
Abstract
Dosage compensation in the nematode Caenorhabditis elegans is achieved by the binding of a condensin-like dosage compensation complex (DCC) to both X chromosomes in hermaphrodites to downregulate gene expression two-fold. Condensin IDC, a sub-part of the DCC, differs from the mitotic condensin I complex by a single subunit, strengthening the connection between dosage compensation and mitotic chromosome condensation. The DCC is targeted to X chromosomes by initial binding to a number of recruiting elements, followed by dispersal or spreading to secondary sites. While the complex is greatly enriched on the X chromosomes, many sites on autosomes also bind the complex. DCC binding does not correlate with DCC-mediated repression, suggesting that the complex acts in a chromosome-wide manner, rather than on a gene-by-gene basis. Worm dosage compensation represents an excellent model system to study how condensin-mediated changes in higher order chromatin organization affect gene expression.
Similar content being viewed by others


Chromosome-based mechanisms of sex determination employed by many higher eukaryotes lead to a sex chromosome-linked gene dosage imbalance between the sexes. Different organisms evolved unique mechanisms to restore this balance—a process referred to as dosage compensation. The problem to be solved is two-fold. First, there is a dosage imbalance between the sex chromosomes and the autosomes in the heterogametic sex. While the ratio of genes located on two different autosomes is 1:1, the ratio of sex-linked genes to autosomal genes is 1:2. Second, there is a difference in sex-linked gene dosage between the sexes, with the homogametic sex possessing twice as many copies of sex-linked genes as the heterogametic sex. This review focuses on our current understanding of the mechanism of dosage compensation in the nematode Caenorhabditis elegans.
C. elegans exists as two sexes: hermaphrodites and males. Hermaphrodites produce both sperm and oocytes and can reproduce by self-fertilization. Males only produce sperm. In addition to five pairs of autosomes, hermaphrodites have two X chromosomes and males have one (Fig. 1a) (Hodgkin 2005). Unlike in mammals, where a sex-determining gene located on the Y chromosome determines maleness, the primary sex determination signal in worms is the ratio of X chromosomes to sets of autosomes (Zarkower 2006).

The mechanism of dosage compensation in worms. a Hermaphrodite worms possess two X chromosomes and two sets of autosomes, while males posses one X chromosome to two sets of autosomes. These chromosomal differences create an X to autosome imbalance in males, and an X chromosome imbalance between the sexes. b Some evidence suggests that the expression level of the X chromosome is increased compared to autosomal gene expression, to compensate for X monosomy in males. However, this upregulation creates overly high levels of X-linked gene expression in hermaphrodites. c In a hermaphrodite-specific process, both X chromosomes are downregulated, which restores gene dosage balance
Balancing the X chromosome and autosomes
The XX vs. XO chromosomal sex determination system results in a functional imbalance of X to autosomal gene dose in XO males. How is this X monosomy tolerated? Recent studies suggested that this dilemma is overcome by hyperactivation of the X chromosome (Gupta et al. 2006). Microarray analysis of gene expression revealed an overall level of X-linked gene expression from the single male X that is comparable to gene expression levels from autosomes, which are present in two copies. These results led to the hypothesis that the problem of X chromosome aneuploidy in C. elegans males has been solved over the course of evolutionary time by increasing transcription rates of X-linked genes roughly two-fold (Gupta et al. 2006) (Fig. 1b).
Upregulation of the male X to achieve expression levels equivalent to autosomes appears to be an evolutionarily conserved mechanism. X chromosome hyperactivity has been studied more extensively in mammals and in flies (Adler et al. 1997; Gupta et al. 2006; Lin et al. 2007; Nguyen and Disteche 2006; Sharova et al. 2007). Interestingly, in mammals, the male X in undifferentiated cells is initially upregulated only about 1.4 to 1.6-fold. Upregulation reaches 2-fold only after differentiation (Lin et al. 2007). A similar analysis of X:A expression before and after the onset of dosage compensation has not yet been conducted in C. elegans, but it would provide necessary strength to the argument that the X chromosomes are hypertranscribed with respect to autosomes.
The mechanism of X-upregulation in worms remains a mystery. Whether the special feature of the X chromosome is different promoter strengths for individual genes or a cellular machinery responsible for chromosome-wide upregulation is yet to be determined. Potential mechanisms could involve an X chromosome specific activator, a genome-wide activator that is enriched on the X chromosome, or a repressor that is depleted on the X. It will also be interesting to determine whether this upregulation affects all X-linked genes or only a subset of genes for which functional monosomy would be deleterious.
X chromosome dosage compensation between the sexes
The second dilemma posed by chromosome-based sex determination mechanisms is the sex-linked gene dosage imbalance between males and females or hermaphrodites. In principle, if upregulation of the X chromosome only occurs in males, it restores both the X to autosome balance and X-linked gene dosage balance between the sexes. Indeed, the mechanism of dosage compensation in Drosophila accomplishes these two goals with one mechanism: male specific upregulation of the X chromosome by the MSL complex (see Review in this issue). However, in mammals and worms, X chromosome upregulation seems to occur in both sexes (Gupta et al. 2006; Lin et al. 2007; Nguyen and Disteche 2006). Therefore, the function of dosage compensation is not only to balance X-linked gene expression between the sexes, but also to prevent disproportionate overexpression of the X chromosomes in hermaphrodite worms and in female mammals.
The first hints that X chromosome dosage compensation operates in C. elegans came from observations that worms are able to tolerate X chromosome aneuploidies better than autosomal aneuploidies (Hodgkin 1983). If a sex-specific dosage compensation mechanism exists, mutants in the process would be expected to produce sex-specific phenotypes. The majority of these types of mutations caused hermaphrodite-specific abnormalities or lethality (Hodgkin 1983; Meneely and Wood 1984; Meyer and Casson 1986; Wood et al. 1985), and hermaphrodite-specific elevation of X-linked mRNA levels (Meyer and Casson 1986). These observations led to the model that dosage compensation is accomplished by downregulation of transcript levels in XX animals. A recent study confirmed these earlier observations, and demonstrated that a large number of X-linked genes are overexpressed in dosage compensation mutants (Jans et al. 2009). One possible mechanism of downregulation of X-linked gene expression in XX animals is complete silencing of one of the two X chromosomes, the mechanism employed by mammalian females (See Review in this issue). However, X-inactivation was ruled out as the dosage compensation mechanism in C. elegans for the following two reasons. First, for cell autonomous genes, recessive sex-linked mutations in heterozygous hermaphrodites would be expected to lead to mosaic phenotypes, which are never observed. Second, for non cell-autonomous genes, X-linked loss-of-function mutations should have dominant phenotypes with varying penetrance and expressivity, which has not been observed either (Hodgkin 1983; Meyer 2005). Therefore, an alternative model was proposed, in which dosage compensation downregulates by half the expression of genes on both X chromosomes in hermaphrodites (Meyer and Casson 1986; Plenefisch et al. 1989) (Fig. 1c). Consistent with this model, the amount of RNA produced by worms carrying various X chromosome duplications is always proportional to copy number. Two copies produce twice as much, and three copies produce three times as much mRNA as a single copy, indicating that all copies of the gene contribute equally to mRNA levels (Meyer and Casson 1986). Additionally, cytological studies of dosage compensation proteins demonstrated that the dosage compensation machinery localizes to both X chromosomes in hermaphrodite somatic cells, indicating that dosage compensation affects both chromosomes (Chuang et al. 1994).
