
Summary
Background Docetaxel and prednisolone chemotherapy (DP) extends survival in metastatic castration resistant prostate cancer (mCRPC). However, emergent clinical resistance is almost inevitable. AKT pathway activation is highly prevalent in mCRPC contributing to disease progression and DP resistance. AZD5363 is a potent oral pan-AKT inhibitor with pre-clinical data indicating activity in mCRPC and synergy with docetaxel. Methods This phase I trial was to determine an AZD5363 recommended phase II dose (RP2D) for combination with DP. Eligibility criteria included chemotherapy naive mCRPC, PSA or radiographic disease progression and ECOG performance status 0 or 1. Treatment comprised DP (75 mg/m2, IV, day 1 and 5 mg BID, PO, day 1–21 respectively for ten cycles) and AZD5363 to disease progression for all patients. We utilised a 3 + 3 dose escalation design to determine a maximum tolerated dose according to defined dose limiting toxicity criteria assessed using CTCAE version 4.03. Planned AZD5363 dose levels were 320 mg (DL1), 400 mg (DL2) and 480 mg (DL3), BID, PO, 4 days on/3 days off, from day 2 of each cycle. Results 10 patients were treated. Dose limiting toxicities affected 2 patients (grade 3 rash ≥5 days; grade 3 diarrhoea) in DL2. The commonest grade 3 or 4, AZD5363 related, symptomatic adverse events were rash and diarrhoea. Hyperglycaemia affected all patients but was self-limiting. PSA reduction to <50% at 12 weeks occurred in 7 patients. Conclusions The RP2D for AZD5363 is 320 mg BID, 4 days on/3 days off, in combination with full dose DP for mCRPC.
Similar content being viewed by others


Introduction
There are approximately 47,000 new cases and 11,000 deaths from prostate cancer in the UK per year [1]. Docetaxel and prednisolone chemotherapy (DP) extends survival and maintains quality of life in metastatic castration resistant prostate cancer (mCRPC) [2, 3]. However, despite the advent of chemotherapy utilisation earlier in the treatment pathway for hormone sensitive disease, and the introduction of multiple other life extending therapeutic options, advanced prostate cancer remains incurable [4–12]. From the point of castrate resistance, median survival is in the range of 2–3 years [4–6]. One contributory factor in these outcomes is the common phenomenon of emergent therapeutic resistance either during, or shortly following, the administration of DP for mCRPC. For example, in a 755 patient phase III study evaluating subsequent chemotherapy, three quarters of patients had progressed during or within 3 months of completing DP [8]. A pressing unmet need, therefore, remains to develop therapeutic strategies to address clinical resistance to DP and improve on current outcomes.
Serine/threonine protein kinase AKT (protein kinase B) pathway activation is highly prevalent in prostate cancer. In addition to the loss of PTEN function through deletion, inactivating mutation, or reduced protein expression, the frequency of pathway alteration rises substantially when inactivating PHLPP and INPP4B alterations or activating PI3K component (PIK3R1, PIK3R3, PIK3CA) and AKT isoform mutations are also included as potential causes of pathway activation [13]. Data indicate that the rate of a potentially activating pathway alteration rises to approaching 100% if assessed within metastatic samples. Furthermore, in pre-clinical models AKT pathway activation has been shown to contribute to disease progression and therapeutic resistance to DP [13–15].
AZD5363 is a potent oral pan-AKT inhibitor with activity against AKT 1, 2 and 3 (IC50 < 10 nM). It also inhibits protein kinase A (PKA) and the Rho associated protein kinases (ROCK1 and 2). AZD5363 inhibits phosphorylation of AKT substrates GSK3β and PRAS40 and the downstream biomarker S6 in cell lines, including LNCaP prostate cancer cells and rodent xenografts including from PC3 prostate cancer cells. Pre-clinical data support both single agent activity in mCRPC and synergy with docetaxel [14, 16, 17]. In first in human studies as a single agent, diarrhoea, hyperglycaemia, nausea, and rash were the most common adverse events (AE) [18, 19]. Exploratory data from these single agent trials is consistent with relevant signalling inhibition based on pre- and post-treatment tumour biopsies (increase in phospho-AKT, reduction in phospho-GSK3β and phospho-PRAS40) and clinical activity in patients with PIK3CA-mutant breast cancer and in patients with tumour AKT1 (E17K) mutations [18, 19].
The ProCAID phase I clinical trial was undertaken to establish a DP/AZD5363 combination to allow for subsequent development in mCRPC and potentially also earlier phases of the disease pathway.
Patients and methods
Patients
Eligible patients were 18 years or older with histologically or cytologically proven mCRPC and an Eastern Cooperative Oncology Group performance status of 0 or 1. All patients had disease progression based on PSA and/or radiographic criteria defined by the Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1) and the Prostate Cancer Working Group 2 [20, 21]. Radiologically measurable and/or evaluable disease was acceptable. Patients were required to have a serum testosterone <1.7 nmol/L and ongoing LHRH analogue or antagonist therapy was permitted to maintain a castrate state. Other therapies for prostate cancer, other than ongoing bisphosphonates or denosumab, were discontinued ≥4 weeks prior to commencing study treatment and anti-androgen withdrawal response was excluded where relevant. Haematological parameter requirements were: haemoglobin ≥9 g/dL, platelets ≥100 × 109/L, neutrophils ≥1.5 × 109/L, bilirubin ≤ the institutional upper limit of normal (ULN), alanine (ALT) and aspartate (AST) aminotransferase ≤1.5 x ULN and sodium and potassium within the normal range for the treating institution. Patients were excluded if they had received previous treatment with cytotoxic chemotherapy but were permitted the prior use of second generation hormonal therapies e.g., abiraterone or enzalutamide. Other exclusion criteria included prior malignancy with an estimated ≥30% chance of relapse within 2 years, previously identified brain metastases, or spinal cord compression unless treated with full functional recovery, prior radiotherapy to >30% of bone marrow, another investigational agent within 30 days of study medication, type I or II diabetes mellitus requiring either insulin or oral hypoglycaemics for routine management, gastrointestinal conditions that might affect drug absorption, significant cardiac disease within the last 6 months, a left ventricular ejection fraction < the institutional lower limit of normal, a QTc interval of >480 msec, or recent exposure to potent inhibitors or inducers of CYP3A4 or substrates of CYP3A4 and CYP2D6. The complete eligibility criteria are listed in the Supplementary Appendix.
Treatment
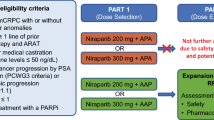
The treatment cycle is summarised in Fig. 1 and comprised DP for up to 10 cycles of 21 days. All patients received docetaxel 75 mg/m2 by one hour intravenous infusion on day 1 and prednisolone 5 mg BID, orally, on days 1 to 21 of each cycle. In addition, patients received AZD5363 which was continued until either disease progression, the commencement of new anti-prostate cancer systemic therapy or unacceptable toxicity. Planned AZD5363 dose levels were 320 mg (dose level 1), 400 mg (dose level 2) and 480 mg (dose level 3), BID, orally, given according to a 4 days on and 3 days off schedule which commenced from cycle 1, day 2. Dexamethasone pre-medication was recommended at 8 mg, orally, at 12, 3 and 1 h prior to each docetaxel infusion and anti-emetics were given according to local institutional protocols. Body surface area calculations and re-calculations due to changes in weight and docetaxel dose banding (up to +/− 5%) were permitted in accordance with local institutional practices.

Schematic representation of the days of dosing (blue crossed boxes) for each of the indicated drugs at the following doses: docetaxel, 75 mg/m2 by one hour intravenous infusion on day 1; dexamethasone 8 mg orally, at 12, 3 and 1 h prior to each docetaxel infusion; AZD5363, dosed according to dose level cohort, BID, orally, taken 4 days on/3 days off, continuously from cycle 1 day 2; prednisolone, 5 mg BID orally days 1–21
Study design
We utilised a conventional 3 + 3 dose escalation phase I clinical trial design. An evaluable patient was defined as one that completed cycle 1 and received at least 80% of the total cumulative doses of AZD5363 and prednisolone, and the full dose of docetaxel, with no more than a 14-day delay in starting cycle 2, or who had experienced a dose limiting toxicity (DLT). AE data was recorded for all patients who commenced study treatment.
The National Cancer Institute Common Toxicity Criteria for Adverse Events version 4.03 (CTCAEv4.03, http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf) was used to characterise AEs. DLTs were predefined and based on recorded AEs as: a greater than 14 day delay in administration of docetaxel for cycle 2 due to drug toxicity; grade 4 neutropenia ≥7 days duration; grade 3 or 4 neutropenia associated with a temperature ≥ 38.5 °C; grade 3 or 4 neutropenia associated with bacteriologically proven sepsis; any grade 4 thrombocytopenia; grade 3 thrombocytopenia associated with non-traumatic bleeding (except where this could be explained by therapeutic anticoagulation); ≥ grade 3 hyperglycaemia for more than 1 week despite optimal intervention; grade 4 hyperglycaemia; AST or ALT >10 x ULN where AZD5363 was considered the most likely cause; AST or ALT >8 x ULN when combined with a doubling of bilirubin from baseline and where AZD5363 was considered the most likely cause; QTc (Fridericia’s or Bazett’s correction) interval > 500 msec or QTc increase >60 msec from baseline on two ECGs at least 30 min apart that could not be attributed to another cause; symptomatic congestive cardiac failure (New York Heart Association class III/IV) and a drop in LVEF that could not be attributed to another cause; a decrease in LVEF of ≥20% to a level below the institutional lower limit of normal range; clinically significant rash that despite optimal treatment remained grade ≥ 3 for 5 days or longer and that could not be attributed to another cause; grade ≥ 3 nausea, vomiting or diarrhoea, despite optimal anti-emetic or anti-diarrhoeal therapy and which could not be attributed to another cause; any other grade ≥ 3 toxicity which in the opinion of the investigator was clinically significant and related to AZD5363. Designation of a DLT excluded isolated laboratory changes of any grade (except as specified above) without clinical sequelae or clinical significance.
Intra-patient dose escalation was not permitted. In addition to protocol defined DLTs, the protocol allowed for the Safety Review Committee to consider other toxicities including those emerging during subsequent treatment cycles in prior patients in making recommendations. Dose escalation was undertaken if none of 3, or 1 of 6, evaluable patients experienced a DLT at the current dose level. If 2 or more patients experienced a DLT then the preceding dose level was established as the maximum tolerated dose (MTD). The recommended phase II dose (RP2D) was the MTD or dose level three if this was tolerated.
The primary endpoint was the determination of an RP2D for AZD5363, using a four days on/three days off administration schedule, in combination with full dose DP. Secondary endpoints included safety and tolerability profiles using CTCAEv4.03 and AZD5363 pharmacokinetics when combined with DP. Results were summarised descriptively.
Trial conduct was consistent with Good Clinical Practice guidelines and the Declaration of Helsinki and following national ethics and regulatory approvals. Informed consent was obtained from all individual participants included in the study. The trial was coordinated by the Cancer Research UK Southampton Clinical Trials Unit and sponsored by University Hospital Southampton NHS Foundation Trust.
Results
Patients
Ten patients with a median age of 67.5 (range 56–72) were recruited from two UK centres with 4 patients in dose level 1, and 6 in dose level 2. Nine patients (90%) had bony metastases and 5 (50%) had visceral metastases. One patient in dose level 1 was considered to be non-evaluable for DLT assessment due to a compliance error, resulting from misunderstanding rather than treatment related AEs, in cycle 1 resulting in only 54.2% of the cumulative AZD5363 dose being taken. This patient subsequently tolerated treatment at full dose from cycle 2 onwards. Patient characteristics are shown in Table 1.
Dose escalation and safety
A median of 8.5 cycles (range 3–10) of DP and 4 cycles (range 1–13) of AZD5363 was administered. The actual number of cycles administered for each patient is shown in Table 2. In dose level 1, all 3 evaluable patients received an equal or greater number of cycles of AZD5363 compared to DP. In dose level 2 however, this was the case for only 2 of 6 patients. The 4 other patients in dose level 2 received only 1 or 2 cycles of AZD5363 whilst receiving between 3 and 10 cycles of docetaxel. No DLTs were seen in dose level 1. Two patients in dose level 2 experienced DLTs. These were due to grade 3 rash for ≥5 days and grade 3 diarrhoea despite optimal anti-diarrhoeal therapy. The severity and number of AEs overall and by dose level is shown in Table 3. Five patients in cycle 1, and 8 (80%) across all cycles had at least one grade 3 or 4 AE.
Rates of specific AEs, occurring in at least one patient, are shown for cycle 1 (Table 4) and across all cycles (Table 5). Across all cycles, the most common grade 3 or 4 AEs considered by the investigator to be related to AZD5363 were maculopapular rash and diarrhoea (3 patients and 2 patients respectively). In most cases of rash and diarrhoea, utilisation of systemic anti-histamines and loperamide respectively allowed for successful management and to allow ongoing administration of study medication. Febrile neutropenia occurred in 2 (20%) of patients across all treatment cycles. There were no treatment related deaths.
Transient hyperglycaemia is an expected AE for both AZD5363 and corticosteroids. Dexamethasone was used as chemotherapy pre-medication and prednisolone is routinely administered with docetaxel for mCRPC [2]. We saw hyperglycaemia in every patient. The mean random glucose on cycle 1 day 2 (first dose of AZD5363) was 6.0 mmol/L pre-dose and then 8.7 mmol/L at 2 h, 9.5 mmol/L at 4 h, and 6.5 mmol/L at 8 h post AZD5363 dose. Insulin and C-peptide levels also rose and fell in parallel to these glucose changes in all patients. Individual patient data are shown in Fig. 2. No patient experienced symptomatic complications as a result of hyperglycaemia or required intervention to correct a high glucose level (an algorithm, including criteria for metformin administration, was provided within the trial protocol).

Individual patient data for a plasma glucose, b insulin and (c) C-peptide levels at the indicated time points during cycle 1 of treatment. Samples on day 3, 9 and 16 of the cycle were taken prior to the morning AZD5363 dose
Pharmacokinetics
Pharmacokinetic analyses indicated that AZD5363 exposure for patients receiving concomitant DP chemotherapy was in keeping with that found in patients receiving mono-therapy in the AZD5363 development programme.
Treatment efficacy
PSA reduction from baseline level to <50% (PSA50) at 12 weeks of treatment was seen in 7 (70%) of patients (Fig. 3).

Percentage change from baseline of individual patient prostate specific antigen (PSA) level after 12 weeks of therapy
Discussion
We undertook a phase I, open-label, combination dose escalation trial to determine an appropriate dose for AZD5363 to be used in combination with conventional doses of DP chemotherapy for use in mCRPC. The resulting RP2D for AZD5363 in this combination is 320 mg, BID, PO, given according to a 4 days on and 3 days off schedule and commencing from cycle 1, day 2 based on pre-defined criteria for dose limiting toxicity. In addition we found that the higher AZD5363 dose level of 400 mg BID resulted in 4 of 6 patients discontinuing AZD5363 within the first 2 cycles of treatment despite being able to tolerate further DP cycles. This supports the selection of the 320 mg BID dose level for this combination, which by comparison was tolerable at a level broadly consistent with the use of DP alone in this setting over multiple cycles [2, 3].
The most common AEs which were considered to be associated with the addition of AZD5363 to chemotherapy were diarrhoea, rash, neutropenia and hyperglycaemia in keeping with the experience to date of single agent AZD5363 administration [18, 19]. Diarrhoea and rash responded in most patients to management with loperamide and systemic anti-histamines with most patients able to continue therapy in the face of these AEs. We did not see evidence of increased nausea, at least above that experienced with DP, which has been an AE in single agent studies.
Hyperglycaemia occurred in all patients who received AZD5363 but was transient and self-limiting within 8 h of the first AZD5363 dose. We had chosen a 4 days on 3 days off schedule for dosing of AZD5363. This was one approach under evaluation within single agent AZD5363 trials at the point that our trial was designed. Our choice of this schedule was driven, in part, to allow us to separate temporally the administration of AZD5363 from the relatively high doses of dexamethasone pre-medication administered prior to docetaxel in an attempt to reduce the potential for interaction to drive hyperglycaemia. This strategy appears to have been successful and despite the concurrent use of prednisolone at comparatively lower doses. Of note, our study excluded patients with type I or II diabetes mellitus requiring either insulin or oral hypoglycaemics for routine management which should be borne in mind in the subsequent development of this approach.
This study was not designed to evaluate the efficacy of this combination at this stage of development. The PSA50 response rate of 70% of patients, and the fact that we administered a median of 8.5 cycles of chemotherapy is broadly consistent with prior data from large randomised trials of DP in this setting [2, 8]. However, we are unable to determine on this current experience what additional benefit AZD5363 might add to DP alone in this setting which will form the basis of future development of this combination. On the basis of our results, recruitment is currently ongoing to a placebo-controlled randomised phase II trial in mCRPC to evaluate the impact on progression free survival of the addition of AZD5363 to DP compared to DP alone. Collection of tissue samples, and blood sampling for circulating biomarkers will allow us to undertake exploratory analyses within this latter trial for patient subsets that might benefit based on activation of the PI3K/AKT signalling pathway.
Our determination of a proposed RP2D at 320 mg BID on a 4 day on/3 day off schedule for combination with DP was based on the treatment of 4 patients at this dose level, of which 3 were fully evaluable. Within a standalone trial this would have required a dose level expansion cohort to confirm tolerability. Our approach instead has been to confirm tolerability through direct progression to a placebo controlled, randomised, phase II component of the trial which is currently recruiting. Patients are randomised (1:1) to DP/AZD5363 or DP/placebo. Following recruitment of the first 20 randomised patients, a prospectively planned review by a fully independent Data Monitoring and Ethics Committee (DMEC) has considered toxicity and tolerability data to which the DMEC were unblinded. On the basis of this review, the committee have recommended ongoing recruitment. This approach to undertaking a dose level expansion cohort was chosen to provide efficiencies in moving through the development of this treatment approach such that these patients will also contribute to the subsequent phase II efficacy assessment based on a primary endpoint of progression free survival.
AKT inhibition utilising AZD5363 is also being developed in other combination approaches. Of note, RE-AKT is an ongoing multicentre, randomised, phase I/II trial in mCRPC, evaluating a combination of AZD5363 with the androgen receptor antagonist enzalutamide (https://clinicaltrials.gov/ct2/show/NCT02525068) [22]. In addition, analogous data for a taxane combination has been reported in breast cancer for AZD5363 plus paclitaxel, with the same 4 day on/3 day off AZD5363 schedule [23]. Toxicities were broadly similar, as was the RP2D, although at a dose level higher, at 400 mg BID, giving further reassurance that our RP2D of 320 mg BID is likely to prove tolerable.
In conclusion, AZD5363 at a dose of 320 mg BID on a 4 day on/3 day off schedule is recommended for further evaluation in combination with full dose DP for mCRPC.
References
Cancer Research UK. Cancer Stats: Cancer Statistics for the UK. http://www.cancerresearchuk.org/cancer-info/cancerstats/
Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, Oudard S, Theodore C, James ND, Turesson I, Rosenthal MA, Eisenberger MA, Investigators TAX (2004) Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 351(15):1502–1512. doi:10.1056/NEJMoa040720
Petrylak DP, Tangen CM, Hussain MH, Lara PN Jr, Jones JA, Taplin ME, Burch PA, Berry D, Moinpour C, Kohli M, Benson MC, Small EJ, Raghavan D, Crawford ED (2004) Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med 351(15):1513–1520. doi:10.1056/NEJMoa041318
James ND, Spears MR, Clarke NW, Dearnaley DP, De Bono JS, Gale J, Hetherington J, Hoskin PJ, Jones RJ, Laing R, Lester JF, McLaren D, Parker CC, Parmar MK, Ritchie AW, Russell JM, Strebel RT, Thalmann GN, Mason MD, Sydes MR (2015) Survival with newly diagnosed metastatic prostate cancer in the "docetaxel era": data from 917 patients in the control arm of the STAMPEDE trial (MRC PR08, CRUK/06/019). Eur Urol 67(6):1028–1038. doi:10.1016/j.eururo.2014.09.032
Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, Iversen P, Bhattacharya S, Carles J, Chowdhury S, Davis ID, de Bono JS, Evans CP, Fizazi K, Joshua AM, Kim CS, Kimura G, Mainwaring P, Mansbach H, Miller K, Noonberg SB, Perabo F, Phung D, Saad F, Scher HI, Taplin ME, Venner PM, Tombal B, Investigators P (2014) Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med 371(5):424–433. doi:10.1056/NEJMoa1405095
Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, Fizazi K, Mainwaring P, Piulats JM, Ng S, Carles J, Mulders PF, Basch E, Small EJ, Saad F, Schrijvers D, Van Poppel H, Mukherjee SD, Suttmann H, Gerritsen WR, Flaig TW, George DJ, Yu EY, Efstathiou E, Pantuck A, Winquist E, Higano CS, Taplin ME, Park Y, Kheoh T, Griffin T, Scher HI, Rathkopf DE, Investigators C-A (2013) Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med 368(2):138–148. doi:10.1056/NEJMoa1209096
de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB Jr, Saad F, Staffurth JN, Mainwaring P, Harland S, Flaig TW, Hutson TE, Cheng T, Patterson H, Hainsworth JD, Ryan CJ, Sternberg CN, Ellard SL, Flechon A, Saleh M, Scholz M, Efstathiou E, Zivi A, Bianchini D, Loriot Y, Chieffo N, Kheoh T, Haqq CM, Scher HI, Investigators C-A (2011) Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med 364(21):1995–2005. doi:10.1056/NEJMoa1014618
de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, Kocak I, Gravis G, Bodrogi I, Mackenzie MJ, Shen L, Roessner M, Gupta S, Sartor AO, Investigators T (2010) Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet 376(9747):1147–1154. doi:10.1016/S0140-6736(10)61389-X
Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, Xu Y, Frohlich MW, Schellhammer PF, Investigators IS (2010) Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 363(5):411–422. doi:10.1056/NEJMoa1001294
Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, Armstrong AJ, Flaig TW, Flechon A, Mainwaring P, Fleming M, Hainsworth JD, Hirmand M, Selby B, Seely L, de Bono JS, Investigators A (2012) Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 367(13):1187–1197. doi:10.1056/NEJMoa1207506
Sweeney CJ, Chen YH, Carducci M, Liu G, Jarrard DF, Eisenberger M, Wong YN, Hahn N, Kohli M, Cooney MM, Dreicer R, Vogelzang NJ, Picus J, Shevrin D, Hussain M, Garcia JA, DiPaola RS (2015) Chemohormonal therapy in metastatic hormone-sensitive prostate cancer. N Engl J Med 373(8):737–746. doi:10.1056/NEJMoa1503747
James ND, Sydes MR, Clarke NW, Mason MD, Dearnaley DP, Spears MR, Ritchie AW, Parker CC, Russell JM, Attard G, de Bono J, Cross W, Jones RJ, Thalmann G, Amos C, Matheson D, Millman R, Alzouebi M, Beesley S, Birtle AJ, Brock S, Cathomas R, Chakraborti P, Chowdhury S, Cook A, Elliott T, Gale J, Gibbs S, Graham JD, Hetherington J, Hughes R, Laing R, McKinna F, McLaren DB, O'Sullivan JM, Parikh O, Peedell C, Protheroe A, Robinson AJ, Srihari N, Srinivasan R, Staffurth J, Sundar S, Tolan S, Tsang D, Wagstaff J, Parmar MK, Investigators S (2015) Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet. doi:10.1016/S0140-6736(15)01037-5
Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, Socci ND, Lash AE, Heguy A, Eastham JA, Scher HI, Reuter VE, Scardino PT, Sander C, Sawyers CL, Gerald WL (2010) Integrative genomic profiling of human prostate cancer. Cancer Cell 18(1):11–22. doi:10.1016/j.ccr.2010.05.026
Kosaka T, Miyajima A, Shirotake S, Suzuki E, Kikuchi E, Oya M (2011) Long-term androgen ablation and docetaxel up-regulate phosphorylated Akt in castration resistant prostate cancer. J Urol 185(6):2376–2381. doi:10.1016/j.juro.2011.02.016
Zhong B, Sallman DA, Gilvary DL, Pernazza D, Sahakian E, Fritz D, Cheng JQ, Trougakos I, Wei S, Djeu JY (2010) Induction of clusterin by AKT--role in cytoprotection against docetaxel in prostate tumor cells. Mol Cancer Ther 9(6):1831–1841. doi:10.1158/1535-7163.MCT-09-0880
Davies BR, Greenwood H, Dudley P, Crafter C, Yu DH, Zhang J, Li J, Gao B, Ji Q, Maynard J, Ricketts SA, Cross D, Cosulich S, Chresta CC, Page K, Yates J, Lane C, Watson R, Luke R, Ogilvie D, Pass M (2012) Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol Cancer Ther 11(4):873–887. doi:10.1158/1535-7163.MCT-11-0824-T
Marques RB, Aghai A, de Ridder CM, Stuurman D, Hoeben S, Boer A, Ellston RP, Barry ST, Davies BR, Trapman J, van Weerden WM (2015) High efficacy of combination therapy using PI3K/AKT inhibitors with androgen deprivation in prostate cancer preclinical models. Eur Urol 67(6):1177–1185. doi:10.1016/j.eururo.2014.08.053
Tamura K, Hashimoto J, Tanabe Y, Kodaira M, Yonemori K, Seto T, Hirai F, Arita S, Toyokawa G, Chen L, Yamamoto H, Kawata T, Lindemann J, Esaki T (2016) Safety and tolerability of AZD5363 in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol 77(4):787–795. doi:10.1007/s00280-016-2987-9
Banerji U, Dean EJ, Perez-Fidalgo JA, Batist G, Bedard PL, You B, Westin SN, Kabos P, Davies B, Elvin P, Lawrence P, Yates JWT, Ambrose H, Rugman P, Foxley A, Salim S, Casson E, Lindemann JPO, Schellens JHM (2015) A pharmacokinetically (PK) and pharmacodynamically (PD) driven phase I trial of the pan-AKT inhibitor AZD5363 with expansion cohorts in PIK3CA mutant breast and gynecological cancers. ASCO Meeting Abstracts 33(15_suppl):2500
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45(2):228–247. doi:10.1016/j.ejca.2008.10.026
Scher HI, Halabi S, Tannock I, Morris M, Sternberg CN, Carducci MA, Eisenberger MA, Higano C, Bubley GJ, Dreicer R, Petrylak D, Kantoff P, Basch E, Kelly WK, Figg WD, Small EJ, Beer TM, Wilding G, Martin A, Hussain M, Prostate Cancer Clinical Trials Working G (2008) Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the prostate cancer clinical trials working group. J Clin Oncol 26(7):1148–1159. doi:10.1200/JCO.2007.12.4487
ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02525068. Accessed 12 January 2017
Turner NC, Oliveira M, Armstrong A, Sablin M-P, Perez-Fidalgo JA, Herebien S, Garcia-Murillas I, Johnson S, Foxley A, Mahmood A, Lindemann JP (2015) Abstract CT331: “BEECH”, a phase I/II study of the AKT inhibitor AZD5363 combined with paclitaxel in patients with advanced or metastatic breast cancer: results from the dose-finding study, including quantitative assessment of circulating tumor DNA as a s. Cancer Res 75(15 Supplement):CT331–CT331. doi:10.1158/1538-7445.am2015-ct331
Acknowledgments
This work was supported by Cancer Research UK [C9317/A16029, CRUK/12/042] and Cancer Research UK core funding from the Southampton Clinical Trials Unit. This research was conducted within the National Institute for Health Research (NIHR) Industry Alliance with AstraZeneca. We thank the patients who entered into the trial and the NIHR Clinical Research Network: Cancer who supported the trial. Clinical trial information: NCT02121639
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
SJC has undertaken consulting or advisory roles for Bayer, Sanofi, Astellas, Janssen, Pfizer and Roche and has received research funding from AstraZeneca, Astex Pharmaceuticals and Plexxikon. GG has undertaken consulting or advisory roles for Sirtex, GlaxoSmithKline and Pfizer. All other authors declare that they have no conflicts of interest.
Funding
This work was supported by Cancer Research UK [C9317/A16029, CRUK/12/042], Cancer Research UK core funding to the Southampton Clinical Trials Unit and AstraZeneca.
Ethical approval
All procedures performed in this clinical trial were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in this study.
Electronic supplementary material
ESM 1
(DOCX 20 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Crabb, S.J., Birtle, A.J., Martin, K. et al. ProCAID: a phase I clinical trial to combine the AKT inhibitor AZD5363 with docetaxel and prednisolone chemotherapy for metastatic castration resistant prostate cancer. Invest New Drugs 35, 599–607 (2017). https://doi.org/10.1007/s10637-017-0433-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-017-0433-4