
Abstract
LOX-1 is an endothelial receptor for oxidized low-density lipoprotein (oxLDL), a key molecule in the pathogenesis of atherosclerosis.The basal expression of LOX-1 is low but highly induced under the influence of proinflammatory and prooxidative stimuli in vascular endothelial cells, smooth muscle cells, macrophages, platelets and cardiomyocytes. Multiple lines of in vitro and in vivo studies have provided compelling evidence that LOX-1 promotes endothelial dysfunction and atherogenesis induced by oxLDL. The roles of LOX-1 in the development of atherosclerosis, however, are not simple as it had been considered. Evidence has been accumulating that LOX-1 recognizes not only oxLDL but other atherogenic lipoproteins, platelets, leukocytes and CRP. As results, LOX-1 not only mediates endothelial dysfunction but contributes to atherosclerotic plaque formation, thrombogenesis, leukocyte infiltration and myocardial infarction, which determine mortality and morbidity from atherosclerosis. Moreover, our recent epidemiological study has highlighted the involvement of LOX-1 in human cardiovascular diseases. Further understandings of LOX-1 and its ligands as well as its versatile functions will direct us to ways to find novel diagnostic and therapeutic approaches to cardiovascular disease.
Similar content being viewed by others

Introduction
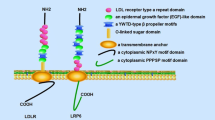
The lectin-like oxidized LDL receptor-1 (LOX-1) was identified as an oxidized LDL (oxLDL) receptor expressed in vascular endothelium. It is a 50 kDa type II transmembrane glycoprotein that structurally belongs to the C-type lectin family [1]. It contains a short N-terminal cytoplasmic domain, a single transmembrane domain and an extracellular domain comprising a neck domain followed by a C-terminal C-type lectin-like domain. Since the discovery of LOX-1, we have been enthusiastically pursuing its roles in cardiovascular diseases including atherosclerosis. LOX-1 is highly induced in the cardiovascular system by a number of proinflammatory and prooxidative stimuli and recognizes various structurally-unrelated molecules or even certain types of cell as a ligand. It has been now established that LOX-1 actively contributes to the development of atherosclerosis. Furthermore, emerging evidence has been implicating pathological roles of LOX-1 in humans. In this review, we will describe the roles of LOX-1 in cardiovascular diseases particularly focusing on atherosclerosis, its versatile functions as a multiligand receptor and the clinical potential for diagnostic and therapeutic targets.
LOX-1 in cardiovascular systems
Discovery of LOX-1
Atherosclerosis is a chronic, multifactorial inflammatory disease and a leading cause of coronary heart diseases and cerebrovascular diseases. Over the decades, there have been numerous efforts to explain the pathogenesis of atherosclerosis. The response-to-injury hypothesis proposed by Ross [2, 3] assumes that injury of endothelium (endothelial dysfunction) leads to a number of compensatory responses that alter normal vascular homeostasis. The response-to-retention hypothesis depicts that LDL is delivered to the subendothelial space, binds to proteoglycans, aggregates and is taken up by macrophages leading to foam cell formation [4, 5]. The oxidative modification hypothesis assumes that LDL must undergo oxidative modification to be proatherogenic because macrophages avidly take up oxidatively modified LDL but not native LDL to be foam cells.
The above hypotheses may not be mutually exclusive but rather emphasize different concepts as necessary steps for the initiation of atherosclerosis. OxLDL recruits circulating monocytes into the intimal space, inhibits them to leave the intima and generates foam cells but the proatherogenic properties of oxLDL are not limited to monocytes/macrophages. In vascular endothelial cells, oxLDL induces the expression and release of monocyte chemoattractant protein-1 (MCP-1), inhibits release and/or function of nitric oxide (NO) and increases the expression of adhesion molecules such as vascular adhesion molecule-1 (VCAM-1), which suggests a link between the response-to-injury and oxidative modification hypothesis. Moreover, it is known that vascular endothelial cells also internalize oxLDL through a putative receptor-mediated pathway that does not involve the macrophage scavenger receptors [6]. In light of these findings, we attempted to identify an oxLDL receptor from vascular endothelial cells.
Expression cloning strategy was employed to identify an endothelial oxLDL receptor using a cDNA library of cultured bovine aortic endothelial cells. After multiple rounds of transfection-recovery, we successfully identified a cDNA encoding an endothelial receptor for oxLDL and designated LOX-1 in 1997 [1]. Chinese hamster ovary cells expressing LOX-1 (LOX-1-CHO) efficiently bound and took up [125I]-labeled oxLDL, which was not observed in wild type CHO cells. The degradation of [125I]-labeled oxLDL was suppressed by unlabeled oxLDL but not native LDL, indicating that native LDL is not a ligand for LOX-1. In bovine aortic endothelial cells, an anti-LOX-1 antibody significantly inhibited [125I]-labeled oxLDL binding, indicating that LOX-1 is a functional oxLDL receptor in vascular endothelial cells.
Amino acid sequences of LOX-1
Human LOX-1 is encoded by OLR1 gene located on chromosome 12p12-p13. LOX-1 consists of four functional domains, a lectin-like domain located at the C-terminal region, a connecting neck domain, a transmembrane domain and an intracellular domain located at the N-terminal region. The amino acid sequences of LOX-1 are well conserved among mammals [7].The amino acid sequences of LOX-1 are, however, distinct from other known scavenger receptors although they recognize common ligands such as oxLDL, bacteria and apoptotic cells [8]. This is likely an example of convergent evolution where different proteins have been adapted to recognize common ligands. LOX-1 seems to have evolved in the context of innate immunity system since the gene clusters together with a subfamily of C-type lectin receptor (CLEC) such as dectin-1(CLEC-7A) and CLEC-1 that act as pattern recognition receptors [9–12] (Fig. 1).The amino acid sequence homology of LOX-1 and other CLEC family proteins is shown in Table 1.

Genomic organization of LOX-1 and other CLEC family genes on human chromosome 12
Cellular responses involving LOX-1
Multiple lines of evidence have demonstrated that activation of endothelial LOX-1 results in endothelial dysfunction, characterized by reduced vasodilation, proinflammatory state and prothrombotic properties. Activation of LOX-1 with oxLDL induces cell injury and apoptosis in human coronary artery endothelial cells [13, 14]. It is associated with increased MCP-1 protein expression and enhanced monocyte adhesion to endothelial cells through the activation of p42/44 mitogen-activated protein kinase (MAPK) [13]. LOX-1 activation with oxLDL increases the expressions CD40 and CD40 ligand and subsequent generation of tumor necrosis factor-α (TNF-α) and P-selectin via protein kinase C (PKC) activation in human coronary artery endothelial cells [15]. OxLDL binding to LOX-1 also immediately increases reactive oxygen species (ROS) formation and reduces NO availability with concomitant activation of NF-κB in bovine aortic endothelial cells [16, 17]. In isolated aortas, oxLDL impairs endothelium-dependent relaxation, which is restored by the treatment of anti-LOX-1 antibody and in LOX-1 gene deficient (LOX-1 KO) mice [18]. The intercellular signal transductions following the activation of LOX-1 have been elusive as the intercellular domain of LOX-1 consists of only 30 amino acids. Recent evidence has shown that a complex formed by LOX-1 and membrane type 1 matrix metalloproteinase (MT1-MMP) has been suggested to underlie the ROS formation and endothelial nitric oxide synthase (eNOS) downregulation [19].
LOX-1 is also expressed in vascular smooth muscle cells, differentiated macrophages and platelets. Low doses of oxLDL increase proliferation of vascular smooth muscle cells while high doses of oxLDL induce apoptosis in vascular smooth muscle cells, both of which could be inhibited by anti-LOX-1 antibody [20, 21]. LOX-1 is absent in human monocytes but induced in differentiated macrophages and substantial amount of oxLDL is taken up via LOX-1 in macrophages stimulated with lisophosphatidylcholine or palmitic acids [22–24].In platelets, LOX-1 is exposed on the surface in an activation-dependent manner and LOX-1 inhibition suppresses adenosine diphosphate (ADP)-induced platelet aggregation [25, 26]. Therefore, LOX-1 is likely to participate in the development of atherosclerosis from an early to late stage of the process.
Regulated expression of LOX-1 in disease states
In vitro, the basal expression of LOX-1 is low but it has been consistently observed that the expression is highly induced by proinflammatory and prooxidative stimuli in endothelial cells, smooth muscle cells and macrophages. The stimuli include TNFα,interleukin-1β (IL-1β),transforming growth factor-β1 (TGF-β1), superoxide anions, hydrogen peroxide, 8-iso-prostaglandin F2α, and lysophosphatidylcholine [27–35]. The expression is also potentiated by other stimuli such as glucose, free fatty acids, angiotensin II, endothelin-1 and sheer stress [24, 29, 36–43]. Each of these is actively involved in the pathogenesis of cardiovascular diseases including atherosclerosis. Interestingly, several of the stimuli are also released in response to LOX-1 activation such as endothelin-1 and TNF-α and oxLDL itself increases LOX-1 expression, suggesting that the LOX-1 axis composes a vicious cycle for the development of atherosclerosis [15, 29, 36, 44].
In line with a number of in vitro results, in vivo LOX-1 expression is also highly upregulated under the influence of proinflammatory and prooxidative stimuli. Vascular LOX-1 gene expression is markedly enhanced in hypertensive rats [28, 45–48], hyperlipidemic rabbits [49] and diabetic rats [50]. Ischemia-reperfusion also increases the LOX-1 expression in myocardium and kidney [51–54]. Recently, it has been demonstrated that LOX-1 is highly induced in adipose tissues in response to high fat diet feeding [55]. Moreover, adipose tissues collected from LOX-1 KO mice were associated with less expression of inflammatory cytokines such as MCP-1, macrophage inflammatory protein-1α and interleukin-6 (IL-6) despite a comparable level of obesity to wild type mice. As adipose inflammation in obese states has been suggested to lead to systemic insulin resistance and atherosclerosis [56], increased LOX-1 expression in obese adipose tissues may also promote the development of atherosclerosis.
Conversely, the expression of vascular LOX-1is inhibited by the several clinical drugs. These include anti-hypertensive (angiotensin II receptor antagonists, calcium channel blockers, angiotensin-converting enzyme inhibitors), anti-hyperlipidemic (statins), anti-diabetic (sulfonyl urea, biguanide, peroxisome proliferator-activated receptor-γ/PPARγ agonist)and anti-thrombotic (aspirin) agents, potentially suggesting that the reduced expression of LOX-1 may partially contribute to the therapeutic outcomes of those drugs (see also section 5.Clinical significance of LOX-1 in Cardiovascular Diseases [57–64]). Interestingly, Chui et al. have reported that treatment of obese diabetic animals with PPARγ agonists resulted in a marked increase in adipose LOX-1 expression with a concomitant reduction of serum oxLDL level [65]. The authors speculated that PPARγ agonists may exhibit anti-atherosclerotic effects partly by trapping circulating oxLDL in adipocytes.
LOX-1 ligands
Modified lipoproteins
LOX-1 was identified as a receptor for oxLDL that was prepared by oxidizing LDL chemically with copper sulfate in vitro. Early studies have demonstrated that LOX-1 also recognizes other chemically modified lipoproteins such as acetyl LDL and hypochlorite-modified high-density lipoproteins [66–68].
In vitro oxidation of LDL yields lipid peroxides such as oxidized phosphatidylcholine, acrolein, 4-hydroxynonenal and malondialdehyde and subsequent modification of apoB protein [69]. Although such oxLDL is an artificial molecule, the presence of such materials that chemically and biochemically resemble the oxLDL has been known in atherosclerotic lesions in humans and rabbits [70]. Later, a series of studies using anti-oxLDL specific monoclonal antibodies have clearly demonstrated the presence of oxLDL in atherosclerotic lesions and in the circulation of humans and animals [71–75]. As expected, however, several of these studies have also illustrated that oxLDL generated in vivo is heterogeneous with distinctive patterns of modification on lipid and protein moieties, implicating the potential that oxLDL with antigenicity to a specific monoclonal antibody does not necessarily bind to and elicit biological actions via LOX-1.
In order to demonstrate the presence of oxLDL that could be a ligand for LOX-1 in vivo, we have developed a sandwich enzyme-linked immunosorbent assay (ELISA)to measure LOX-1 ligand containing apoB (LAB) with anti-apoB antibody and immobilized LOX-1. The plasma level of LAB was significantly higher in Watanabe heritable hyperlipidemic rabbits (WHHL) compared to Japanese white rabbits [76, 77]. Higher concentration of plasma LAB was also demonstrated in apoE-deficient (apoE KO) mice, another atherosclerosis-prone animal model [78]. Moreover, aortic root of apoE KO mice accumulated significant amount of LAB, as evidenced by immunohistochemical staining using extracellular domain of LOX-1 as a probe [78]. Therefore, oxLDL that could be a ligand for LOX-1 is highly present in circulation and lesions of atherosclerosis-prone animals.
Recent evidence has shown that other atherogenic lipoproteins such as carbamylated LDL, remnant-like lipoprotein particle and electronegative LDL mediate proatherogenic properties by binding to LOX-1 in vitro [79–82]. Therefore, LAB is likely to include not only oxLDL that could be a ligand for LOX-1 but other proatherogenic modified LDL, suggesting that the level of LAB may reflect proatherogenic states better than measuring the absolute amount of oxLDL with specific epitope by anti-oxLDL antibody (also see section 5.3 LOX Index). Interestingly, monocyte adhesion to human coronary arterial endothelial cells induced by carbamylated LDL was dependent on LOX-1 whereas the uptake of carbamylated LDL was equivalently contributed to by LOX-1 and other scavenger receptors such as scavenger receptor expressed by endothelial cell-1 (SREC-1), CD36 and scavenger receptor-A (SR-A) [79].
Cellular ligands
LOX-1 recognizes other ligands with no structural similarities to lipoproteins. These include polyanionic chemicals, anionic phospholipids and cellular ligands [66, 83–87]. The reported ligands for LOX-1 are summarized in Table 2. The pathological significance of LOX-1 as a cell adhesion molecule has been demonstrated as follows.
Activated but not non-activated platelets increase endothelin-1 release and superoxide production from endothelial cells upon ligation to LOX-1, suggesting that not only oxLDL but activated platelets induce endothelial dysfunction through LOX-1 [44, 84, 86]. Importantly, co-incubation of activated platelets with endothelial cells almost completely diminishes endothelial NO release by bradykinin, indicating that a sufficient amount of superoxide is generated to disrupt vascular protective effects of NO. Phosphatidylserine (PS) exposed on activated platelets seems to work as a ligand for LOX-1 because 1) superoxide generation is significantly inhibited by annexin V, a PS binding protein, 2) PS liposome, a ligand for LOX-1 also increases superoxide and 3) platelets expose PS upon activation.
Incubation of human aortic endothelial cells with high glucose or C-reactive protein (CRP) increases LOX-1 expression and monocyte adhesion, and the adhesion is suppressed by anti-LOX-1 antibody [41, 88]. Monocyte adhesion also initiates ROS generation and NF-κB phosphorylation in endothelial cells [89]. In vivo adhesion of leukocytes to LOX-1 has been demonstrated in endotoxin-induced inflammation in rats [90]. Flow chamber analysis demonstrated that leukocytes directly bind to LOX-1. As adhesion of mononuclear leukocytes to vascular endothelium cells is also considered as an early step in atherogenesis, LOX-1 may also support leukocyte adhesion during the development of atherosclerosis.
LOX-1 expressed on vascular endothelial cells binds to and phagocytoses aged/apoptotic red blood cells [66]. Again, PS on cellular membranes seems to be a key molecule in the cell recognition by LOX-1 because PS is exposed on the outer membrane of aged/apoptotic cells and PS liposome efficiently displaces the binding. Although there is no evidence showing that endothelial LOX-1 promotes the clearance of aged/apoptotic cells in vivo, LOX-1 may enhance procoagulant activity by trapping aged/apoptotic cells on the vascular surface since apoptotic cells exposing PS are potent procoagulant molecules [91].
C-reactive protein
CRP is an acute-phase protein released from hepatocytes in response to inflammation and tissue damage. It binds to various ligands exposed on damaged tissues or bacteria, promoting phagocytosis and complement activation [92, 93] and has long been used as an inflammatory biomarker [94]. Moreover, although still being debated, accumulating literature reports that CRP induces endothelial dysfunction [95]. Based on the overlapping biological functions and clinical implications, we hypothesized that CRP might be a ligand for LOX-1. Indeed, CRP bound to LOX-1-CHO cells but not wild type CHO cells [96]. Similar results were also reported from another group showing that CRP bound to LOX-1 expressed on bovine aortic endothelial cells [97]. A mutant LOX-1 protein, in which arginine residues at 208, 229, 231 and 248 were displaced with glutamine, still bound to CRP but not to oxLDL [97, 98], suggesting that the binding modes of CRP and oxLDL to LOX-1 are distinctive. The Kd for the binding of CRP to LOX-1 was 1.6 × 10−6 M [96], which is within the dynamic range of CRP in response to inflammatory stimuli (> 10−6 M). CRP indeed increased the expressions of IL-8, ICAM-1 and VCAM-1 mRNA through the binding to LOX-1 in transformed human aortic endothelial cells [97].
Although the ligand specificity of the scavenger receptor family considerably overlaps, we have recently found that CRP is recognized by LOX-1 and SR-A but not by other scavenger receptors such as CD36, SR-B1 and CD68 [99]. In vivo, exogenously added CRP enhances the vascular permeability through binding to LOX-1in rats, implicating the importance of LOX-1 as a CRP receptor in the cardiovascular system [96].Recent epidemiological studies have shown that even a slight increase in the serum concentration of CRP can be a major risk indicator for ischemic heart diseases [100–102]. Of particular interest, the JUPITER (Justification for the Use of Statins in Primary Prevention: an Intervention Trial Evaluating Rosuvastatin) study revealed that rosuvastatin significantly reduced the incidence of major cardiovascular events in people without hyperlipidemia but with elevated high sensitivity (hsCRP) [103]. Since statins reportedly reduce the expression of LOX-1 [104], inhibition of the interplay of CRP and LOX-1 may underlie the pleiotropic effects of statin.
Other LOX-1 ligands
Heat shock proteins (HSP) were originally described for their role as chaperones induced by temperature shock as well as various other kinds of stress and later works have identified certain types of HRPs including HSP90, HSP70, HSP60 and HSP27 that were released outside the cell [105]. LOX-1 recognizes HSP60 and HSP70 in dendritic cells, where it works as a receptor for antigen cross-presentation [106, 107]. Moreover, targeting a tumor antigen to LOX-1 elicits protective immune responses against antigen-expressing tumor cells in vivo [106].
Chronic hyperglycemia and increased oxidative stress generate advanced glycation end products (AGEs), non-enzymatic reaction products between reducing sugars and amine residues on protein, lipoproteins and nucleic acids [108]. The pathological roles of AGEs in diabetic nephropathy have been well elucidated but AGEs also seem to play multiple roles in the development of atherosclerosis such as endothelial dysfunction and inhibition of hepatic LDL clearance. The receptor for AGE (RAGE) is the most studied receptor for AGEs. Jono et al. have demonstrated that LOX-1 functions as a receptor for AGEs and a substantial part of the specific binding of AGE-albumin on bovine aortic endothelial cells is mediated by LOX-1 [109]. VLDL/LDL collected from diabetic rats contains a larger amount of LOX-1 ligand activity compared to that from normoglycemic rats and AGE-albumin increases LOX-1 expression in bovine aortic endothelial cells, suggesting that LOX-1 also plays a role in AGE-mediated endothelial dysfunction [50].
Uchida’s group has recently identified that lipid peroxidation products, trans-2-nonenal and 4-oxo-2(E)-nonenal, covalently bind to protein in vivo and that the modified proteins induce proinflammatory events via LOX-1 in human macrophages [110, 111].
Pathological roles of LOX-1; from endothelial dysfunction to restenosis
Initiation of atherosclerosis
Immunohistochemical studies and gain-of-function and loss-of-function approaches have provided compelling evidence that LOX-1 is involved in the development of atherosclerosis in vivo. LOX-1 is predominantly expressed in the endothelial cells and to a lesser degree in infiltrated macrophages of early atherosclerotic lesions in WHHL rabbits. Importantly, the expression is higher even in non-lesion areas of the rabbits compared to their wild type counterpart [112], suggesting that LOX-1 is involved in the initiation of atherosclerosis. Immunohistochemical studies also support the involvement of LOX-1 in human. LOX-1 expression is highly expressed on luminal endothelial cells, especially in the early stage of atherosclerosis and macrophages and smooth muscle cells accumulated into the intima [113]. Evidence is also available showing that LOX-1 is functionally involved in the initiation of atherosclerosis. Mice overexpressing LOX-1 (LOX-1 TG) fed high fat diet display impaired endothelium-dependent relaxation in mesenteric arteries at a time when significant atherosclerotic lesions are absent [114]. Interestingly, we have recently found that oxLDL increases vascular permeability via LOX-1 and thereby enhances lipid retention under subendothelial space in hypertensive rats [48], which seems to provide a novel link between the oxidative modification hypothesis and the response-to-retention hypothesis.
Genetically-modified mice have also pointed out the roles of LOX-1in the long-term progression of atherosclerosis. LOX-1 TG mice crossed with apoE KO mice (LOX-1TG/apoE KO) on high fat diet display pronounced lipid accumulation, oxidative stress and increased infiltration of macrophages in the heart and vessels [115]. In LOX-1 KO mice crossed with LDL receptor KO mice (LOX-1/LDLR KO), the formation of atherosclerotic lesions is significantly reduced compared to LDLR KO mice with a comparable serum LDL level [18]. Ectopic expression of LOX-1 in the liver of apoE KO mice ameliorates the development of atherosclerotic lesions with a reduction in plasma LAB [116]. This study is particularly important, indicating that 1) proatherogenic LAB is present in vivo, 2) LAB/LOX-1 axis promotes atherosclerosis in vivo and 3) eliminating the circulating LAB is sufficient for preventing atherosclerosis.
Advanced atherosclerosis
The disruption of unstable atherosclerotic plaques and subsequent formation of occlusive thrombi are currently considered as the primary cause of acute coronary syndrome. Apoptotic death of smooth muscle cells in the fibrous cap of the atherosclerotic plaques in addition to the degradation of extracellular matrix proteins by matrix metalloproteinases (MMPs)are considered to be involved in plaque rupture [117]. Circumstantial evidence support that LOX-1 regulates atherosclerotic plaque stability. Thrombosis accompanied in atheroma collected from patients with unstable angina is clearly stained with anti-LOX-1 antibody [25]. Saji’s group has reported that LOX-1 expression is intensive in atherosclerotic plaque with thinner fibromuscular cap and macrophage-rich lipid core area in WHHL rabbits with advanced atherosclerotic plaques [49, 118]. In human advanced atherosclerotic plaques, macrophages and smooth muscle cells in the intima dominantly express LOX-1, which is co-localized with Bax, a proapoptoic factor [20, 113].
Role of LOX-1 after the cardiovascular events
Myocardial reperfusion by thrombolysis or primary angioplasty has been widely performed for the management of acute myocardial infarction. Although early reperfusion can salvage jeopardized myocardium, reperfusion itself causes myocardial cell injury [119]. Moreover coronary artery restenosis after angioplasty occurs in ~30% of all patients [120, 121]. Interestingly, LOX-1 also seems to play a significant role in a series of events after myocardial infarction. We observed that the expression of LOX-1 was significantly increased in medial to intimal smooth muscle cells after balloon injury of the carotid artery in rats. An anti-LOX-1 antibody administered after the injury suppressed intimal hyperplasia, oxidative stress and leukocyte infiltration [122].Similar results were obtained using a gene silencer targeting LOX-1 [123]. In ischemic myocardium, the basal expression of LOX-1 is low, however, highly induced following reperfusion and the administration of anti-LOX-1 antibody halved myocardial infarction size, adhesion molecule expression and leukocyte recruitment in rats [51, 52].
Clinical significance of LOX-1 in cardiovascular diseases
Genetics
Several groups have looked at single nucleotide polymorphisms (SNPs) in the human LOX-1 gene to determine whether they are associated with cardiovascular diseases. Tatsuguchi et al. have identified an SNP, G501C (K167N), a missense mutation of LOX-1 protein [124]. Discordant results have been reported on the association of the SNP and cardiovascular risk [124–128]. Biochemically, the binding and uptake of oxLDL were lower in cells expressing LOX-1 with K167N mutation than native LOX-1 [129, 130], suggesting that LOX-1 with K167N mutation may be protective for atherogenesis.
Mango et al. have examined 253 individuals (150 individuals with acute myocardial infarction and 103 healthy controls) and identified 7 SNPs within intron 4, 5, exon 5 and the 3′-untranslated region (UTR) [125]. Individuals having a C-to-T change in 3′-UTR exhibit a significant association with acute myocardial infarction with an odds ratio of 3.74. They have also demonstrated that the SNP generated a new splicing isoform of LOX-1 that lacks exon 5 (LOXIN) and that LOXIN have a protective role in LOX-1 mediated apoptosis [131].
Soluble LOX-1 (sLOX-1)
A number of membrane proteins are subjected to proteolysis at the membrane-proximal site of the extracellular domain. Elevated levels of soluble forms of membrane proteins in circulating blood have been suggested to reflect increased expression of the protein and disease states [132–135]. LOX-1 is also cleaved and released as a soluble form (sLOX-1) into culture media following treatment with TNFα and IL-18 and inhibition of ADAM10, a member of a disintegrin and metalloproteinase (ADAM) family, suppressed the shedding [136, 137]. Hayashida et al. have developed a sandwich ELISA to measure sLOX-1 using two different rabbit antibodies for human LOX-1 and have demonstrated that serum levels ofsLOX-1are prognostic biomarkers of early acute coronary syndromes [138–140]. Compared to troponin T and hsCRP, an established diagnostic biomarker and a predictor for acute coronary syndrome, respectively, sLOX-1 seems to reflect the vulnerable atherosclerotic plaque better than the two. They speculated that LOX-1, highly expressed in macrophages and smooth muscle found in rupture-prone plaque, was cleaved under the condition in which protease activity is elevated.
LOX index as a predictive marker for stroke and coronary heart disease
We measured LAB of more than 2,000 healthy subjects in a community-based cohort study [141].During the 11-year follow-up period, the hazard ratio (HR) of ischemic stroke was highest in the highest LAB quartile with statistical significance. In this study, we also measured sLOX-1 by sandwich ELISA using mouse monoclonal anti-human LOX-1 antibody and chicken monoclonal anti-human LOX-1 antibody and calculated a parameter “LOX Index”, a multiplication of LAB and sLOX-1, which we assumed reflects the biological activity of LOX-1 ligands. The results demonstrated that LOX Index is a novel predictive marker for particularly ischemic stroke, and to a lesser extent coronary heart disease. The multivariable-adjusted HR for ischemic stroke from second to the top quartile of LOX Index was constantly 3-fold higher than that of the bottom quartile after multivariable adjustment. An association of LOX-1 with ischemic stroke has been also reported in experimental animals [142].
It is known that cholesterol is a strong risk factor for ischemic heart disease but not for ischemic stroke although atherothrombosis is a common underlying event [143–147]. In accordance to this, in another baseline survey using the same cohort as our report [141], there was no association between LDL cholesterol or non-HDL cholesterol concentrations and the incidence of ischemic stroke [148]. The paradox of cholesterol and stroke is also seen in the effect of statins on stroke incidence; the greater the reduction of LDL with statin treatment, the lower the risk of stroke despite the absence of association of baseline LDL and stroke incidents [149]. In hypercholesteromic patients, baseline LAB levels did not correlate with LDL cholesterol and was significantly lowered by pitavastatin [150]. In WHHL rabbits, LAB as well as the expression of LOX-1 are also reduced by the treatment of statin [60, 77]. Thus, stroke morbidity and the statins’ benefit might be explained in the context of LOX-1.
Therapeutic potential of LOX-1 inhibition for cardiovascular diseases
A number of animal and human studies implicate that LOX-1 is an attractive therapeutic target for human cardiovascular diseases. LOX-1 is a unique membrane receptor in terms that it seems to elicit biological functions via diverse mechanisms; LOX-1 uptakes oxLDL as an endocytotic receptor, tethers leukocytes and platelets as an adhesion molecule, and immediately generates ROS through the activation of intercellular molecules. Moreover, LOX-1 recognizes diverse ligands with no structural similarities and importantly, many of the ligands are also implicated in cardiovascular diseases. To date, we have developed a mouse monoclonal antibody developed for LOX-1, which inhibits ligand binding and/or LOX-1 activation with modified lipoproteins, cellular ligands and proteins in vitro [1, 16, 17, 80, 84–86, 89, 96, 151, 152]. In vivo, the antibody suppressed arterial lipid accumulation [48], neointimal hyperplasia in response to arterial injury [122], CRP-induced vascular hyperpermeability [96], neutrophil invasion after lipopolysaccharide challenge in rats [90] and myocardial ischemia-reperfusion injury [51]. Human monoclonal antibody for human LOX-1 has also been developed using Xenomouse and it inhibits oxLDL-induced ROS formation, RhoA/Rac1 activation and MCP-1 expression [19, 89, 153], suggesting that antibody therapy is feasible. LOX-1 inhibition with small-molecule compounds (drug-like compound) is more useful in terms of oral availability and medical costs but may be challenging since LOX-1 seems somewhat different from “druggable” targets such as G protein-coupled receptors, channels, enzymes and proteases [154]. However, several small-molecule inhibitors for SR-A and SR-B1 have been identified using lipoprotein as a ligand [155, 156]. Moreover, we have recently identified procyanidin, a polyphenol constituent abundant in red wine and apples which potently inhibits oxLDL binding to LOX-1 and lipid accumulation in rats [157], further implicating that the inhibition of oxLDL to LOX-1 with small molecules might be feasible. Disruption of CRP-LOX-1 interaction is also an attractive target, although it would be much more challenging since inhibition of protein-protein interaction with small-molecule compounds is considerably difficult [158–160].Further studies are necessary to investigate the binding modes of each ligand to LOX-1, and which of the ligand interactions underlies the specific disease phenotypes.
Concluding remarks
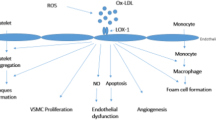
LOX-1 was originally discovered as a receptor for oxLDL that mediates endothelial dysfunction. The roles of LOX-1 in the initiation and the development of atherosclerosis have been almost established. The underlying mechanisms seem far more complex than originally thought as LOX-1 mediates the foam cell formation, proliferation and apoptosis of smooth muscle cells, platelet aggregation and cell adhesion. The recognition of diverse ligands and inducible expression pattern of LOX-1 in different types of cells are likely to explain the participation of LOX-1 in multiple stages of atherosclerosis from endothelial dysfunction to restenosis. Furthermore, it is intriguing that the major three hypotheses of atherogenesis could be explained by the versatile functions of LOX-1 (Fig. 2).

LOX-1 and the hypotheses of atherogenesis
Hyperlipidemia, diabetes and hypertension, referred to as lifestyle-related diseases, are leading causes for atherosclerosis and subsequent events such as coronary heart disease and cerebrovascular disease. In addition to these factors, other factors such as aging, gender and cigarette smoking further increase the risk of the cardiovascular diseases. LOX-1 originally may have worked as a host-defense molecule during the evolution of recognizing bacteria and other organisms, binding them and excluding them, as in the case of dectin-1. In the current lifestyle and environment of developed countries, however, excess amounts of waste materials are generated by the metabolism of excess amounts of nutrients, some of which behave like the organisms which must be excluded. LOX-1 is, then, activated to exclude such pseudo-organisms, e.g. oxLDL. This may induce a dysregulated host-defense signal, resulting in chronic inflammation leading to the promotion of lifestyle-related disease. We believe that further understanding of the versatile functions of LOX-1 would help establish a novel diagnostic method and therapeutic strategy for the treatment and/or prevention of cardiovascular diseases.
References
Sawamura T, Kume N, Aoyama T, Moriwaki H, Hoshikawa H, Aiba Y, et al. An endothelial receptor for oxidized low-density lipoprotein. Nature. 1997;386:73–7.
Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–9.
Ross R. Atherosclerosis—an inflammatory disease. New Engl J of Med. 1999;340:115–26.
Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15:551–61.
Williams KJ, Tabas I. The response-to-retention hypothesis of atherogenesis reinforced. Curr Opin Lipidol. 1998;9:471–4.
Krieger M, Acton S, Ashkenas J, Pearson A, Penman M, Resnick D. Molecular flypaper, host defense, and atherosclerosis. Structure, binding properties, and functions of macrophage scavenger receptors. J of. Biol Chem. 1993;268:4569–72.
Chen M, Narumiya S, Masaki T, Sawamura T. Conserved C-terminal residues within the lectin-like domain of LOX-1 are essential for oxidized low-density-lipoprotein binding. Biochem J. 2001;355:289–96.
Murphy JE, Tedbury PR, Homer-Vanniasinkam S, Walker JH, Ponnambalam S. Biochemistry and cell biology of mammalian scavenger receptors. Atherosclerosis. 2005;182:1–15.
Aoyama T, Sawamura T, Furutani Y, Matsuoka R, Yoshida MC, Fujiwara H, et al. Structure and chromosomal assignment of the human lectin-like oxidized low-density-lipoprotein receptor-1 (LOX-1) gene. Biochem J. 1999;339:177–84.
Yamanaka S, Zhang XY, Miura K, Kim S, Iwao H. The human gene encoding the lectin-type oxidized LDL receptor (OLR1) is a novel member of the natural killer gene complex with a unique expression profile. Genomics. 1998;54:191–9.
Sobanov Y, Bernreiter A, Derdak S, Mechtcheriakova D, Schweighofer B, Düchler M, et al. A novel cluster of lectin-like receptor genes expressed in monocytic, dendritic and endothelial cells maps close to the NK receptor genes in the human NK gene complex. Eur J Immunol. 2001;31:3493–503.
Brown GD. Dectin-1: a signalling non-TLR pattern-recognition receptor. Nat Rev Immunol. 2006;6:33–43.
Li D, Mehta JL. Upregulation of endothelial receptor for oxidized LDL (LOX-1) by oxidized LDL and implications in apoptosis of human coronary artery endothelial cells: evidence from use of antisense LOX-1 mRNA and chemical inhibitors. Arterioscler Thromb Vasc Biol. 2000;20:1116–22.
Li DY, Chen HJ, Staples ED, Ozaki K, Annex B, Singh BK, et al. Oxidized low-density lipoprotein receptor LOX-1 and apoptosis in human atherosclerotic lesions. J Cardiovasc Pharmacol Ther. 2002;7:147–53.
Li D, Liu L, Chen H, Sawamura T, Mehta JL. LOX-1, an oxidized LDL endothelial receptor, induces CD40/CD40L signaling in human coronary artery endothelial cells. Arterioscler Thromb Vasc Biol. 2003;23:816–21.
Cominacini L, Pasini AF, Garbin U, Davoli A, Tosetti ML, Campagnola M, et al. Oxidized low density lipoprotein (ox-LDL) binding to ox-LDL receptor-1 in endothelial cells induces the activation of NF-kappaB through an increased production of intracellular reactive oxygen species. J Biol Chem. 2000;275:12633–8.
Cominacini L, Rigoni A, Pasini AF, Garbin U, Davoli A, Campagnola M, et al. The binding of oxidized low density lipoprotein (ox-LDL) to ox-LDL receptor-1 reduces the intracellular concentration of nitric oxide in endothelial cells through an increased production of superoxide. J Biol Chem. 2001;276:13750–5.
Mehta JL, Sanada N, Hu CP, Chen J, Dandapat A, Sugawara F, et al. Deletion of LOX-1 reduces atherogenesis in LDLR knockout mice fed high cholesterol diet. Circ Res. 2007;100:1634–42.
Sugimoto K, Ishibashi T, Sawamura T, Inoue N, Kamioka M, Uekita H, et al. LOX-1-MT1-MMP axis is crucial for RhoA and Rac1 activation induced by oxidized low-density lipoprotein in endothelial cells. Cardiovasc Res. 2009;84:127–36.
Kataoka H, Kume N, Miyamoto S, Minami M, Morimoto M, Hayashida K, et al. Oxidized LDL modulates Bax/Bcl-2 through the lectinlike Ox-LDL receptor-1 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2001;21:955–60.
Eto H, Miyata M, Kume N, Minami M, Itabe H, Orihara K, et al. Expression of lectin-like oxidized LDL receptor-1 in smooth muscle cells after vascular injury. Biochem Biophys Res Commun. 2006;341:591–8.
Yoshida H, Kondratenko N, Green S, Steinberg D, Quehenberger O. Identification of the lectin-like receptor for oxidized low-density lipoprotein in human macrophages and its potential role as a scavenger receptor. Biochem J. 1998;334:9–13.
Schaeffer DF, Riazy M, Parhar KS, Chen JH, Duronio V, Sawamura T et al. LOX-1 augments oxLDL uptake by lysophosphatidylcholine-stimulated murine macrophages but is not required for oxLDL clearance from plasma. J Lipid Res. 2009;1676–84.
Ishiyama J, Taguchi R, Yamamoto A, Murakami K. Palmitic acid enhances lectin-like oxidized LDL receptor (LOX-1) expression and promotes uptake of oxidized LDL in macrophage cells. Atherosclerosis. 2010;209:118–24.
Chen M, Kakutani M, Naruko T, Ueda M, Narumiya S, Masaki T, et al. Activation-dependent surface expression of LOX-1 in human platelets. Biochem Biophys Res Commun. 2001;282:153–8.
Marwali MR, Hu CP, Mohandas B, Dandapat A, Deonikar P, Chen J, et al. Modulation of ADP-induced platelet activation by aspirin and pravastatin: role of lectin-like oxidized low-density lipoprotein receptor-1, nitric oxide, oxidative stress, and inside-out integrin signaling. J Pharmacol Exp Ther. 2007;322:1324–32.
Kume N, Murase T, Moriwaki H, Aoyama T, Sawamura T, Masaki T, et al. Inducible expression of lectin-like oxidized LDL receptor-1 in vascular endothelial cells. Circ Res. 1998;83:322–7.
Nagase M, Abe J, Takahashi K, Ando J, Hirose S, Fujita T. Genomic organization and regulation of expression of the lectin-like oxidized low-density lipoprotein receptor (LOX-1) gene. J Biol Chem. 1998;273:33702–7.
Aoyama T, Fujiwara H, Masaki T, Sawamura T. Induction of lectin-like oxidized LDL receptor by oxidized LDL and lysophosphatidylcholine in cultured endothelial cells. J Mol Cell Cardiol. 1999;31:2101–14.
Aoyama T, Chen M, Fujiwara H, Masaki T, Sawamura T. LOX-1 mediates lysophosphatidylcholine-induced oxidized LDL uptake in smooth muscle cells. FEBS Lett. 2000;467:217–20.
Minami M, Kume N, Kataoka H, Morimoto M, Hayashida K, Sawamura T, et al. Transforming growth factor-[beta]1 increases the expression of lectin-like oxidized low-density lipoprotein receptor-1. Biochem Biophys Res Comm. 2000;272:357–61.
Iwashima Y, Eto M, Hata A, Kaku K, Horiuchi S, Ushikubi F, et al. Advanced glycation end products-induced gene expression of scavenger receptors in cultured human monocyte-derived macrophages. Biochem Biophys Res Commun. 2000;277:368–80.
Nagase M, Ando K, Nagase T, Kaname S, Sawamura T, Fujita T. Redox-sensitive regulation of lox-1 gene expression in vascular endothelium. Biochem Biophys Res Commun. 2001;281:720–5.
Halvorsen B, Staff AC, Henriksen T, Sawamura T, Ranheim T. 8-iso-prostaglandin F(2alpha) increases expression of LOX-1 in JAR cells. Hypertension. 2001;37:1184–90.
Chen H, Li D, Saldeen T, Mehta JL. Transforming growth factor-beta(1) modulates oxidatively modified LDL-induced expression of adhesion molecules: role of LOX-1. Circ Res. 2001;89:1155–60.
Mehta JL, Li DY. Identification and autoregulation of receptor for OX-LDL in cultured human coronary artery endothelial cells. Biochem Biophys Res Commun. 1998;248:511–4.
Murase T, Kume N, Korenaga R, Ando J, Sawamura T, Masaki T, et al. Fluid shear stress transcriptionally induces lectin-like oxidized LDL receptor-1 in vascular endothelial cells. Circ Res. 1998;83:328–33.
Li DY, Zhang YC, Philips MI, Sawamura T, Mehta JL. Upregulation of endothelial receptor for oxidized low-density lipoprotein (LOX-1) in cultured human coronary artery endothelial cells by angiotensin II type 1 receptor activation. Circ Res. 1999;84:1043–9.
Morawietz H, Rueckschloss U, Niemann B, Duerrschmidt N, Galle J, Hakim K, et al. Angiotensin II induces LOX-1, the human endothelial receptor for oxidized low-density lipoprotein. Circulation. 1999;100:899–902.
Morawietz H, Duerrschmidt N, Niemann B, Galle J, Sawamura T, Holtz J. Induction of the oxLDL receptor LOX-1 by endothelin-1 in human endothelial cells. Biochem Biophys Res Commun. 2001;284:961–5.
Li L, Sawamura T, Renier G. Glucose enhances endothelial LOX-1 expression: role for LOX-1 in glucose-induced human monocyte adhesion to endothelium. Diabetes. 2003;52:1843–50.
Li L, Sawamura T, Renier G. Glucose enhances human macrophage LOX-1 expression: role for LOX-1 in glucose-induced macrophage foam cell formation. Circ Res. 2004;94:892–901.
Maingrette F, Renier G. Linoleic acid increases lectin-like oxidized LDL receptor-1 (LOX-1) expression in human aortic endothelial cells. Diabetes. 2005;54:1506–13.
Sakurai K, Cominacini L, Garbin U, Fratta Pasini A, Sasaki N, Takuwa Y, et al. Induction of endothelin-1 production in endothelial cells via co-operative action between CD40 and lectin-like oxidized LDL receptor (LOX-1). J Cardiovasc Pharmacol. 2004;44 Suppl 1:S173–80.
Nagase M, Hirose S, Sawamura T, Masaki T, Fujita T. Enhanced expression of endothelial oxidized low-density lipoprotein receptor (LOX-1) in hypertensive rats. Biochem Biophys Res Commun. 1997;237:496–8.
Nagase M, Hirose S, Fujita T. Unique repetitive sequence and unexpected regulation of expression of rat endothelial receptor for oxidized low-density lipoprotein (LOX-1). Biochem J. 1998;330:1417–22.
Nagase M, Kaname S, Nagase T, Wang G, Ando K, Sawamura T, et al. Expression of LOX-1, an oxidized low-density lipoprotein receptor, in experimental hypertensive glomerulosclerosis. J Am Soc Nephrol. 2000;11:1826–36.
Nakano A, Inoue N, Sato Y, Nishimichi N, Takikawa K, Fujita Y, et al. LOX-1 mediates vascular lipid retention under hypertensive state. J Hypertension. 2010;28:1273–80.
Kuge Y, Kume N, Ishino S, Takai N, Ogawa Y, Mukai T, et al. Prominent lectin-like oxidized low density lipoprotein (LDL) receptor-1 (LOX-1) expression in atherosclerotic lesions is associated with tissue factor expression and apoptosis in hypercholesterolemic rabbits. Biol Pharm Bull. 2008;31:1475–82.
Chen M, Nagase M, Fujita T, Narumiya S, Masaki T, Sawamura T. Diabetes enhances lectin-like oxidized LDL receptor-1 (LOX-1) expression in the vascular endothelium: possible role of LOX-1 ligand and AGE. Biochem Biophys Res Commun. 2001;287:962–8.
Li D, Williams V, Liu L, Chen H, Sawamura T, Antakli T, et al. LOX-1 inhibition in myocardial ischemia-reperfusion injury: modulation of MMP-1 and inflammation. Am J Physiol Heart Circ Physiol. 2002;283:H1795–801.
Kataoka K, Hasegawa K, Sawamura T, Fujita M, Yanazume T, Iwai-Kanai E, et al. LOX-1 pathway affects the extent of myocardial ischemia-reperfusion injury. Biochem Biophys Res Commun. 2003;300:656–60.
Li D, Williams V, Liu L, Chen H, Sawamura T, Romeo F, et al. Expression of lectin-like oxidized low-density lipoprotein receptors during ischemia-reperfusion and its role in determination of apoptosis and left ventricular dysfunction. J Am Coll Cardiol. 2003;41:1048–55.
Kosaka H, Yoneyama H, Zhang L, Fujii S, Yamamoto A, Igarashi J. Induction of LOX-1 and iNOS expressions by ischemia-reperfusion of rat kidney and the opposing effect of L-arginine. FASEB J. 2003;17:636–43.
Takanabe-Mori R, