Activation of Akt during simulated ischemia/reperfusion in cardiac myocytes. Biochem Biophys Res Commun 270:947–952
Yamashita K, Kajstura J, Discher DJ et al (2001) Reperfusion-activated Akt kinase prevents apoptosis in transgenic mouse hearts overexpressing insulin-like growth factor-1. Circ Res 88:609–614
Omura T, Yoshiyama M, Shimada T et al (1999) Activation of mitogen-activated protein kinases in in vivo ischemia/reperfused myocardium in rats. J Mol Cell Cardiol 31:1269–1279
Bogoyevitch MA, Gillespie-Brown J, Ketterman AJ et al (1996) Stimulation of the stress-activated mitogen-activated protein kinase subfamilies in perfused heart. p38/RK mitogen-activated protein kinases and c-Jun N-terminal kinases are activated by ischemia/reperfusion. Circ Res 79:162–173
Matsui T, Li L, Wu JC et al (2002) Phenotypic spectrum caused by transgenic overexpression of activated Akt in the heart. J Biol Chem 277:22896–22901
Miao W, Luo Z, Kitsis RN, Walsh K (2000) Intracoronary, adenovirus-mediated Akt gene transfer in heart limits infarct size following ischemia-reperfusion injury in vivo. J Mol Cell Cardiol 32:2397–2402
Yue TL, Wang C, Gu JL et al (2000) Inhibition of extracellular signal-regulated kinase enhances Ischemia/Reoxygenation-induced apoptosis in cultured cardiac myocytes and exaggerates reperfusion injury in isolated perfused heart. Circ Res 86:692–699
Baxter GF, Mocanu MM, Brar BK, Latchman DS, Yellon DM (2001) Cardioprotective effects of transforming growth factor-beta1 during early reoxygenation or reperfusion are mediated by p42/p44 MAPK. J Cardiovasc Pharmacol 38:930–939
Jonassen AK, Brar BK, Mjos OD, Sack MN, Latchman DS, Yellon DM (2000) Insulin administered at reoxygenation exerts a cardioprotective effect in myocytes by a possible anti-apoptotic mechanism. J Mol Cell Cardiol 32:757–764
Jonassen AK, Sack MN, Mjos OD, Yellon DM (2001) Myocardial protection by insulin at reperfusion requires early administration and is mediated via Akt and p70s6 kinase cell-survival signaling. Circ Res 89:1191–1198
Gao F, Gao E, Yue TL et al (2002) Nitric oxide mediates the antiapoptotic effect of insulin in myocardial ischemia-reperfusion: the roles of PI3-kinase, Akt, and endothelial nitric oxide synthase phosphorylation. Circulation 105:1497–1502
Otani H, Yamamura T, Nakao Y et al (2000) Insulin-like growth factor-I improves recovery of cardiac performance during reperfusion in isolated rat heart by a wortmannin-sensitive mechanism. J Cardiovasc Pharmacol 35:275–281
Brar BK, Stephanou A, Pennica D, Latchman DS (2001) CT-1 mediated cardioprotection against ischaemic re-oxygenation injury is mediated by PI3 kinase, Akt and MEK1/2 pathways. Cytokine 16:93–96
Brar BK, Stephanou A, Liao Z et al (2001) Cardiotrophin-1 can protect cardiac myocytes from injury when added both prior to simulated ischaemia and at reoxygenation. Cardiovasc Res 51:265–274
Liao Z, Brar BK, Cai Q et al (2002) Cardiotrophin-1 (CT-1) can protect the adult heart from injury when added both prior to ischaemia and at reperfusion. Cardiovasc Res 53:902–910
Jiang ZS, Padua RR, Ju H et al (2002) Acute protection of ischemic heart by FGF-2: involvement of FGF-2 receptors and protein kinase C. Am J Physiol Heart Circ Physiol 282:H1071–H1080
Hanlon PR, Fu P, Wright GL, Steenbergen C, Arcasoy MO, Murphy E (2005) Mechanisms of erythropoietin-mediated cardioprotection during ischemia-reperfusion injury: role of protein kinase C and phosphatidylinositol 3-kinase signaling. FASEB J 19:1323–1325
Bullard AJ, Govewalla P, Yellon DM (2005) Erythropoietin protects the myocardium against reperfusion injury in vitro and in vivo. Basic Res Cardiol 100:397–493
Ueda K, Takano H, Hasegawa H et al (2006) Granulocyte colony stimulating factor directly inhibits myocardial ischemia-reperfusion injury through Akt-endothelial NO synthase pathway. Arterioscler Thromb Vasc Biol 26:e108–e113
Smith CC, Mocanu MM, Davidson SM, Wynne AM, Simpkin JC, Yellon DM (2006) Leptin, the obesity-associated hormone, exhibits direct cardioprotective effects. Br J Pharmacol 149:5–13
Brar BK, Stephanou A, Knight R, Latchman DS (2002) Activation of protein kinase B/Akt by urocortin is essential for its ability to protect cardiac cells against hypoxia/reoxygenation-induced cell death. J Mol Cell Cardiol 34:483–492
Chanalaris A, Lawrence KM, Stephanou A et al (2003) Protective effects of the urocortin homologues stresscopin (SCP) and stresscopin-related peptide (SRP) against hypoxia/reoxygenation injury in rat neonatal cardiomyocytes. J Mol Cell Cardiol 35:1295–1305
Brar BK, Jonassen AK, Stephanou A et al (2000) Urocortin protects against ischemic and reperfusion injury via a MAPK-dependent pathway. J Biol Chem 275:8508–8514
Schulman D, Latchman DS, Yellon DM (2002) Urocortin protects the heart from reperfusion injury via upregulation of p42/p44 MAPK signaling pathway. Am J Physiol Heart Circ Physiol 283:H1481–H1488
Valentim L, Laurence KM, Townsend PA et al (2006) Urocortin inhibits Beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. J Mol Cell Cardiol 40:846–852
Yang XM, Krieg T, Cui L, Downey JM, Cohen MV (2004) NECA and bradykinin at reperfusion reduce infarction in rabbit hearts by signaling through PI3K, ERK, and NO. J Mol Cell Cardiol 36:411–421
Kis A, Baxter GF, Yellon DM (2003) Limitation of myocardial reperfusion injury by AMP579, an adenosine A1/A2A receptor agonist: role of A2A receptor and Erk1/2. Cardiovasc Drugs Ther 17:415–425
Park SS, Zhao H, Jang Y, Mueller RA, Xu Z (2006) N6-(3-iodobenzyl)-adenosine-5’-N-methylcarboxamide confers cardioprotection at reperfusion by inhibiting mitochondrial permeability transition pore opening via glycogen synthase kinase 3 beta. J Pharmacol Exp Ther 318:124–131
Bell RM, Yellon DM (2003) Bradykinin limits infarction when administered as an adjunct to reperfusion in mouse heart: the role of PI3K, Akt and eNOS. J Mol Cell Cardiol 35:185–193
Gross ER, Hsu AK, Gross GJ (2004) Opioid-induced cardioprotection occurs via glycogen synthase kinase beta inhibition during reperfusion in intact rat hearts. Circ Res 94:960–966
Hamid SA, Baxter GF (2005) Adrenomedullin limits reperfusion injury in experimental myocardial infarction. Basic Res Cardiol 100:387–396
Bose AK, Mocanu MM, Carr RD, Yellon DM (2005) Glucagon like peptide-1 is protective against myocardial ischemia/reperfusion injury when given either as a preconditioning mimetic or at reperfusion in an isolated rat heart model. Cardiovasc Drugs Ther 19:9–11
Bose AK, Mocanu MM, Carr RD, Brand CL, Yellon DM (2005) Glucagon-like peptide 1 can directly protect the heart against ischemia/reperfusion Injury. Diabetes 54:146–151
Yang XM, Philipp S, Downey JM, Cohen MV (2006) Atrial natriuretic peptide administered just prior to reperfusion limits infarction in rabbit hearts. Basic Res Cardiol 101:311–318
Tissier R, Waintraub X, Couvreur N et al (2007) Pharmacological postconditioning with the phytoestrogen genistein. J Mol Cell Cardiol 42:79–87
Zhang SJ, Yang XM, Liu GS, Cohen MV, Pemberton K, Downey JM (2003) CGX-1051, a peptide from Conus snail venom, attenuates infarction in rabbit hearts when administered at reperfusion. J Cardiovasc Pharmacol 42:764–771
Chiari PC, Bienengraeber MW, Pagel PS, Krolikowski JG, Kersten JR, Warltier DC (2005) Isoflurane protects against myocardial infarction during early reperfusion by activation of phosphatidylinositol-3-kinase signal transduction: evidence for anesthetic-induced postconditioning in rabbits. Anesthesiology 102:102–109
Feng J, Lucchinetti E, Ahuja P, Pasch T, Perriard JC, Zaugg M (2005) Isoflurane postconditioning prevents opening of the mitochondrial permeability transition pore through inhibition of glycogen synthase kinase 3beta. Anesthesiology 103:987–995
Pagel PS, Krolikowski JG, Neff DA et al (2006) Inhibition of glycogen synthase kinase enhances isoflurane-induced protection against myocardial infarction during early reperfusion in vivo. Anesth Analg 102:1348–1354
Feng J, Fischer G, Lucchinetti E et al (2006) Infarct-remodeled myocardium is receptive to protection by isoflurane postconditioning: role of protein kinase B/Akt signaling. Anesthesiology 104:1004–1014
Weihrauch D, Krolikowski JG, Bienengraeber M, Kersten JR, Warltier DC, Pagel PS (2005) Morphine enhances isoflurane-induced postconditioning against myocardial infarction: the role of phosphatidylinositol-3-kinase and opioid receptors in rabbits. Anesth Analg 101:942–9, table
Krolikowski JG, Weihrauch D, Bienengraeber M, Kersten JR, Warltier DC, Pagel PS (2006) Role of Erk1/2, p70s6K, and eNOS in isoflurane-induced cardioprotection during early reperfusion in vivo. Can J Anaesth 53:174–182
Wang C, Neff DA, Krolikowski JG et al (2006) The influence of B-cell lymphoma 2 protein, an antiapoptotic regulator of mitochondrial permeability transition, on isoflurane-induced and ischemic postconditioning in rabbits. Anesth Analg 102:1355–1360
Bell RM, Yellon DM (2003) Atorvastatin, administered at the onset of reperfusion, and independent of lipid lowering, protects the myocardium by up-regulating a pro-survival pathway. J Am Coll Cardiol 41:508–515
Efthymiou CA, Mocanu MM, Yellon DM (2005) Atorvastatin and myocardial reperfusion injury: new pleiotropic effect implicating multiple prosurvival signaling. J Cardiovasc Pharmacol 45:247–252
Bopassa JC, Ferrera R, Gateau-Roesch O, Couture-Lepetit E, Ovize M (2006) PI 3-kinase regulates the mitochondrial transition pore in controlled reperfusion and postconditioning. Cardiovasc Res 69:178–185
Manintveld OC, Te Lintel HM, van den Bos EJ et al (2006) Cardiac effects of postconditioning depend critically on the duration of index ischemia. Am J Physiol Heart Circ Physiol 292:H1551–H1560
Yang XM, Philipp S, Downey JM, Cohen MV (2005) Postconditioning’s protection is not dependent on circulating blood factors or cells but involves adenosine receptors and requires PI3-kinase and guanylyl cyclase activation. Basic Res Cardiol 100:57–63
Zhu M, Feng J, Lucchinetti E et al (2006) Ischemic postconditioning protects remodeled myocardium via the PI3K-PKB/Akt reperfusion injury salvage kinase pathway. Cardiovasc Res 72:152–162
Peng LY, Ma H, He JG et al (2006) [Ischemic postconditioning attenuates ischemia/reperfusion injury in isolated hypertrophied rat heart]. Zhonghua Xin Xue Guan Bing Za Zhi 34:685–689
Zhao, Z-Q, Wang, N-P, Mykytenko, J., Reeves, J, Deneve, J, Jiang, R, Zatta, AJ, Guyton, RA, Vinten-Johansen, J (2006) Postconditioning attenuates cardiac muscle cell apoptosis via translocation of survival kinases and opening of KATP channels in mitochondria. Circulation 114:II-261
Fujita M, Asanuma H, Hirata A et al (2007) Prolonged transient acidosis during early reperfusion contributes to the cardioprotective effects of postconditioning. Am J Physiol Heart Circ Physiol 292:H2004–H2008
Darling CE, Jiang R, Maynard M, Whittaker P, Vinten-Johansen J, Przyklenk K (2005) ‘Postconditioning’ via Stuttering reperfusion limits myocardial infarct size in rabbit hearts: role of ERK 1/2. Am J Physiol Heart Circ Physiol 289:H1618–H1626
Yang XM, Proctor JB, Cui L, Krieg T, Downey JM, Cohen MV (2004) Multiple, brief coronary occlusions during early reperfusion protect rabbit hearts by targeting cell signaling pathways. J Am Coll Cardiol 44:1103–1110
Sun HY, Wang NP, Halkos M et al (2006) Postconditioning attenuates cardiomyocyte apoptosis via inhibition of JNK and p38 mitogen-activated protein kinase signaling pathways. Apoptosis 11:1583–1593
Philipp S, Yang XM, Cui L, Davis AM, Downey JM, Cohen MV (2006) Postconditioning protects rabbit hearts through a protein kinase C-adenosine A2b receptor cascade. Cardiovasc Res 70:308–314
Penna C, Rastaldo R, Mancardi D et al (2006) Post-conditioning induced cardioprotection requires signaling through a redox-sensitive mechanism, mitochondrial ATP-sensitive K+ channel and protein kinase C activation. Basic Res Cardiol 101:180–189
Fantinelli JC, Mosca SM (2007) Comparative effects of ischemic pre and postconditioning on ischemia-reperfusion injury in spontaneously hypertensive rats (SHR). Mol Cell Biochem 296:45–51
Zatta AJ, Kin H, Lee G et al (2006) Infarct-sparing effect of myocardial postconditioning is dependent on protein kinase C signalling. Cardiovasc Res 70:315–324
Suleman N, Opie L, Lecour S (2006) Ischemic postconditioning confers cardioprotection via phosporylation of STAT-3. J Mol Cell Cardiol 40:155
Argaud L, Gateau-Roesch O, Raisky O, Loufouat J, Robert D, Ovize M (2005) Postconditioning inhibits mitochondrial permeability transition. Circulation 111:194–197
Burley DS, Baxter GF (2005) Post-conditioning is dependent on pkg activation in early reperfusion. J Mol Cell Cardiol 38:28
da Silva R, Grampp T, Pasch T, Schaub MC, Zaugg M (2004) Differential activation of mitogen-activated protein kinases in ischemic and anesthetic preconditioning. Anesthesiology 100:59–69
Fryer RM, Pratt PF, Hsu AK, Gross GJ (2001) Differential activation of extracellular signal regulated kinase isoforms in preconditioning and opioid-induced cardioprotection. J Pharmacol Exp Ther 296:642–649
Fryer RM, Patel HH, Hsu AK, Gross GJ (2001) Stress-activated protein kinase phosphorylation during cardioprotection in the ischemic myocardium. Am J Physiol Heart Circ Physiol 281:H1184–H1192
Sanada S, Asanuma H, Minamino T et al (2004) Optimal windows of statin use for immediate infarct limitation: 5’-nucleotidase as another downstream molecule of phosphatidylinositol 3-kinase. Circulation 110:2143–2149
Wynne AM, Mocanu MM, Yellon DM (2005) Pioglitazone mimics preconditioning in the isolated perfused rat heart: a role for the prosurvival kinases PI3K and P42/44MAPK. J Cardiovasc Pharmacol 46:817–822
Bell RM, Clark JE, Hearse DJ, Shattock MJ (2007) Reperfusion kinase phosphorylation is essential but not sufficient in the mediation of pharmacological preconditioning: characterisation in the bi-phasic profile of early and late protection. Cardiovasc Res 73:153–163
Ping P, Murphy E (2000) Role of p38 mitogen-activated protein kinases in preconditioning: a detrimental factor or a protective kinase? Circ Res 86:921–922
Hausenloy DJ, Yellon DM (2007) Preconditioning and postconditioning: United at reperfusion. Pharmacol Therapeutics (in press)
Kin H, Zhao ZQ, Sun HY et al (2004) Postconditioning attenuates myocardial ischemia-reperfusion injury by inhibiting events in the early minutes of reperfusion. Cardiovasc Res 62:74–85
Griffiths EJ, Halestrap AP (1995) Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J 307( Pt 1):93–98
Di Lisa F, Menabo R, Canton M, Barile M, Bernardi P (2001) Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem 276:2571–2575
Kim JS, Jin Y, Lemasters JJ (2006) Reactive oxygen species, but not Ca2+ overloading, trigger pH- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. Am J Physiol Heart Circ Physiol 290:H2024–H2034
Crompton M (1999) The mitochondrial permeability transition pore and its role in cell death. Biochem J 341(Pt 2):233–249
Hausenloy DJ, Yellon DM (2003) The mitochondrial permeability transition pore: its fundamental role in mediating cell death during ischaemia and reperfusion. J Mol Cell Cardiol 35:339–341
Hausenloy DJ, Duchen MR, Yellon DM (2003) Inhibiting mitochondrial permeability transition pore opening at reperfusion protects against ischaemia-reperfusion injury. Cardiovasc Res 60:617–625
Lefer AM, Tsao P, Aoki N, Palladino MA Jr (1990) Mediation of cardioprotection by transforming growth factor-beta. Science 249:61–64
Kulik G, Klippel A, Weber MJ (1997) Antiapoptotic signalling by the insulin-like growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Mol Cell Biol 17:1595–1606
Parrizas M, Saltiel AR, LeRoith D (1997) Insulin-like growth factor 1 inhibits apoptosis using the phosphatidylinositol 3′-kinase and mitogen-activated protein kinase pathways. J Biol Chem 272:154–161
Fujio Y, Nguyen T, Wencker D, Kitsis RN, Walsh K (2000) Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation 101:660–667
Hong F, Kwon SJ, Jhun BS et al (2001) Insulin-like growth factor-1 protects H9c2 cardiac myoblasts from oxidative stress-induced apoptosis via phosphatidylinositol 3-kinase and extracellular signal-regulated kinase pathways. Life Sci 68:1095–1105
Chao W, Matsui T, Novikov MS et al (2003) Strategic advantages of insulin-like growth factor-I expression for cardioprotection. J Gene Med 5:277–286
Wang L, Ma W, Markovich R, Chen JW, Wang PH (1998) Regulation of cardiomyocyte apoptotic signaling by insulin-like growth factor I. Circ Res 83:516–522
Yamamura T, Otani H, Nakao Y, Hattori R, Osako M, Imamura H (2001) IGF-I differentially regulates Bcl-xL and Bax and confers myocardial protection in the rat heart. Am J Physiol Heart Circ Physiol 280:H1191–H1200
Pennica D, King KL, Shaw KJ et al (1995) Expression cloning of cardiotrophin 1, a cytokine that induces cardiac myocyte hypertrophy. Proc Natl Acad Sci USA 92:1142–1146
Sheng Z, Pennica D, Wood WI, Chien KR (1996) Cardiotrophin-1 displays early expression in the murine heart tube and promotes cardiac myocyte survival. Development 122:419–428
Sheng Z, Knowlton K, Chen J, Hoshijima M, Brown JH, Chien KR (1997) Cardiotrophin 1 (CT-1) inhibition of cardiac myocyte apoptosis via a mitogen-activated protein kinase-dependent pathway. Divergence from downstream CT-1 signals for myocardial cell hypertrophy J Biol Chem 272:5783–5791
Buehler A, Martire A, Strohm C et al (2002) Angiogenesis-independent cardioprotection in FGF-1 transgenic mice. Cardiovasc Res 55:768–777
Heusch G (2006) Obesity–a risk factor or a RISK factor for myocardial infarction? Br J Pharmacol 149:1–3
Harada M, Qin Y, Takano H et al (2005) G-CSF prevents cardiac remodeling after myocardial infarction by activating the Jak-Stat pathway in cardiomyocytes. Nat Med 11:305–311
Kitamura K, Kangawa K, Kawamoto M et al (1993) Adrenomedullin: a novel hypotensive peptide isolated from human pheochromocytoma. Biochem Biophys Res Commun 192:553–560
Kato K, Yin H, Agata J, Yoshida H, Chao L, Chao J (2003) Adrenomedullin gene delivery attenuates myocardial infarction and apoptosis after ischemia and reperfusion. Am J Physiol Heart Circ Physiol 285:H1506–H1514
Yin H, Chao L, Chao J (2004) Adrenomedullin protects against myocardial apoptosis after ischemia/reperfusion through activation of Akt-GSK signaling. Hypertension 43:109–116
Okumura H, Nagaya N, Itoh T et al (2004) Adrenomedullin infusion attenuates myocardial ischemia/reperfusion injury through the phosphatidylinositol 3-kinase/Akt-dependent pathway. Circulation 109:242–248
Deacon CF (2004) Therapeutic strategies based on glucagon-like peptide 1. Diabetes 53:2181–2189
D’Souza SP, Yellon DM, Martin C et al (2003) B-type natriuretic peptide limits infarct size in rat isolated hearts via KATP channel opening. Am J Physiol Heart Circ Physiol 284:H1592–H1600
Sangawa K, Nakanishi K, Ishino K, Inoue M, Kawada M, Sano S (2004) Atrial natriuretic peptide protects against ischemia-reperfusion injury in the isolated rat heart. Ann Thorac Surg 77:233–237
Patten RD, Pourati I, Aronovitz MJ et al (2004) 17beta-estradiol reduces cardiomyocyte apoptosis in vivo and in vitro via activation of phospho-inositide-3 kinase/Akt signaling. Circ Res 95:692–699
Lee TM, Lin MS, Chou TF, Tsai CH, Chang NC (2004) Adjunctive 17beta-estradiol administration reduces infarct size by altered expression of canine myocardial connexin43 protein. Cardiovasc Res 63:109–117
Stefano GB, Prevot V, Beauvillain JC et al (2000) Cell-surface estrogen receptors mediate calcium-dependent nitric oxide release in human endothelia. Circulation 101:1594–1597
Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK (2000) Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature 407:538–541
Piriou V, Chiari P, Gateau-Roesch O et al (2004) Desflurane-induced preconditioning alters calcium-induced mitochondrial permeability transition. Anesthesiology 100:581–588
Raphael J, Abedat S, Rivo J et al (2006) Volatile anesthetic preconditioning attenuates myocardial apoptosis in rabbits after regional ischemia and reperfusion via Akt signaling and modulation of Bcl-2 family proteins. J Pharmacol Exp Ther 318:186–194
Schlack W, Preckel B, Stunneck D, Thamer V (1998) Effects of halothane, enflurane, isoflurane, sevoflurane and desflurane on myocardial reperfusion injury in the isolated rat heart. Br J Anaesth 81:913–919
Preckel B, Schlack W, Comfere T, Obal D, Barthel H, Thamer V (1998) Effects of enflurane, isoflurane, sevoflurane and desflurane on reperfusion injury after regional myocardial ischaemia in the rabbit heart in vivo. Br J Anaesth 81:905–912
Tessier-Vetzel D, Tissier R, Waintraub X, Ghaleh B, Berdeaux A (2006) Isoflurane inhaled at the onset of reperfusion potentiates the cardioprotective effect of ischemic postconditioning through a NO-dependent mechanism. J Cardiovasc Pharmacol 47:487–492
Venkatapuram S, Wang C, Krolikowski JG et al (2006) Inhibition of apoptotic protein p53 lowers the threshold of isoflurane-induced cardioprotection during early reperfusion in rabbits. Anesth Analg 103:1400–1405
Di Napoli P, Antonio TA, Grilli A et al (2001) Simvastatin reduces reperfusion injury by modulating nitric oxide synthase expression: an ex vivo study in isolated working rat hearts. Cardiovasc Res 51:283–293
Kureishi Y, Luo Z, Shiojima I et al (2000) The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med 6:1004–1010
Schwartz LM, Lagranha CJ (2006) Ischemic postconditioning during reperfusion activates Akt and ERK without protecting against lethal myocardial ischemia-reperfusion injury in pigs. Am J Physiol Heart Circ Physiol 290:H1011–H1018
Heusch G (2004) Postconditioning: old wine in a new bottle? J Am Coll Cardiol 44:1111–1112
Kin H, Zatta AJ, Lofye MT et al (2005) Postconditioning reduces infarct size via adenosine receptor activation by endogenous adenosine. Cardiovasc Res 67:124–133
Kin H, Zatta AJ, Jiang R, Reeves JG, Mykytenko J, Sorescu GP, Zhao Z-Q, Wang NP, Guyton RA, Vinten-Johansen J (2005) Activation of opioid receptors mediates the infarct size reduction by Postconditioning. J Mol Cell Cardiol 38:827
Marber MS, Latchman DS, Walker JM, Yellon DM (1993) Cardiac stress protein elevation 24 hours after brief ischemia or heat stress is associated with resistance to myocardial infarction. Circulation 88:1264–1272
Harada H, Andersen JS, Mann M, Terada N, Korsmeyer SJ (2001) p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc Natl Acad Sci USA 98:9666–9670
Juhaszova M, Zorov DB, Kim SH et al (2004) Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest 113:1535–1549
Davidson SM, Hausenloy D, Duchen MR, Yellon DM (2006) Signalling via the reperfusion injury signalling kinase (RISK) pathway links closure of the mitochondrial permeability transition pore to cardioprotection. Int J Biochem Cell Biol 38:414–419
Costa AD, Garlid KD, West IC et al (2005) Protein kinase G transmits the cardioprotective signal from cytosol to mitochondria. Circ Res 97:329–336
Andrukhiv A, Costa AD, West IC, Garlid KD (2006) Opening mitoKATP increases superoxide generation from complex I of the electron transport chain. Am J Physiol Heart Circ Physiol 291:H2067–H2074
Costa AD, Jakob R, Costa CL, Andrukhiv K, West IC, Garlid KD (2006) The mechanism by which the mitochondrial ATP-sensitive K+ channel opening and H2O2 inhibit the mitochondrial permeability transition. J Biol Chem 281:20801–20808