The dosage compensation machinery
Genes that regulate dosage compensation
The X:A ratio that determines sex in worms also controls dosage compensation by regulating the master sex switch gene, xol-1 (Fig. 2). A low X:A ratio leads to activation of xol-1 in males, while a high X:A ratio leads to repression of xol-1 in hermaphrodites. (Miller et al. 1988; Rhind et al. 1995). In molecular terms, a set of X-linked “numerator” genes (XSEs, X signal elements) code for proteins that repress xol-1. Conversely, a set of genes on the autosomes (ASEs, autosomal signal elements) code for proteins that activate xol-1 (Reviewed in (Meyer 2005; Zarkower 2006)). XSEs include the nuclear hormone receptor SEX-1 (Carmi et al. 1998), the RNA binding protein FOX-1 (Hodgkin et al. 1994; Skipper et al. 1999), the ONECUT Homeodomain protein CEH-39 (Gladden and Meyer 2007), and SEX-2 (Gladden et al. 2007). SEX-1 and CEH-39 repress xol-1 primarily at the level of transcription, while FOX-1 acts posttranscriptionally (Akerib and Meyer 1994; Carmi et al. 1998; Carmi and Meyer 1999; Gladden and Meyer 2007; Nicoll et al. 1997). In addition to its role as an XSE, SEX-1 also functions downstream of xol-1 to promote dosage compensation (Gladden et al. 2007). The ASE SEA-1 leads to an increase in xol-1 transcript levels (Powell et al. 2005), while an additional autosomal signal element, SEA-2, has not been described in detail (Meyer 2005). By the combined action of XSEs and ASEs, the X:A ratio is translated into the activity state of xol-1 in a remarkably precise manner. In XX:AAA animals (X:A ratio of 0.67), XOL-1 levels are high and the animals develop as males, while in XXX:AAAA animals (X:A ratio of 0.75), XOL-1 levels are low and they develop as hermaphrodites (Madl and Herman 1979).

The genetic pathway regulating sex determination and dosage compensation. Autosomal signal elements (ASEs) activate, while X signal elements (XSEs) repress the master switch gene xol-1. xol-1 represses the sdc genes, which in turn both repress her-1 and promote the activity of dosage compensation genes. her-1 promotes male development, while dosage compensation genes downregulate expression of genes on hermaphrodite X chromosomes. In males, xol-1 is turned on, which leads to male development and inhibition of dosage compensation. In hermaphrodites, xol-1 is turned off, which leads to hermaphrodite development and X chromosome dosage compensation
In addition to its sex-determination role, XOL-1 also regulates dosage compensation. High XOL-1 levels in males turn off dosage compensation, while low XOL-1 levels in hermaphrodites trigger dosage compensation (Rhind et al. 1995). The molecular mechanism of this switch is unclear. XOL-1 is similar to GHMP kinases, although it does not appear to bind ATP and therefore likely acts by a mechanism distinct from other GHMP kinases (Luz et al. 2003). Mutations in xol-1 cause XO-specific lethality and feminization (Miller et al. 1988), due to an inability to repress genes that execute dosage compensation and sex determination. This property of xol-1 was later exploited in genetic screens to identify mutations in dosage compensation genes.
A large number of gene products are needed to execute dosage compensation (Fig. 2). dpy-21, -26, -27, and -28 were originally described as mutations causing an XX-specific Dpy phenotype (dumpy, short and fat) or XX-specific lethality (Hodgkin 1983; Meneely and Wood 1984; Meyer and Casson 1986; Wood et al. 1985). Mutations in sdc-1 were originally identified due to an egg-laying defect phenotype (Trent et al. 1983), but later were shown to cause both sexual transformation and dosage compensation defects in hermaphrodites (Villeneuve and Meyer 1987). sdc-2 was isolated in a screen for sex-specific lethal mutations (Nusbaum and Meyer 1989). Screens aimed at finding suppressors of the XO-specific lethality caused by xol-1 mutations uncovered sdc-3 (DeLong et al. 1993; Plenefisch et al. 1989), dpy-30 (Hsu and Meyer 1994), as well as additional alleles of sdc-2 (Nusbaum and Meyer 1989), dpy-27, dpy-26, and dpy-21 (Plenefisch et al. 1989). Additional members of the dosage compensation machinery, MIX-1 (Lieb et al. 1998) and CAPG-1 (Csankovszki et al. 2009) were found by subsequent biochemical analysis.
sdc-2, together with sdc-1 and sdc-3, regulates both sex determination and dosage compensation (Chu et al. 2002; Davis and Meyer 1997; Dawes et al. 1999; Nusbaum and Meyer 1989; Villeneuve and Meyer 1987; Villeneuve and Meyer 1990; Yonker and Meyer 2003). In the dosage compensation pathway, the sdc genes promote the activity of dpy-21, -26, -27, -28, and -30, as well as mix-1 and capg-1. In the sex determination pathway, the sdc genes repress her-1, a gene needed for male development. In hermaphrodites, XOL-1 levels are low, therefore the sdc genes are on and they repress her-1 and trigger dosage compensation. In males, due to high XOL-1 levels, the sdc genes are off, therefore her-1 is expressed and dosage compensation is turned off (Dawes et al. 1999). Most dosage compensation proteins are supplied maternally in the oocyte, and are initially present in both male and hermaphrodite embryos (Reviewed in (Meyer 2005)). The only exception is SDC-2, which is transcribed and translated exclusively in hermaphrodites, and is needed to recruit all other DCC proteins to the X chromosome (Dawes et al. 1999). Therefore, SDC-2 confers both sex-specificity and chromosome-specificity to the process of dosage compensation.
The worm dosage compensation complex
Biochemical analysis of the DCC revealed that the complex consists of two subparts (Fig. 3a). The SDC-1, SDC-2 and SDC-3 proteins form one sub-complex (Chu et al. 2002). SDC-1 contains seven zinc fingers (Nonet and Meyer 1991), SDC-2 is a highly charged protein with a coiled coil motif (Dawes et al. 1999), and SDC-3 contains a pair of zinc-finger motifs and a motif similar to myosin heavy chain (Davis and Meyer 1997; Klein and Meyer 1993). Five additional proteins, DPY-26, DPY-27, DPY-28, MIX-1, and CAPG-1, form the other sub-complex, condensin IDC, a homolog of the evolutionarily conserved condensin complex involved in mitotic and meiotic chromosome segregation and compaction (Chuang et al. 1994; Chuang et al. 1996; Csankovszki et al. 2009; Lieb et al. 1998; Lieb et al. 1996; Tsai et al. 2008). In addition, DPY-21, a novel, but conserved, protein with a proline-rich N terminus, interacts with members of both subcomplexes, albeit less robustly (Yonker and Meyer 2003). DPY-30 is an additional DCC component, but its physical interactions with other DCC members have not been described in detail (Hsu et al. 1995; Hsu and Meyer 1994).

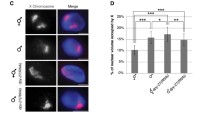
The dosage compensation complex (DCC). a Five subunits of the DCC (DPY-27, MIX-1, DPY-26, DPY-28, CAPG-1) form the condensin IDC subcomplex, a complex similar to mitotic and meiotic condensin complexes. The SDC-1, -2, -3 proteins, together with DPY-21 and DPY-30, form another sub-complex of the DCC. b The DCC localizes to both X chromosomes in hermaphrodites. A single hermaphrodite intestinal nucleus is shown (DAPI, blue). The X chromosome territories are marked by fluorescent in situ hybridization using an X paint probe (red). The region in the nucleus occupied by the DCC is marked by immunofluorescence with DPY-27-specific antibodies (green). Scale bar = 1 µm
As part of their sex determination function, the SDC proteins also repress the autosomal gene her-1, a gene required for male development (Chu et al. 2002; Davis and Meyer 1997; Dawes et al. 1999; Nusbaum and Meyer 1989; Villeneuve and Meyer 1987; Villeneuve and Meyer 1990). The SDC proteins recruit all other members of the DCC (except for DPY-21) to the her-1 locus, even though genetically these other proteins play less significant roles in her-1 repression (Chu et al. 2002; Yonker and Meyer 2003). It should be noted that these two repressive functions of the dosage compensation complex are very different. At the her-1 locus, the complex accomplishes localized twenty-fold repression (Trent et al. 1991), while on the X it causes chromosome-wide two-fold repression (Meyer and Casson 1986). How the DCC accomplishes these two modes of repression remains to be determined. One clue may lie in DPY-21, as this protein localizes to the X chromosome but not to the her-1 locus (Yonker and Meyer 2003).
Several DCC subunits function in cellular processes outside of their role in dosage compensation. The best-studied example of this is the roles played by subunits of the condensin IDC subcomplex in mitosis and meiosis (see below) (Csankovszki et al. 2009; Lieb et al. 1998; Lieb et al. 1996; Tsai et al. 2008). An additional member of the DCC with an outside function is DPY-30 (Hsu et al. 1995; Hsu and Meyer 1994). DPY-30 is homologous to a subunit of the histone H3 lysine 4 methyltransferase complex COMPASS (Krogan et al. 2002; Miller et al. 2001; Nagy et al. 2002). In fact, DPY-30 is expressed in both males and hermaphrodites, and its localization appears to be diffuse nuclear, not strictly X-specific, consistent with a more ubiquitous role in gene regulation (Hsu et al. 1995; Hsu and Meyer 1994).
Chromosome specificity of DCC binding
Recruitment of the DCC to the X chromosome
Analysis of DCC localization by fluorescent microscopy demonstrated that the complex associates with both X chromosomes in somatic nuclei of hermaphrodite worms (Chuang et al. 1994; Csankovszki et al. 2009; Davis and Meyer 1997; Dawes et al. 1999; Lieb et al. 1998; Lieb et al. 1996; Tsai et al. 2008; Yonker and Meyer 2003) (Fig. 3b). DCC chromatin immunoprecipitation studies confirmed X-chromosome enrichment of the complex (Ercan et al. 2007; Jans et al. 2009). These studies also revealed that the complex does not bind the X chromosomes uniformly. Instead, about 1500 DCC peaks are scattered along the length of the X chromosome, compared to 200–300 peaks on each autosome. Peak heights vary greatly. About 50 peaks on the X chromosome are of exceptionally high amplitude and are designated “DCC foci.” Peaks on both the X and the autosomes are preferentially found in promoter regions (Ercan et al. 2007; Jans et al. 2009). A key question in the field is how the DCC achieves X chromosome enrichment.
There are several possible models for targeting the DCC to the X chromosome. At one extreme, the DCC may individually recognize and bind each of its target sites. This model predicts the existence of a DNA element, which is enriched on the X chromosome, and is distributed all over its length. The DCC would then bind each of these elements to achieve chromosome-wide targeting. In some ways, this model is reminiscent of target recognition by sequence-specific DNA-binding proteins, such as transcription factors, which have many targets in the genome. The difference is that transcription factor targets are scattered on several chromosomes, while DCC targets are greatly enriched on the X. At the other extreme, the X chromosome may contain a single site that initially recruits the machinery and mediates spreading along the entire length of the chromosome. This mechanism does not necessitate chromosome-specific elements to be distributed along the length of the chromosome, but it requires a mechanism for long-range spreading of the complex. Mechanisms that incorporate aspects of both of these models are also possible, with binding sites of various affinities and/or recruiting abilities being distributed along the length of the chromosome, which may or may not mediate spreading of the complex into local chromatin. Indeed, this last model appears to be the case in worms.
Studies of DCC binding to duplications of various X chromosome fragments demonstrated that multiple regions of the chromosome are able to recruit the complex when detached from the X (Csankovszki et al. 2004; Lieb et al. 2000). In later studies, these large “recruiting” regions were narrowed down to short DNA fragments (Jans et al. 2009; McDonel et al. 2006). These sites were named rex (recruiting element on X) sites, based on their ability to recruit the DCC in the context of extrachromosomal transgenic arrays. Additional rex sites were discovered when DNA corresponding to sites of DCC binding on the X chromosome were tested in extrachromosomal array assays (Jans et al. 2009). A total of 38 rex sites have been identified to date. The availability of a large number of rex sites made it possible to identify a 12-base pair sequence motif (MEX, motif enriched on X) present within these sites (Jans et al. 2009). This motif is enriched on the X chromosome by 4-fold to 25-fold, depending on quality of the match to the consensus motif. The MEX motif is an extension of one of two motifs described earlier based on the analysis of a small number of rex sites (McDonel et al. 2006), and of a motif obtained by genome-wide analysis of DNA sequences bound by the DCC (Ercan et al. 2007). The importance of this motif for recruitment was demonstrated by mutational analysis (Jans et al. 2009; McDonel et al. 2006). The authors estimate the number of rex sites on the X chromosome to be about 200 (Jans et al. 2009), which is comparable to the estimate given for the number of so-called chromatin entry sites/high affinity sites on the Drosophila X (estimated at 150) (Alekseyenko et al. 2008; Straub et al. 2008).
Multiple classes of DCC binding elements
Not all sites bound by the DCC on the native X are able to maintain their ability to attract the complex when detached from the X. Indeed, there are about 1500 peaks of DCC binding on the X chromosome, but only an estimated 200 of these sites are rex sites (Jans et al. 2009). Sites that fail to attract the complex in the context of extrachromosomal arrays, but are bound when located on the X chromosome, are called dox sites (dependent on X) (Jans et al. 2009). Sites that fail to attract large quantities of the DCC on duplicated fragments of the X chromosome, but form DCC foci on the native X, are referred to as “way stations” (Blauwkamp and Csankovszki 2009).
Interestingly, way stations do not necessarily correspond to dox sites. In fact, some sites that lose their ability to bind large amounts of the DCC on X-duplications are able to recruit the DCC in the context of extrachromosomal arrays (Blauwkamp and Csankovszki 2009; Jans et al. 2009). It is not yet clear why these sites behave differently in these two contexts. One difference is copy number. These sites are present in a single copy on duplications, but are present in many copies on large extrachromosomal arrays. Indeed, copy number was shown to be an important factor in determining the ability of DNA elements to initiate the assembly of the dosage compensation machinery in flies and in mammals (Gilfillan et al. 2007; Heard et al. 1999). Additional possibilities as to the reason for this difference include different chromosomal contexts, different chromatin structures, or different subnuclear localization of duplications versus arrays. Together, these findings suggest that there are various classes of DCC binding sites in worms with a continuum of affinities for the DCC and/or varying degrees of ability to recruit the complex in isolation.
