
Summary
BI 836826 is a chimeric immunoglobulin G1 antibody targeting CD37, a transmembrane protein expressed on normal and malignant B cells. This open-label, phase Ib, dose-escalation study was conducted to determine the recommended phase II dose (RP2D) of BI 836826 + ibrutinib in patients with relapsed/refractory chronic lymphocytic leukemia (CLL). Eligible patients received 420 mg/day of ibrutinib with escalating doses of BI 836826. BI 836826 was administered in 4-week cycles. After Cycle 12, patients achieving complete response (CR), CR with incomplete marrow recovery, or minimal residual disease-negative partial response could continue to receive BI 836826 + ibrutinib every 4 weeks for ≤ 12 additional cycles. Patients received either 100 mg (n = 3) or 200 mg (n = 3) BI 836826 + ibrutinib. In the 100 mg BI 836826 cohort, one patient received two cycles and two patients received 22 cycles of BI 836826. In the 200 mg BI 836826 cohort, patients received 12, 16 and 20 cycles of BI 836826, respectively. All patients discontinued BI 836826 and continued ibrutinib outside the trial. No dose-limiting toxicities were reported in the maximum tolerated dose (MTD) evaluation period. As the trial was discontinued before the MTD was reached, the RP2D was not determined. Grade 3/4 adverse events (AEs) were predominantly hematological. Pseudomonal bacteremia was the only drug-related AE of special interest. BI 836826 + ibrutinib did not exceed the MTD at doses up to 200 mg in patients with CLL. However, RP2D and MTD were not formally established, as the sponsor discontinued the trial.
Similar content being viewed by others


Introduction
The Bruton tyrosine kinase (BTK) inhibitor, ibrutinib, is well established in the treatment of patients with previously untreated and relapsed/refractory chronic lymphocytic leukemia (CLL) [1,2,3]. While ibrutinib monotherapy is associated with impressive response rates in CLL patients, including those with del(17p) [1, 4], deep responses are rare, thus necessitating continuous use. This can result in cumulative toxicities, leading to treatment discontinuation [5]. Furthermore, patients often become resistant to long-term treatment, potentially leading to disease progression or transformation [6]. Therefore, there is clinical rationale for combining ibrutinib with other agents that could increase depth of response and delay development of resistance.
Preclinical and clinical studies have demonstrated that CD37, a tetraspanin B cell surface molecule, is a potential drug target in patients with CLL [7,8,9,10].
BI 836826 is a chimeric mouse–human anti-CD37 monoclonal antibody engineered to enhance binding and effector function that mediates direct antibody-dependent cell-mediated cytotoxicity (ADCC) against CLL cells [11]. A phase I, dose escalation study of BI 836826 in patients with relapsed/refractory CLL demonstrated acceptable tolerability and notable efficacy, particularly in patients with poor-risk features such as del(17p) and TP53 mutations [12]. In this phase Ib, dose-escalation study, we investigated the combination of ibrutinib and BI 836826 in patients with relapsed/refractory CLL.
Patients and methods
Patients
Eligible patients were ≤ 18 years old with relapsed/refractory CLL according to the International Workshop on Chronic Lymphocytic Leukemia (IWCLL) criteria [13]. All patients had received at least one prior line of systemic treatment. Prior BTK inhibitors were not allowed. Other eligibility criteria included Eastern Cooperative Oncology Group (ECOG) Performance Status (PS) of 0–2; clinically quantifiable disease burden (absolute lymphocyte count > 10,000/µL, measurable lymphadenopathy, or quantifiable bone marrow infiltration); adequate organ function; and residual non-hematological toxicity from prior treatment of grade ≤ 1.
Key exclusion criteria were: any CD37-targeting antibody or CD37 antibody-drug conjugate; allogeneic stem cell transplant within 1 year or active graft-versus-host disease; known transformation of CLL to an aggressive B-cell malignancy at the time of screening; history of non-CLL malignancy except for adequately treated in-situ, stage I or II carcinoma in complete response (CR) or any other cancer that had been in CR for ≥ 2 years after the end of cancer treatment; and active uncontrolled autoimmune cytopenia.
The trial was carried out in accordance with the Declaration of Helsinki, Good Clinical Practice Guidelines, applicable regulatory requirements and Boehringer Ingelheim standard operating procedures. The study protocol was approved by the Institutional Review Boards of all participating institutions. Written informed consent was obtained from all patients.