Jaburek M, Costa AD, Burton JR, Costa CL, Garlid KD (2006) Mitochondrial PKC{epsilon} and Mitochondrial ATP-Sensitive K+ Channel Copurify and Coreconstitute to Form a Functioning Signaling Module in Proteoliposomes. Circ Res 99:878–883
Kim JS, Ohshima S, Pediaditakis P, Lemasters JJ (2004) Nitric oxide: a signaling molecule against mitochondrial permeability transition- and pH-dependent cell death after reperfusion. Free Radic Biol Med 37:1943–1950
Yamaguchi H, Wang HG (2001) The protein kinase PKB/Akt regulates cell survival and apoptosis by inhibiting Bax conformational change. Oncogene 20:7779–7786
Zuurbier CJ, Eerbeek O, Meijer AJ (2005) Ischemic preconditioning, insulin, and morphine all cause hexokinase redistribution. Am J Physiol Heart Circ Physiol 289:H496–H499
Pastorino JG, Hoek JB, Shulga N (2005) Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res 65:10545–10554
Abdallah Y, Gkatzoflia A, Gligorievski D et al (2006) Insulin protects cardiomyocytes against reoxygenation-induced hypercontracture by a survival pathway targeting SR Ca2+ storage. Cardiovasc Res 70:346–353
Dosenko VE, Nagibin VS, Tumanovskaya LV, Zagoriy VY, Moibenko AA, Vaage J (2006) Proteasomal proteolysis in anoxia-reoxygenation, preconditioning and postconditioning of isolated cardiomyocytes. Pathophysiology 13:119–125
Hahn HS, Yussman MG, Toyokawa T et al (2002) Ischemic protection and myofibrillar cardiomyopathy: dose-dependent effects of in vivo deltaPKC inhibition. Circ Res 91:741–748
Inagaki K, Hahn HS, Dorn GW, Mochly-Rosen D (2003) Additive protection of the ischemic heart ex vivo by combined treatment with delta-protein kinase C inhibitor and epsilon-protein kinase C activator. Circulation 108:869–875
Noma K, Oyama N, Liao JK (2006) Physiological role of ROCKs in the cardiovascular system. Am J Physiol Cell Physiol 290:C661–C668
Loirand G, Guerin P, Pacaud P (2006) Rho kinases in cardiovascular physiology and pathophysiology. Circ Res 98:322–334
Bao W, Hu E, Tao L et al (2004) Inhibition of Rho-kinase protects the heart against ischemia/reperfusion injury. Cardiovasc Res 61:548–558
Wolfrum S, Dendorfer A, Rikitake Y et al (2004) Inhibition of Rho-kinase leads to rapid activation of phosphatidylinositol 3-kinase/protein kinase Akt and cardiovascular protection. Arterioscler Thromb Vasc Biol 24:1842–1847
Hamid S, Bower HS, Baxter GF (2007) Rho-kinase plays a major role as a mediator of irreversible injury in reperfused myocardium. Am J Physiol Heart Circ Physiol Jan 12; [Epub ahead of print]
Asakura M, Jiyoong K, Minamino T, Shintani Y, Asanuma H, Kitakaze M (2004) Rationale and design of a large-scale trial using atrial natriuretic peptide (ANP) as an adjunct to percutaneous coronary intervention for ST-segment elevation acute myocardial infarction: Japan-Working groups of acute myocardial infarction for the reduction of Necrotic Damage by ANP (J-WIND-ANP). Circ J 68:95–100
Schwartz GG, Olsson AG, Ezekowitz MD et al (2001) Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA 285:1711–1718
Cannon CP, Braunwald E, McCabe CH et al (2004) Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 350:1495–1504
Mahaffey KW, Puma JA, Barbagelata NA et al (1999) Adenosine as an adjunct to thrombolytic therapy for acute myocardial infarction: results of a multicenter, randomized, placebo-controlled trial: the Acute Myocardial Infarction STudy of ADenosine (AMISTAD) trial. J Am Coll Cardiol 34:1711–1720
Ross AM, Gibbons RJ, Stone GW, Kloner RA, Alexander RW (2005) A randomized, double-blinded, placebo-controlled multicenter trial of adenosine as an adjunct to reperfusion in the treatment of acute myocardial infarction (AMISTAD-II). J Am Coll Cardiol 45:1775–1780
Nikolaidis LA, Mankad S, Sokos GG et al (2004) Effects of glucagon-like peptide-1 in patients with acute myocardial infarction and left ventricular dysfunction after successful reperfusion. Circulation 109:962–965
Ehrenreich H, Hasselblatt M, Dembowski C et al (2002) Erythropoietin therapy for acute stroke is both safe and beneficial. Mol Med 8:495–505
Lipsic E, van der MP, Voors AA et al (2006) A single bolus of a long-acting erythropoietin analogue darbepoetin alfa in patients with acute myocardial infarction: a randomized feasibility and safety study. Cardiovasc Drugs Ther 20:135–141
Lee TM, Su SF, Chou TF, Tsai CH (2002) Pharmacologic preconditioning of estrogen by activation of the myocardial adenosine triphosphate-sensitive potassium channel in patients undergoing coronary angioplasty. J Am Coll Cardiol 39:871–877
Shimokawa H, Hiramori K, Iinuma H et al (2002) Anti-anginal effect of fasudil, a Rho-kinase inhibitor, in patients with stable effort angina: a multicenter study. J Cardiovasc Pharmacol 40:751–761
Shibuya M, Hirai S, Seto M, Satoh S, Ohtomo E (2005) Effects of fasudil in acute ischemic stroke: results of a prospective placebo-controlled double-blind trial. J Neurol Sci 238:31–39
Kishi T, Hirooka Y, Masumoto A et al (2005) Rho-kinase inhibitor improves increased vascular resistance and impaired vasodilation of the forearm in patients with heart failure. Circulation 111:2741–2747
Chyrchel M, Rakowski T, Rzeszutko L et al (2006) Effects of high-dose statin administered prior to coronary angioplasty on the incidence of cardiac events in patients with acute coronary syndrome. Kardiol Pol 64:1357–1362
Marzilli M, Orsini E, Marraccini P, Testa R (2000) Beneficial effects of intracoronary adenosine as an adjunct to primary angioplasty in acute myocardial infarction. Circulation 101:2154–2159
Quintana M, Hjemdahl P, Sollevi A et al (2003) Left ventricular function and cardiovascular events following adjuvant therapy with adenosine in acute myocardial infarction treated with thrombolysis, results of the ATTenuation by Adenosine of Cardiac Complications (ATTACC) study. Eur J Clin Pharmacol 59:1–9
Mehta SR, Yusuf S, Diaz R et al (2005) Effect of glucose-insulin-potassium infusion on mortality in patients with acute ST-segment elevation myocardial infarction: the CREATE-ECLA randomized controlled trial. JAMA 293:437–446
Jonassen AK, Aasum E, Riemersma RA, Mjos OD, Larsen TS (2000) Glucose-insulin-potassium reduces infarct size when administered during reperfusion. Cardiovasc Drugs Ther 14:615–623
Apstein CS, Opie LH (2005) A challenge to the metabolic approach to myocardial ischaemia. Eur Heart J 26:956–959
Belhomme D, Peynet J, Louzy M, Launay JM, Kitakaze M, Menasche P (1999) Evidence for preconditioning by isoflurane in coronary artery bypass graft surgery. Circulation 100:II340–II344
Van Der Linden PJ, Daper A, Trenchant A, De Hert SG (2003) Cardioprotective effects of volatile anesthetics in cardiac surgery. Anesthesiology 99:516–517
Symons JA, Myles PS (2006) Myocardial protection with volatile anaesthetic agents during coronary artery bypass surgery: a meta-analysis. Br J Anaesth 97:127–136
Takano H, Hasegawa H, Kuwabara Y et al (2006) Feasibility and safety of granulocyte colony-stimulating factor treatment in patients with acute myocardial infarction. Int J Cardiol
Kang HJ, Kim HS, Zhang SY et al (2004) Effects of intracoronary infusion of peripheral blood stem-cells mobilised with granulocyte-colony stimulating factor on left ventricular systolic function and restenosis after coronary stenting in myocardial infarction: the MAGIC cell randomised clinical trial. Lancet 363:751–756
Hill JM, Syed MA, Arai AE et al (2005) Outcomes and risks of granulocyte colony-stimulating factor in patients with coronary artery disease. J Am Coll Cardiol 46:1643–1648
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hausenloy, D.J., Yellon, D.M. Reperfusion injury salvage kinase signalling: taking a RISK for cardioprotection. Heart Fail Rev 12, 217–234 (2007). https://doi.org/10.1007/s10741-007-9026-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-007-9026-1