What are the defining features of rex sites? The MEX motif is clearly part of the answer. rex sites tend to contain MEX variants, which are a better match to the consensus than variants within dox sites (Jans et al. 2009). However, the MEX motif is not the whole answer. Many rex sites contain MEX variants comparable to variants in dox sites, indicating that something else must distinguish these two classes of sites. In addition, not all occurrences of the MEX motif are bound by the DCC. While most (but not all) sites containing the best MEX variants are bound by the DCC on the X chromosomes, these same quality MEX variants are not bound by the DCC on autosomes (Jans et al. 2009). Clustering of these motifs may provide the needed additional specificity (Jans et al. 2009; McDonel et al. 2006). Another potential feature of rex sites might be high DCC occupancy (Ercan et al. 2007). Indeed, DCC peaks with the 17 highest peak scores correspond to rex sites. However, there are also many rex sites that correspond to smaller DCC peaks (Jans et al. 2009). Future analysis of DCC binding to the X chromosome with limiting concentrations of DCC might help shed light on the nature of these various classes of binding sites.
Taken as a whole, the current model of DCC targeting to the X chromosome involves initial recruitment to a limited number of sites, which is followed by disbursement to other sites on the chromosome with weaker affinity and/or recruiting ability (Blauwkamp and Csankovszki 2009; Csankovszki et al. 2004; Ercan et al. 2007; Jans et al. 2009; McDonel et al. 2006). An unresolved question in the field is how these secondary sites become occupied. Peaks of DCC binding can be as close as 0.9 kb away from each other, or as far as 94 kb (Ercan et al. 2007; Jans et al. 2009). Any model of “spreading” must be able to accommodate uneven or discontinuous distribution of the complex along the chromosome. The fact that dox sites are preferentially located in promoters raises the possibility that transcription plays a role in facilitating binding to these sites (Jans et al. 2009).
DCC binding on autosomes
Recent genome-wide analysis revealed autosomal sites of DCC binding (Ercan et al. 2007; Jans et al. 2009). These sites include the her-1 gene, genes encoding histones, ribosomal proteins, tRNAs and miRNAs. However, different rules seem to dictate binding to autosomes than binding to the X. Occurrences of the MEX motif on autosomes are not bound by the DCC. A different motif (MEA, motif enriched on autosomes) was found within autosomal sites of DCC binding. However, this motif does not seem to play a functional role in DCC recruitment (Jans et al. 2009). The best-studied autosomal DCC binding locus is her-1. The sequence elements needed for DCC binding at her-1 (Chu et al. 2002) differ from the MEX motif (Jans et al. 2009), implying that the mechanism of X chromosome recognition may differ from her-1 binding. Consistent with that, recruitment of the complex to her-1 is somewhat different from recruitment to X. While SDC-2 appears to be pivotal for X recognition, SDC-3 is the driving force behind her-1 binding (Yonker and Meyer 2003). It will be interesting to determine whether other autosomal locations of DCC binding are SDC-2 driven, SDC-3 driven, or use yet another mechanism.
Molecular mechanisms of repression by the DCC
It is not easy to envision a mode of gene regulation that is able to impose a precise two-fold downregulation of gene expression in a chromosome-wide manner. This form of regulation must also be superimposed on the individual temporal and tissue specific regulation of each gene. From studies of steady state RNA levels in dosage compensation mutants, we know that the DCC acts by altering mRNA levels (Meyer and Casson 1986). Therefore, it is assumed that some aspect of transcription and/or RNA processing is affected. A clue as to the mechanism of action of the DCC comes from the similarity of the condensin IDC sub-complex to the conserved condensin complex required for the compaction and segregation of chromosomes during mitosis and meiosis. Thus, a hypothesis was proposed that dosage compensation in C. elegans involves a molecular mechanism similar to the mechanism of mitotic chromosome condensation (Chuang et al. 1994; Csankovszki et al. 2009; Lieb et al. 1998; Lieb et al. 1996; Tsai et al. 2008). The DCC may alter higher order chromosome organization by inducing partial chromosome condensation in interphase, leading to decreased transcription.
Three condensin complexes in the worm
Condensin complexes are conserved five subunit complexes, present from yeast to humans (Reviewed in (Belmont 2006; Hirano 2005)). They consist of two SMC (structural maintenance of chromosomes) proteins of the SCM2 and SMC4 subclasses, and three regulatory subunits (CAPs, chromosome associated polypeptides). While yeast has one condensin complex, higher eukaryotes have two, condensin I and condensin II (Hirota et al. 2004; Ono et al. 2004; Ono et al. 2003). The two complexes contain the same SMC subunits and unique sets of CAP subunits. Condensin I (the complex present in yeast) contains CAP-D2, CAP-G, and CAP-H subunits (CAP I class), while condensin II (specific to higher eukaryotes) contains CAP-D3, CAP-G2, and CAP-H2 subunits (CAP II class).
Recent biochemical and proteomic analysis revealed the existence of not two, but three condensin complexes in C. elegans (Csankovszki et al. 2009). Two of these complexes are homologs of condensin I. One variant of condensin I (condensin IDC), plays a role in dosage compensation as a sub-part of the DCC (Chuang et al. 1994; Csankovszki et al. 2009; Lieb et al. 1998; Lieb et al. 1996; Tsai et al. 2008). The other condensin I variant (referred to as condensin I), as well as condensin II, function in mitosis and meiosis (Chan et al. 2004; Csankovszki et al. 2009; Hagstrom et al. 2002). Condensins I and II contain identical SMC subunits, while condensins IDC and I contain identical CAP subunits (Fig. 4). The mitotic phenotypes associated with condensin I subunits are much less severe than the phenotypes associated with condensin II subunits, indicating that in worms, the function of condensin II predominates in mitosis (Csankovszki et al. 2009). Condensin I also plays a novel role during meiosis in the regulation of crossover number and distribution (Tsai et al. 2008; Mets and Meyer 2009).

Three condensin complexes regulate chromosome dynamics in worms. a Condensin IDC functions in dosage compensation, while condensin I and condensin II function in mitosis and meiosis. The subunits of each complex are indicated. Condensin IDC and condensin I differ by a single subunit, yet associate with chromosomes in distinct patterns and have different functions. b The chromosomal association patterns of the three condensin complexes during interphase in mid-stage embryos, during mitosis in early embryos, and during meiosis I in spermatocytes, are shown. Condensin IDC associates with the X chromosomes during the process of dosage compensation, but the complex does not function in mitosis and meiosis. Condensin I associates with mitotic chromosomes in a diffuse non-uniform manner, and localizes to the points of cohesion between homologous chromosomes during meiosis. By contrast, condensin II is enriched at the centromeres during mitosis and localizes to the core of sister chromatids during meiosis. Scale bar = 2 µm
Interestingly, condensin IDC and condensin I differ in only one of five subunits, the SMC4 subunit (Csankovszki et al. 2009). Condensin I contains SMC-4, and condensin IDC contains DPY-27. Replacing SMC-4 with DPY-27 is sufficient to turn a mitotic/meiotic complex into a gene regulatory complex. Thus, condensin IDC likely evolved by duplication of the ancestral smc-4 gene, and specialization of its function. On the one hand, condensin I is able to bind all chromosomes during mitosis and meiosis, which leads to the formation of compact, individualized chromosomes. On the other hand, condensin IDC is largely X-specific, interacts with the X chromosome throughout the cell cycle, and its binding results in a two-fold downregulation of gene expression. Future studies will reveal how such similar complexes can perform these diverse functions, but the unique SMC4 subunits likely play a key role.
What can we learn about DCC function from studies of condensin?
Condensin complexes purified from several organisms have been biochemically characterized. Xenopus (Bazett-Jones et al. 2002; Kimura and Hirano 1997; Kimura et al. 1999), C. elegans (Hagstrom et al. 2002), and human (Kimura et al. 2001) condensin complexes possess the ability to introduce positive supercoils into naked DNA in vitro in an ATP-dependent manner, and to compact single DNA molecules (Strick et al. 2004). While condensin IDC has not been tested for these in vitro activities, its function in dosage compensation is ATP-dependent. When the ATP binding domain in DPY-27 or MIX-1 is mutated, the protein is no longer able to perform its dosage compensation function (Chuang et al. 1994; Lieb et al. 1998). The importance of the SMC ATP-binding motif in dosage compensation is a clear link between this process and typical condensin ATPase activities, and further strengthens the relationship between dosage compensation and mitotic chromosome condensation.
It is not yet clear how these biochemical in vitro activities relate to in vivo condensation of chromosomes as they enter mitosis or meiosis. There is a disagreement in the field whether condensin is necessary for chromosome compaction per se, or whether it functions in some other aspect of higher order chromosome organization. Depletion of condensin subunits in some systems leads to chromosome condensation defects, but in other systems it does not. Instead, a phenotype that has been universally observed upon condensin depletion is a defect in chromosome segregation (Belmont 2006; Hirano 2005). Therefore the exact contribution of condensin to chromosome compaction during mitosis and meiosis remains unclear.
Condensin I and II make distinct contributions to chromosome architecture during mitosis. Vertebrate condensin II is predominantly nuclear and binds chromosomes as condensation begins in prophase, while condensin I is cytoplasmic in interphase and accesses chromosomes only after nuclear envelope break down. After this point, the two condensins have alternating distributions along the chromosome core (Gerlich et al. 2006; Hirota et al. 2004; Ono et al. 2004; Ono et al. 2003). Condensin II plays a primary role in condensation during prophase (Gerlich et al. 2006; Hagstrom et al. 2002; Hirota et al. 2004; Stear and Roth 2002). In contrast, condensin I appears to provide mechanical stability to mitotic chromosomes(Gerlich et al. 2006). In addition, condensin contributes to centromere organization and kinetochore function (Hagstrom et al. 2002; Ono et al. 2004; Stear and Roth 2002; Wignall et al. 2003). Depletion of condensin I or condensin II alone results in distinct chromosomal defects, and depletion of both condensins simultaneously leads to the most severe defects (Hirota et al. 2004; Ono et al. 2004; Ono et al. 2003).
Similar to other organisms, the various condensin complexes in worms associate with chromosomes in strikingly different patterns (Fig. 4), and have different mutant phenotypes, implying that they perform distinct functions (Csankovszki et al. 2009). Condensin IDC associates with hermaphrodite X chromosomes in somatic tissues after the onset of dosage compensation at about the 50-cell stage (Dawes et al. 1999). After the onset of dosage compensation, condensin IDC remains associated with the X chromosomes throughout the cell cycle. However, condensin IDC does not play a role during mitosis or meiosis. During mitosis, condensin I associates with mitotic chromosomes in a discontinuous and non-uniform manner (Csankovszki et al. 2009; Lieb et al. 1996; Tsai et al. 2008), while condensin II is enriched at the centromeres (Chan et al. 2004; Csankovszki et al. 2009; Hagstrom et al. 2002; Maddox et al. 2006; Stear and Roth 2002). During meiosis, condensin II is enriched within the core of sister chromatids (Chan et al. 2004; Csankovszki et al. 2009). On the other hand, condensin I localizes to sites of cohesion at the interface between homologs (meiosis I) and sister chromatids (meiosis II), which implies that condensin I may play a role in chromosome separation and cohesin removal (Csankovszki et al. 2009). Condensin I in other organisms has been shown to play a role in the establishment of cohesion between sister chromatids (Lam et al. 2006), as well as in the removal of cohesin-mediated linkages between chromosomes in mitosis (Hirota et al. 2004) and meiosis (Yu and Koshland 2005). What dictates these various chromosomal association patterns and how the complexes achieve their diverse functions are not known, but comparative analysis of these complexes will likely shed light on the mechanistic relationship between mitotic/meiotic chromosome segregation and dosage compensation.
Interestingly, condensins I and II in other organisms have also been implicated in gene regulation. In yeast, condensin I appears to play a role in silencing at the mating type locus (Bhalla et al. 2002), in rDNA silencing (Machin et al. 2004) and in tRNA-gene mediated silencing (Haeusler et al. 2008). In Drosophila, mutations in condensin I subunits affect repression of transgenes inserted in heterochromatin (Cobbe et al. 2006; Dej et al. 2004). A murine condensin II (CAP-G2) subunit physically interacts with two transcription factors and antagonizes their activation of reporter genes (Xu et al. 2006). The bacterial SMC complex also has roles in gene regulation (Dervyn et al. 2004). These observations imply that the mechanistic link between condensin-mediated gene regulation and chromosome segregation is evolutionarily conserved.
How does DCC binding lead to repression?
Despite recent advances in the study of condensin function, the molecular mechanism of condensin-mediated gene repression remains a mystery. According to the model, condensin binding induces changes in higher order chromosome structure, which affects some aspect of RNA production. Enrichment of the DCC at promoters of X-linked genes originally suggested that the complex may achieve repression by directly binding regulated genes (Ercan et al. 2007). However, analyzing X-linked gene expression levels in dosage compensation mutants revealed a more complicated picture (Jans et al. 2009). This study demonstrated that many X-linked genes are indeed subject to dosage compensation, but many other X-linked genes are not. Dosage compensated genes are interspersed with non-dosage compensated genes. Genes of all expression levels can be dosage compensated and the magnitude of gene expression changes varies from less than two-fold to over 5-fold. Interestingly, there is no correlation between DCC binding and gene expression changes in dosage compensation mutants (Jans et al. 2009). In these mutants, many genes normally bound by the DCC are overexpressed, but many are not. Conversely, many genes normally not DCC-bound are also overexpressed in the mutants. The authors suggested that the DCC does not repress genes by direct binding, but rather it acts at a distance. The mechanism may involve looping of chromatin fibers to bring regulated genes to the vicinity of DCC bound sites (Jans et al. 2009). Alternatively, gene expression changes of DCC-unbound genes can also be attributed to indirect effects. Consistent with that, many genes on autosomes are also misregulated in the dosage compensation mutants (see below, Jans et al. 2009).
By contrast, genome-wide DCC binding and gene expression studies in flies showed a correlation between binding and repression. In flies, genes most affected by depletion of the MSL complex are those normally bound directly by the DCC (Alekseyenko et al. 2006; Hamada et al. 2005). The difference between the Drosophila model of gene-by-gene regulation and the C. elegans model of chromosome-wide regulation may lie in the differences between the mechanisms of action of the two dosage compensation machineries. In Drosophila, the machinery alters the structure of the nucleosome fiber by acetylation of lysine 16 on histone H4, while in worms the machinery is thought to act at the level of higher order structures, not at the level of nucleosomes.
Some features of repression by the DCC are reminiscent of the role of condensin function in tRNA mediated gene silencing (tgm silencing) in the yeast S. cerevisiae. During tgm silencing, condensin binds tRNA genes via a physical interaction with TFIIIC and TFIIIB. The tRNA genes cluster near the nucleolus in a condensin-dependent manner, which leads to silencing of nearby RNA Pol II transcribed genes (Haeusler et al. 2008). Interestingly, in worms, a large fraction of tRNA genes are bound by condensin IDC, both on the autosomes and on the X chromosome (Jans et al. 2009). In tgm silencing, condensin binds the tRNA genes, which leads to silencing of other nearby genes not bound by condensin (Haeusler et al. 2008). Similarly, in dosage compensation, condensin IDC binding leads to repression of genes, many of which are not bound by the complex (Jans et al. 2009). In tgm silencing, as in dosage compensation, repressed genes are interspersed with non-repressed genes. Perhaps spatial clustering of distant chromosomal sites is involved in both processes. These parallels raise the possibility that features of condensin-mediated repression are conserved between different species.
Other repression mechanisms?
Dosage compensation in other organisms involves modulation of the structure of the nucleosome fiber to achieve gene silencing (in mammals) or two-fold upregulation (in flies). Are chromatin changes involved in worm dosage compensation? DPY-30, in addition to functioning in dosage compensation, is also a subunit of the COMPASS histone methyltransferase complex (Meyer 2005). However, this function of DPY-30 has not yet been linked to dosage compensation. A recent study reported that the histone variant HTZ-1/H2A.Z is underrepresented on X chromosomes in worms, but the authors did not find any evidence for a direct role for HTZ-1 in dosage compensation (Whittle et al. 2008). There are well-studied mechanisms of changes in chromatin structure leading to gene silencing or gene activation, but perhaps a two-fold repression is not easily achieved by changes in the structure of the nucleosome fiber.
Nevertheless, the action of condensin IDC and a mechanism related to mitotic chromosome condensation are unlikely to be the whole story. DCC components, such as the SDC proteins, as well as DPY-21 and DPY-30, are not condensin subunit homologs, yet their function contributes to repression. Part of their role is in DCC recruitment. SDC-2 and SDC-3 are needed to recruit all the other members of the DCC to the X chromosome (Davis and Meyer 1997; Dawes et al. 1999). DPY-30 also plays a role in recruitment (Hsu et al. 1995; Hsu and Meyer 1994). However, other functions, beyond recruitment, cannot be excluded. In addition, DPY-21 must play a role different from recruitment and/or stability of the DCC. dpy-21 mutants display normal localization of the DCC to the X chromosomes, yet dosage compensation function is compromised (Yonker and Meyer 2003). Future functional studies of these DCC members will reveal their contribution to the dosage compensation process.
DCC action on autosomes
Perhaps the most surprising recent finding in the field is that the DCC seems to affect gene expression on the autosomes in addition to regulating X-linked genes. In dosage compensation mutants, expression of nearly 30% of autosomal genes decreases, while expression of X-linked genes increases (Jans et al. 2009). Just like on the X chromosome, DCC bound genes are not necessarily the ones whose expression changes in the mutants. While some of the autosomal gene expression changes observed in these mutants could be due to indirect effects, the clear trend of X overexpression and autosome underexpression makes it likely that more direct effects are also involved. Perhaps dosage compensation in hermaphrodites involves establishing an intricate balance of X to autosome gene expression ratio by downregulating X chromosome gene expression and upregulating autosomal gene expression at the same time. How the DCC could be involved in these two processes remains to be determined.
Abbreviations
- ASE:
-
autosomal signal element
- C. elegans :
-
Caenorhabditis elegans
- CAP:
-
chromosome associated polypeptide
- DCC:
-
dosage compensation complex
- dox:
-
dependent on X
- Dpy:
-
dumpy
- MEA:
-
motif enriched on autosomes
- MEX:
-
motif enriched on X
- rex:
-
recruitment element on X
- SMC:
-
structural maintenance of chromosomes
- tgm:
-
tRNA gene-mediated
- XSE:
-
X signal element
References
Adler DA, Rugarli EI, Lingenfelter PA, Tsuchiya K, Poslinski D, Liggitt HD, Chapman VM, Elliott RW, Ballabio A, Disteche CM (1997) Evidence of evolutionary up-regulation of the single active X chromosome in mammals based on Clc4 expression levels in Mus spretus and Mus musculus. Proc Natl Acad Sci U S A 94:9244–9248
Akerib CC, Meyer BJ (1994) Identification of X chromosome regions in Caenorhabditis elegans that contain sex-determination signal elements. Genetics 138:1105–1125
Alekseyenko AA, Larschan E, Lai WR, Park PJ, Kuroda MI (2006) High-resolution ChIP-chip analysis reveals that the Drosophila MSL complex selectively identifies active genes on the male X chromosome. Genes Dev 20:848–857
Alekseyenko AA, Peng S, Larschan E, Gorchakov AA, Lee OK, Kharchenko P, McGrath SD, Wang CI, Mardis ER, Park PJ, Kuroda MI (2008) A sequence motif within chromatin entry sites directs MSL establishment on the Drosophila X chromosome. Cell 134:599–609
Bazett-Jones DP, Kimura K, Hirano T (2002) Efficient supercoiling of DNA by a single condensin complex as revealed by electron spectroscopic imaging. Mol Cell 9:1183–1190
Belmont AS (2006) Mitotic chromosome structure and condensation. Curr Opin Cell Biol 18:632–638
Bhalla N, Biggins S, Murray AW (2002) Mutation of YCS4, a budding yeast condensin subunit, affects mitotic and nonmitotic chromosome behavior. Mol Biol Cell 13:632–645
Blauwkamp TA, Csankovszki G (2009) Two classes of DCC binding elements facilitate binding and spreading of the DCC Along C. elegans X chromosomes. Mol Cell Biol
Carmi I, Meyer BJ (1999) The primary sex determination signal of Caenorhabditis elegans. Genetics 152:999–1015
Carmi I, Kopczynski JB, Meyer BJ (1998) The nuclear hormone receptor SEX-1 is an X-chromosome signal that determines nematode sex. Nature 396:168–173
Chan RC, Severson AF, Meyer BJ (2004) Condensin restructures chromosomes in preparation for meiotic divisions. J Cell Biol 167:613–625
Chu DS, Dawes HE, Lieb JD, Chan RC, Kuo AF, Meyer BJ (2002) A molecular link between gene-specific and chromosome-wide transcriptional repression. Genes Dev 16:796–805
Chuang PT, Albertson DG, Meyer BJ (1994) DPY-27: a chromosome condensation protein homolog that regulates C. elegans dosage compensation through association with the X chromosome. Cell 79:459–474
Chuang PT, Lieb JD, Meyer BJ (1996) Sex-specific assembly of a dosage compensation complex on the nematode X chromosome. Science 274:1736–1739
Cobbe N, Savvidou E, Heck MM (2006) Diverse mitotic and interphase functions of condensins in Drosophila. Genetics 172:991–1008
Csankovszki G, McDonel P, Meyer BJ (2004) Recruitment and spreading of the C. elegans dosage compensation complex along X chromosomes. Science 303:1182–1185
Csankovszki G, Collette K, Spahl K, Carey J, Snyder M, Petty E, Patel U, Tabuchi T, Liu H, McLeod I et al (2009) Three distinct condensin complexes control C. elegans chromosome dynamics. Curr Biol 19:9–19
Davis TL, Meyer BJ (1997) SDC-3 coordinates the assembly of a dosage compensation complex on the nematode X chromosome. Development 124:1019–1031
Dawes HE, Berlin DS, Lapidus DM, Nusbaum C, Davis TL, Meyer BJ (1999) Dosage compensation proteins targeted to X chromosomes by a determinant of hermaphrodite fate. Science 284:1800–1804
Dej KJ, Ahn C, Orr-Weaver TL (2004) Mutations in the Drosophila condensin subunit dCAP-G: defining the role of condensin for chromosome condensation in mitosis and gene expression in interphase. Genetics 168:895–906
DeLong L, Plenefisch JD, Klein RD, Meyer BJ (1993) Feedback control of sex determination by dosage compensation revealed through Caenorhabditis elegans sdc-3 mutations. Genetics 133:875–896
Dervyn E, Noirot-Gros MF, Mervelet P, McGovern S, Ehrlich SD, Polard P, Noirot P (2004) The bacterial condensin/cohesin-like protein complex acts in DNA repair and regulation of gene expression. Mol Microbiol 51:1629–1640
Ercan S, Giresi PG, Whittle CM, Zhang X, Green RD, Lieb JD (2007) X chromosome repression by localization of the C. elegans dosage compensation machinery to sites of transcription initiation. Nat Genet 39:403–408
Gerlich D, Hirota T, Koch B, Peters JM, Ellenberg J (2006) Condensin I stabilizes chromosomes mechanically through a dynamic interaction in live cells. Curr Biol 16:333–344
Gilfillan GD, Konig C, Dahlsveen IK, Prakoura N, Straub T, Lamm R, Fauth T, Becker PB (2007) Cumulative contributions of weak DNA determinants to targeting the Drosophila dosage compensation complex. Nucleic Acids Res 35:3561–3572
Gladden JM, Meyer BJ (2007) A ONECUT homeodomain protein communicates X chromosome dose to specify Caenorhabditis elegans sexual fate by repressing a sex switch gene. Genetics 177:1621–1637
Gladden JM, Farboud B, Meyer BJ (2007) Revisiting the X:A signal that specifies Caenorhabditis elegans sexual fate. Genetics 177:1639–1654
Gupta V, Parisi M, Sturgill D, Nuttall R, Doctolero M, Dudko OK, Malley JD, Eastman PS, Oliver B (2006) Global analysis of X-chromosome dosage compensation. J Biol 5:3
Haeusler RA, Pratt-Hyatt M, Good PD, Gipson TA, Engelke DR (2008) Clustering of yeast tRNA genes is mediated by specific association of condensin with tRNA gene transcription complexes. Genes Dev 22:2204–2214
Hagstrom KA, Holmes VF, Cozzarelli NR, Meyer BJ (2002) C. elegans condensin promotes mitotic chromosome architecture, centromere organization, and sister chromatid segregation during mitosis and meiosis. Genes Dev 16:729–742
Hamada FN, Park PJ, Gordadze PR, Kuroda MI (2005) Global regulation of X chromosomal genes by the MSL complex in Drosophila melanogaster. Genes Dev 19:2289–2294
Heard E, Mongelard F, Arnaud D, Avner P (1999) Xist yeast artificial chromosome transgenes function as X-inactivation centers only in multicopy arrays and not as single copies. Mol Cell Biol 19:3156–3166
Hirano T (2005) Condensins: organizing and segregating the genome. Curr Biol 15:R265–R275
Hirota T, Gerlich D, Koch B, Ellenberg J, Peters JM (2004) Distinct functions of condensin I and II in mitotic chromosome assembly. J Cell Sci 117:6435–6445
Hodgkin J (1983) X chromosome dosage and gene expression in Caenorhabditis elegans: two unusual dumpy genes. Mol Gen Genet 192:452–458
Hodgkin J (2005) Karyotype, ploidy, and gene dosage. WormBook, 1–9
Hodgkin J, Zellan JD, Albertson DG (1994) Identification of a candidate primary sex determination locus, fox-1, on the X chromosome of Caenorhabditis elegans. Development 120:3681–3689
Hsu DR, Meyer BJ (1994) The dpy-30 gene encodes an essential component of the Caenorhabditis elegans dosage compensation machinery. Genetics 137:999–1018
Hsu DR, Chuang PT, Meyer BJ (1995) DPY-30, a nuclear protein essential early in embryogenesis for Caenorhabditis elegans dosage compensation. Development 121:3323–3334
Jans J, Gladden JM, Ralston EJ, Pickle CS, Michel AH, Pferdehirt R, Eisen MB, Meyer BJ (2009) A condensin-like dosage compensation complex acts at a distance to control expression throughout the genome. Genes Dev, in press
Kimura K, Hirano T (1997) ATP-dependent positive supercoiling of DNA by 13S condensin: a biochemical implication for chromosome condensation. Cell 90:625–634
Kimura K, Rybenkov VV, Crisona NJ, Hirano T, Cozzarelli NR (1999) 13S condensin actively reconfigures DNA by introducing global positive writhe: implications for chromosome condensation. Cell 98:239–248
Kimura K, Cuvier O, Hirano T (2001) Chromosome condensation by a human condensin complex in Xenopus egg extracts. J Biol Chem 276:5417–5420
Klein RD, Meyer BJ (1993) Independent domains of the Sdc-3 protein control sex determination and dosage compensation in C. elegans. Cell 72:349–364
Krogan NJ, Dover J, Khorrami S, Greenblatt JF, Schneider J, Johnston M, Shilatifard A (2002) COMPASS, a histone H3 (Lysine 4) methyltransferase required for telomeric silencing of gene expression. J Biol Chem 277:10753–10755
Lam WW, Peterson EA, Yeung M, Lavoie BD (2006) Condensin is required for chromosome arm cohesion during mitosis. Genes Dev 20:2973–2984
Lieb JD, Capowski EE, Meneely P, Meyer BJ (1996) DPY-26, a link between dosage compensation and meiotic chromosome segregation in the nematode. Science 274:1732–1736
Lieb JD, Albrecht MR, Chuang PT, Meyer BJ (1998) MIX-1: an essential component of the C. elegans mitotic machinery executes X chromosome dosage compensation. Cell 92:265–277
Lieb JD, de Solorzano CO, Rodriguez EG, Jones A, Angelo M, Lockett S, Meyer BJ (2000) The Caenorhabditis elegans dosage compensation machinery is recruited to X chromosome DNA attached to an autosome. Genetics 156:1603–1621
Lin H, Gupta V, Vermilyea MD, Falciani F, Lee JT, O’Neill LP, Turner BM (2007) Dosage compensation in the mouse balances up-regulation and silencing of X-linked genes. PLoS Biol 5:e326
Luz JG, Hassig CA, Pickle C, Godzik A, Meyer BJ, Wilson IA (2003) XOL-1, primary determinant of sexual fate in C. elegans, is a GHMP kinase family member and a structural prototype for a class of developmental regulators. Genes Dev 17:977–990
Machin F, Paschos K, Jarmuz A, Torres-Rosell J, Pade C, Aragon L (2004) Condensin regulates rDNA silencing by modulating nucleolar Sir2p. Curr Biol 14:125–130
Maddox PS, Portier N, Desai A, Oegema K (2006) Molecular analysis of mitotic chromosome condensation using a quantitative time-resolved fluorescence microscopy assay. Proc Natl Acad Sci U S A 103:15097–15102
Madl JE, Herman RK (1979) Polyploids and sex determination in Caenorhabditis elegans. Genetics 93:393–402
McDonel P, Jans J, Peterson BK, Meyer BJ (2006) Clustered DNA motifs mark X chromosomes for repression by a dosage compensation complex. Nature 444:614–618
Meneely PM, Wood WB (1984) An autosomal gene that affects X chromosome expression and sex determination in Caenorhabditis elegans. Genetics 106:29–44
Mets DG, Meyer BJ (2009) Condensins regulate meiotic DNA break distribution, thus crossover frequency, by controlling chromosome structure. Cell (in press)
Meyer BJ (2005) X-Chromosome dosage compensation. WormBook, 1–14
Meyer BJ, Casson LP (1986) Caenorhabditis elegans compensates for the difference in X chromosome dosage between the sexes by regulating transcript levels. Cell 47:871–881
Miller LM, Plenefisch JD, Casson LP, Meyer BJ (1988) xol-1: a gene that controls the male modes of both sex determination and X chromosome dosage compensation in C. elegans. Cell 55:167–183
Miller T, Krogan NJ, Dover J, Erdjument-Bromage H, Tempst P, Johnston M, Greenblatt JF, Shilatifard A (2001) COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc Natl Acad Sci U S A 98:12902–12907
Nagy PL, Griesenbeck J, Kornberg RD, Cleary ML (2002) A trithorax-group complex purified from Saccharomyces cerevisiae is required for methylation of histone H3. Proc Natl Acad Sci U S A 99:90–94
Nguyen DK, Disteche CM (2006) Dosage compensation of the active X chromosome in mammals. Nat Genet 38:47–53
Nicoll M, Akerib CC, Meyer BJ (1997) X-chromosome-counting mechanisms that determine nematode sex. Nature 388:200–204
Nonet ML, Meyer BJ (1991) Early aspects of Caenorhabditis elegans sex determination and dosage compensation are regulated by a zinc-finger protein. Nature 351:65–68
Nusbaum C, Meyer BJ (1989) The Caenorhabditis elegans gene sdc-2 controls sex determination and dosage compensation in XX animals. Genetics 122:579–593
Ono T, Losada A, Hirano M, Myers MP, Neuwald AF, Hirano T (2003) Differential contributions of condensin I and condensin II to mitotic chromosome architecture in vertebrate cells. Cell 115:109–121
Ono T, Fang Y, Spector DL, Hirano T (2004) Spatial and temporal regulation of Condensins I and II in mitotic chromosome assembly in human cells. Mol Biol Cell 15:3296–3308
Plenefisch JD, DeLong L, Meyer BJ (1989) Genes that implement the hermaphrodite mode of dosage compensation in Caenorhabditis elegans. Genetics 121:57–76
Powell JR, Jow MM, Meyer BJ (2005) The T-box transcription factor SEA-1 is an autosomal element of the X:A signal that determines C. elegans sex. Dev Cell 9:339–349
Rhind NR, Miller LM, Kopczynski JB, Meyer BJ (1995) xol-1 acts as an early switch in the C. elegans male/hermaphrodite decision. Cell 80:71–82
Sharova LV, Sharov AA, Piao Y, Shaik N, Sullivan T, Stewart CL, Hogan BL, Ko MS (2007) Global gene expression profiling reveals similarities and differences among mouse pluripotent stem cells of different origins and strains. Dev Biol 307:446–459
Skipper M, Milne CA, Hodgkin J (1999) Genetic and molecular analysis of fox-1, a numerator element involved in Caenorhabditis elegans primary sex determination. Genetics 151:617–631
Stear JH, Roth MB (2002) Characterization of HCP-6, a C. elegans protein required to prevent chromosome twisting and merotelic attachment. Genes Dev 16:1498–1508
Straub T, Grimaud C, Gilfillan GD, Mitterweger A, Becker PB (2008) The chromosomal high-affinity binding sites for the Drosophila dosage compensation complex. PLoS Genet 4:e1000302
Strick TR, Kawaguchi T, Hirano T (2004) Real-time detection of single-molecule DNA compaction by condensin I. Curr Biol 14:874–880
Trent C, Tsuing N, Horvitz HR (1983) Egg-laying defective mutants of the nematode Caenorhabditis elegans. Genetics 104:619–647
Trent C, Purnell B, Gavinski S, Hageman J, Chamblin C, Wood WB (1991) Sex-specific transcriptional regulation of the C. elegans sex-determining gene her-1. Mech Dev 34:43–55
Tsai CJ, Mets DG, Albrecht MR, Nix P, Chan A, Meyer BJ (2008) Meiotic crossover number and distribution are regulated by a dosage compensation protein that resembles a condensin subunit. Genes Dev 22:194–211
Villeneuve AM, Meyer BJ (1987) sdc-1: a link between sex determination and dosage compensation in C. elegans. Cell 48:25–37
Villeneuve AM, Meyer BJ (1990) The role of sdc-1 in the sex determination and dosage compensation decisions in Caenorhabditis elegans. Genetics 124:91–114
Whittle CM, McClinic KN, Ercan S, Zhang X, Green RD, Kelly WG, Lieb JD (2008) The genomic distribution and function of histone variant HTZ-1 during C. elegans embryogenesis. PLoS Genet 4:e1000187
Wignall SM, Deehan R, Maresca TJ, Heald R (2003) The condensin complex is required for proper spindle assembly and chromosome segregation in Xenopus egg extracts. J Cell Biol 161:1041–1051
Wood WB, Meneely P, Schedin P, Donahue L (1985) Aspects of dosage compensation and sex determination in Caenorhabditis elegans. Cold Spring Harb Symp Quant Biol 50:575–583
Xu Y, Leung CG, Lee DC, Kennedy BK, Crispino JD (2006) MTB, the murine homolog of condensin II subunit CAP-G2, represses transcription and promotes erythroid cell differentiation. Leukemia 20:1261–1269
Yonker SA, Meyer BJ (2003) Recruitment of C. elegans dosage compensation proteins for gene-specific versus chromosome-wide repression. Development 130:6519–6532
Yu HG, Koshland D (2005) Chromosome morphogenesis: condensin-dependent cohesin removal during meiosis. Cell 123:397–407
Zarkower D (2006) Somatic sex determination. WormBook, 1–12
Acknowledgments
We would like to thank Martha Snyder, Michael Wells, and members of the Csankovszki laboratory for critical reading of the manuscript. Work in the lab is supported by a National Institute of Health Grant (NIH R01 GM079533) to G.C. and a Predoctoral Training Grant in Genetics (NIH T32 GM07544) to E. L. P. and K. S. C.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Edith Heard.
Rights and permissions
About this article
Cite this article
Csankovszki, G., Petty, E.L. & Collette, K.S. The worm solution: a chromosome-full of condensin helps gene expression go down. Chromosome Res 17, 621–635 (2009). https://doi.org/10.1007/s10577-009-9061-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10577-009-9061-y