Study design and treatment
The primary objectives of this single arm, open-label, dose escalation phase Ib study were to determine the recommended phase II dose (RP2D) of BI 836826 plus ibrutinib in patients with relapsed/refractory CLL, and the number of patients with dose-limiting toxicities (DLTs) during the maximum tolerated dose (MTD) evaluation period (Cycle 1). Other objectives were the determination of the MTD, safety and efficacy of the combination.
Eligible patients underwent a two-week run-in phase with ibrutinib and remained on ibrutinib throughout the trial at a constant dose of 420 mg daily. BI 836826 was planned at dose levels of 100, 200, 400, 600, 800 and 1400 mg. Mandatory pre-medication (antihistamine, analgesic and glucocorticoid) to mitigate the risk of infusion-related reactions (IRRs) was given 30–120 min prior to BI 836826 administration. BI 836826 was then administered via rate-controlled intravenous infusion in Cycle 1 as a 10 mg dose on Day 1, on Days 2 and 8 at 50% of the assigned dose on each day, and on Day 15 at 100% of the assigned dose. In Cycles 2–4, BI 836826 was administered at the assigned dose on Days 1 and 15 of each cycle. In Cycles 5–12, BI 836826 was administered at the assigned dose on Day 1 only. BI 836826 was administered in 4-week cycles. At the end of Cycle 12, bone marrow biopsies were performed to evaluate response. Patients with a CR, CR with incomplete marrow recovery (CRi), or minimal residual disease (MRD)-negative partial response (PR) could choose to receive 12 additional treatment cycles of the combination. Following completion of 12 or 24 cycles, or discontinuation due to disease progression, unacceptable toxicity, or withdrawal of consent, patients could continue to receive ibrutinib outside of the study at the discretion of the treating investigator.
Study assessments
Safety was assessed by determining the incidence and severity of adverse events, graded according to the Common Terminology Criteria for Adverse Events (CTCAE), version 4.0. Assessment of laboratory parameters, vital signs, physical examination and ECG was also undertaken. DLTs were defined as: any non-hematologic adverse event of grade ≥ 3 related to BI 836826 or ibrutinib, except IRRs and grade 3 alanine aminotransferase and/or aspartate aminotransferase elevation without concomitant bilirubin elevation or any other asymptomatic grade 3 laboratory abnormality with spontaneous recovery within 1 week; hematologic adverse events related to BI 836826 or ibrutinib including grade 4 neutropenia with concomitant infection; grade 4 febrile neutropenia, and grade 3 febrile neutropenia not resolving within 72 hours; grade 4 thrombocytopenia with clinically significant bleeding; grade 4 anemia; any grade 5 hematologic adverse event. Adverse events of special interest (AESIs) included DLTs, infusion-related reactions of grade ≥ 3, late-onset infections, events indicative of drug-induced liver injury, and tumor lysis syndrome. Adverse events consistent with the definition of a DLT but occurring after the MTD evaluation period were also considered AESIs.
Overall response, duration of response, MRD and reduction in size of tumor lymph nodes were exploratory endpoints. Overall response was defined as patients who achieved CR, CRi, PR or PR with lymphocytosis (PR-L) according to modified IWCLL guidelines [13]. MRD was evaluated in both blood and bone marrow samples and was defined as < 1 leukemic cell per 10,000 leukocytes detected by multi-parameter flow cytometry.
Statistical methods
All analyses were descriptive and exploratory. No formal statistical analysis was conducted. Dose escalation was guided by a Bayesian 5-parameter logistic regression model (BLRM) with overdose control [14, 15] that was fitted to binary toxicity outcomes.
Results
Patients and treatment exposure
Between July 7, 2016 and July 9, 2019, 10 patients were screened, of whom 7 entered the run-in period with ibrutinib. Six patients were treated with BI 836826 plus ibrutinib across three sites in the United States (100 mg BI 836826: n = 3; 200 mg BI 836826: n = 3). The median age among treated patients was 71.0 years (range 57–76 years); all patients were Caucasian (Table 1).
In the 100 mg BI 836826 cohort, one patient received two cycles and two patients received 22 cycles of BI 836826. The patients in the 200 mg cohort received 12, 16 and 20 cycles of BI 836826, respectively. All six patients discontinued treatment with BI 836826 (disease progression, n = 1; investigator discretion, n = 3; lack of response by cycle 12, n = 2). Mean duration of BI 836826 exposure was 440.2 days. There were no dose reductions. Two patients in the 100 mg BI 836826 cohort had at least one important protocol deviation. Both patients continued treatment with BI 836826 beyond cycle 12 without having a CR, CRi, or MRD-negative PR. One of these patients did not receive pre-medication with glucocorticoid in Cycle 3, as required per protocol.
DLTs during the MTD evaluation period, MTD and RP2D
No DLTs were reported during the MTD evaluation period with 100 or 200 mg BI 836826. However, the trial was discontinued before patients were treated with 400 mg BI 836826, and the MTD and RP2D were therefore not determined.
Safety
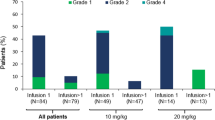
All six patients had at least one adverse event that was considered by the investigator to be related to BI 836826. The most common BI 836826-related adverse events (any grade/grade ≥ 33) were IRR (67%/17%), neutropenia (50%/33%), anemia (33%/33%), lymphopenia (33%/33%) and fatigue (33%/0%; Table 2). All IRRs (ten in four patients) were grade ≤ 2 except for one grade 3 IRR. The majority of IRRs occurred within the first two cycles and were resolved within a day.
Five patients had serious adverse events (considered related to study drug in 4 patients), including neutropenia, acute coronary syndrome, cellulitis, pseudomonal bacteremia and Bowen’s disease. No serious adverse event was experienced by more than one patient. There were no fatal adverse events, and no dose reductions or permanent discontinuations of BI 836826 due to adverse events. One patient had an ibrutinib dose reduction due to grade 2 neutropenia. After the MTD evaluation period, two patients in the 200 mg cohort reported grade 3 DLTs: a duodenal ulcer occurring on Day 325 and pseudomonal bacteremia (considered as related to study drug) on Day 280.
Three AESIs were recorded: the two DLTs reported after the MTD evaluation period and grade 2 basal cell carcinoma (Day 622).
Based on laboratory data, grade 4 neutropenia was reported for two patients (one patient in each dose cohort). Neither episode was associated with a concomitant infection. One patient in the 100 mg dose cohort had an episode of grade 4 thrombocytopenia, which was not associated with concomitant bleeding. Two patients (one in each dose cohort) had an episode of grade ≥ 3 decreased CD4 + T-cell count but there were no concomitant CD4+-related specific infections.
Efficacy
Overall response rate was 83% among the 6 evaluable patients (one CR and four PRs; Table 3) The ORR in the 200 mg cohort was 100% (one CR, two PR). Of five patients who underwent peripheral blood-based MRD analysis, two were MRD-negative, one in each cohort including the patient who achieved CR. Three patients had bone marrow aspirate samples analyzed for MRD. None were MRD-negative but one patient developed MRD-negative CR a month after study completion.
All five patients were censored for the analysis of duration of overall response since they had discontinued before progression or death was observed. Mean (standard deviation [SD]) duration of response was 538.0 (31.1) days and 312.0 (143.96) days in the 100 mg and 200 mg BI 836826 cohorts, respectively. The median best percentage change from baseline in the SPD of lymph nodes was -81.2% (range -84% to -51%) in the 100 mg dose group and -79.2% (range -92% to -59%) in the 200 mg dose group (Fig. 1). Tumor size was reduced in all six patients.

Maximum change in SPD of lymph node lesions at best response
Discussion
In this phase Ib, dose-escalation study of BI 836826 plus ibrutinib in patients with relapsed/refractory CLL, no DLTs were observed during the MTD evaluation period in either the 100 mg or 200 mg BI 836320 cohorts. Despite these promising findings, the sponsor took the strategic decision to discontinue the clinical development of BI 836826, due to the rapidly evolving CLL treatment landscape. Accordingly, the MTD was not reached and the RP2D was not determined. There were no safety reasons to terminate the program. Nevertheless, the results from this study suggest that BI 836826 can be safely combined with ibrutinib, with no apparent additive toxicity. Adverse events associated with the combination were consistent with previous clinical experience with BI 836826 [12] or ibrutinib monotherapy [4].
The efficacy of ibrutinib in relapsed/refractory CLL is well established [4, 17, 18], with an objective response rate of 63% observed in the phase III study, RESONATE [4]. Of note, overall response rate to ibrutinib generally deepens over time [17, 19], and had increased to 91% by the final analysis of RESONATE [2, 20]. Our preliminary findings (83% overall response rate) suggest that the addition of anti-CD37 agents to ibrutinib could potentially improve efficacy. While the sample size is limited, the promising efficacy of this combination especially at the 200 mg dose (100%), supports ongoing exploration of agents, including those that target CD37, for fixed-duration therapy in CLL.
A number of CD37-based therapeutics are currently undergoing investigation in CLL, including otlertuzumab, an anti-CD37 mono-specific ADAPTIR therapeutic protein [8]. Notably, in phase II studies, the addition of otlertuzumab to bendamustine greatly enhanced response compared with bendamustine alone in relapsed CLL patients [21]. Additionally, 212Pb-NNV003, a CD37-targeted radioimmunotherapy, has demonstrated notable anti-tumor effects in an animal model of CLL [22].
In conclusion, BI 836826 plus ibrutinib did not exceed the MTD at doses up to 200 mg in patients with CLL, and no DLTs were reported in the MTD evaluation period. While the RPTD and MTD were not formally established, our findings suggest that an anti-CD37 antibody may be combined with ibrutinib, or potentially other BTK inhibitors, for the treatment of relapsed/refractory CLL. Since continuous therapy with BTK inhibitors leads to toxicities, therapeutic resistance and is associated with high costs, exploration of novel combinations involving active agents with different therapeutic targets remains an unmet medical need.
Data availability
The clinical study report (including appendices, but without line listings) and other clinical documents related to this study may be accessed on request. Prior to providing access, the documents and data will be examined, and, if necessary, redacted and de-identified to protect the personal data of study participants and personnel, and to respect the boundaries of the informed consent of the study participants. See https://trials.boehringer-ingelheim.com/data_sharing/sharing.html#accordion-1-2 for further details. Bona fide, qualified scientific and medical researchers may request access to de-identified, analyzable patient-level study data, together with documentation describing the structure and content of the datasets. Researchers should use https://clinicalstudydatarequest.com/ to request access to raw data from this study.
References
Burger JA, Barr PM, Robak T, Owen C, Ghia P, Tedeschi A, Bairey O, Hillmen P, Coutre SE, Devereux S, Grosicki S, McCarthy H, Simpson D, Offner F, Moreno C, Dai S, Lal I, Dean JP, Kipps TJ (2019) Long-term efficacy and safety of first-line ibrutinib treatment for patients with CLL/SLL: 5 years of follow-up from the phase 3 RESONATE-2 study. Leukemia 34(3):787–798. https://doi.org/10.1038/s41375-019-0602-x
Byrd JC, Hillmen P, O’Brien S, Barrientos JC, Reddy NM, Coutre S, Tam CS, Mulligan SP, Jaeger U, Barr PM, Furman RR, Kipps TJ, Thornton P, Moreno C, Montillo M, Pagel JM, Burger JA, Woyach JA, Dai S, Vezan R, James DF, Brown JR (2019) Long-term follow-up of the RESONATE phase 3 trial of ibrutinib vs ofatumumab. Blood 133(19):2031–2042. https://doi.org/10.1182/blood-2018-08-870238
Hallek M (2019) Chronic lymphocytic leukemia: 2020 update on diagnosis, risk stratification and treatment. Am J Hematol 94(11):1266–1287. https://doi.org/10.1002/ajh.25595
Byrd JC, Brown JR, O’Brien S, Barrientos JC, Kay NE, Reddy NM, Coutre S, Tam CS, Mulligan SP, Jaeger U, Devereux S, Barr PM, Furman RR, Kipps TJ, Cymbalista F, Pocock C, Thornton P, Caligaris-Cappio F, Robak T, Delgado J, Schuster SJ, Montillo M, Schuh A, de Vos S, Gill D, Bloor A, Dearden C, Moreno C, Jones JJ, Chu AD, Fardis M, McGreivy J, Clow F, James DF, Hillmen P, Investigators R (2014) Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med 371(3):213–223. https://doi.org/10.1056/NEJMoa1400376
Gordon MJ, Churnetski M, Alqahtani H, Rivera X, Kittai A, Amrock SM, James S, Hoff S, Manda S, Spurgeon SE, Choi M, Cohen JB, Persky D, Danilov AV (2018) Comorbidities predict inferior outcomes in chronic lymphocytic leukemia treated with ibrutinib. Cancer 124(15):3192–3200. https://doi.org/10.1002/cncr.31554
Pula B, Golos A, Gorniak P, Jamroziak K (2019) Overcoming Ibrutinib Resistance in Chronic Lymphocytic Leukemia. Cancers (Basel) 11(12). https://doi.org/10.3390/cancers11121834
Beckwith KA, Frissora FW, Stefanovski MR, Towns WH, Cheney C, Mo X, Deckert J, Croce CM, Flynn JM, Andritsos LA, Jones JA, Maddocks KJ, Lozanski G, Byrd JC, Muthusamy N (2014) The CD37-targeted antibody-drug conjugate IMGN529 is highly active against human CLL and in a novel CD37 transgenic murine leukemia model. Leukemia 28(7):1501–1510. https://doi.org/10.1038/leu.2014.32
Byrd JC, Pagel JM, Awan FT, Forero A, Flinn IW, Deauna-Limayo DP, Spurgeon SE, Andritsos LA, Gopal AK, Leonard JP, Eisenfeld AJ, Bannink JE, Stromatt SC, Furman RR (2014) A phase 1 study evaluating the safety and tolerability of otlertuzumab, an anti-CD37 mono-specific ADAPTIR therapeutic protein in chronic lymphocytic leukemia. Blood 123(9):1302–1308. https://doi.org/10.1182/blood-2013-07-512137
Maaland AF, Heyerdahl H, O’Shea A, Eiriksdottir B, Pascal V, Andersen JT, Kolstad A, Dahle J (2019) Targeting B-cell malignancies with the beta-emitting anti-CD37 radioimmunoconjugate (177)Lu-NNV003. Eur J Nucl Med Mol Imaging 46(11):2311–2321. https://doi.org/10.1007/s00259-019-04417-1
Sawas A, Savage KJ, Perez RP, Advani RH, Zaine JM, Lackey JM Trave F, Anand B, Chu R, Reyno LM, O'Connor OA (2017) A phase 1 study of the anti-CD37 antibody-drug conjugate AGS67E in advanced lymphoid malignancies. Interim results. Presented at the 14th International Conference on Malignant Lymphoma, Lugano, Switzerland, 14–17 June, 2017. Hematol Oncol 35(S2)
Heider KH, Kiefer K, Zenz T, Volden M, Stilgenbauer S, Ostermann E, Baum A, Lamche H, Kupcu Z, Jacobi A, Muller S, Hirt U, Adolf GR, Borges E (2011) A novel Fc-engineered monoclonal antibody to CD37 with enhanced ADCC and high proapoptotic activity for treatment of B-cell malignancies. Blood 118(15):4159–4168. https://doi.org/10.1182/blood-2011-04-351932
Stilgenbauer S, Aurran Schleinitz T, Eichhorst B, Lang F, Offner F, Rossi JF, Schroyens W, Van Den Neste E, Ysebaert L, von Wangenheim U, Ursula Kress U, Blum P, Zenz T (2019) Phase 1 first-in-human trial of the anti-CD37 antibody BI 836826 in relapsed/refractory chronic lymphocytic leukemia. Leukemia 33(10):2531–2535. https://doi.org/10.1038/s41375-019-0475-z
Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, Hillmen P, Keating MJ, Montserrat E, Rai KR, Kipps TJ, International Workshop on Chronic Lymphocytic L (2008) Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 111 (12):5446–5456. https://doi.org/10.1182/blood-2007-06-093906
Babb J, Rogatko A, Zacks S (1998) Cancer phase I clinical trials: efficient dose escalation with overdose control. Stat Med 17 (10):1103–1120. https://doi.org/10.1002/(sici)1097-0258(19980530)17:10<1103::aid-sim793>3.0.co;2-9
Neuenschwander B, Branson M, Gsponer T (2008) Critical aspects of the Bayesian approach to phase I cancer trials. Stat Med 27(13):2420–2439. https://doi.org/10.1002/sim.3230
Rai KR, Sawitsky A, Cronkite EP, Chanana AD, Levy RN, Pasternack BS (1975) Clinical staging of chronic lymphocytic leukemia. Blood 46(2):219–234
O’Brien S, Furman RR, Coutre S, Flinn IW, Burger JA, Blum K, Sharman J, Wierda W, Jones J, Zhao W, Heerema NA, Johnson AJ, Luan Y, James DF, Chu AD, Byrd JC (2018) Single-agent ibrutinib in treatment-naive and relapsed/refractory chronic lymphocytic leukemia: a 5-year experience. Blood 131(17):1910–1919. https://doi.org/10.1182/blood-2017-10-810044
Huang X, Qiu L, Jin J, Zhou D, Chen X, Hou M, Hu J, Hu Y, Ke X, Li J, Liang Y, Liu T, Lv Y, Ren H, Sun A, Wang J, Zhao C, Salman M, Sun S, Howes A, Wang J, Wu P, Li J (2018) Ibrutinib versus rituximab in relapsed or refractory chronic lymphocytic leukemia or small lymphocytic lymphoma: a randomized, open-label phase 3 study. Cancer Med 7(4):1043–1055. https://doi.org/10.1002/cam4.1337
Byrd JC, Furman RR, Coutre SE, Burger JA, Blum KA, Coleman M, Wierda WG, Jones JA, Zhao W, Heerema NA, Johnson AJ, Shaw Y, Bilotti E, Zhou C, James DF, O’Brien S (2015) Three-year follow-up of treatment-naive and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood 125(16):2497–2506. https://doi.org/10.1182/blood-2014-10-606038
Munir T, Brown JR, O’Brien S, Barrientos JC, Barr PM, Reddy NM, Coutre S, Tam CS, Mulligan SP, Jaeger U, Kipps TJ, Moreno C, Montillo M, Burger JA, Byrd JC, Hillmen P, Dai S, Szoke A, Dean JP, Woyach JA (2019) Final analysis from RESONATE: Up to six years of follow-up on ibrutinib in patients with previously treated chronic lymphocytic leukemia or small lymphocytic lymphoma. Am J Hematol 94(12):1353–1363. https://doi.org/10.1002/ajh.25638
Robak T, Hellmann A, Kloczko J, Loscertales J, Lech-Maranda E, Pagel JM, Mato A, Byrd JC, Awan FT, Hebart H, Garcia-Marco JA, Hill BT, Hallek M, Eisenfeld AJ, Stromatt SC, Jaeger U (2017) Randomized phase 2 study of otlertuzumab and bendamustine versus bendamustine in patients with relapsed chronic lymphocytic leukaemia. Br J Haematol 176(4):618–628. https://doi.org/10.1111/bjh.14464
Saidi A, Maaland A, Torgue J, Heyerdah H, Dahle J (2019) 212Pb-NNV003 as a novel targeted alpha therapy for CD37 positive B-cell chronic lymphocytic leukemia (CLL) and non-hodgkin lymphoma (NHL). J Med Imaging Radiat Sci 50(1):S8. https://doi.org/10.1016/j.jmir.2019.03.025
Acknowledgements
We thank the patients, their families, and all the investigators who participated in these studies. The authors were fully responsible for all content and editorial decisions, were involved at all stages of manuscript development, and have approved the final version.
Funding
The conduct of this research, study design, data collection, and analysis was financially supported by Boehringer Ingelheim. Medical writing assistance, funded by Boehringer Ingelheim, was provided by Lynn Pritchard, DPhil, of GeoMed, an Ashfield company, part of UDG Healthcare plc, during the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
AVD has received consulting fees from AstraZeneca, Abbvie, BeiGene, Genentech, TG Therapeutics, Janssen, Rigel Pharmaceutical, Nurix, Bayer Oncology, Karyopharm and Pharmacyclics and has ongoing research funding from AstraZeneca, Takeda Oncology, Gilead Sciences, Bayer Oncology, Genentech, Verastem Oncology, MEI and Bristol Myers Squibb.
SES reports grants from Boehringer Ingelheim during the conduct of the study and personal fees from Pharmacyclics outside the submitted work.
TS reports institutional funds fees from Boehringer Ingelheim during the conduct of the study; personal fees from AstraZeneca, PCYC, Janssen, Juno, Celgene, BeiGene and Kite Pharma outside the submitted work.
AMQ, DM and DS are employees of Boehringer Ingelheim.
JRB reports personal fees from Abbvie, Acerta/AstraZeneca, Astellas, BeiGene, Catapult Therapeutics, Dynamo Therapeutics, Genentech/Roche, Janssen, Juno/Celgene, Kite, MEI Pharma, Nextcea, Novartis, Octapharma, Pfizer, Pharmacyclics, Redx, Rigel, Sun, Sunesis, Teva, and TG Therapeutics; grants and personal fees from Gilead, Loxo/Lilly and Verastem outside the submitted work. She has sat on Data Safety Monitoring Boards for Invectys and Morphosys.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Danilov, A.V., Spurgeon, S.E., Siddiqi, T. et al. A phase Ib, open label, dose escalation trial of the anti-CD37 monoclonal antibody, BI 836826, in combination with ibrutinib in patients with relapsed/refractory chronic lymphocytic leukemia. Invest New Drugs 39, 1099–1105 (2021). https://doi.org/10.1007/s10637-020-01056-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-020-01056-4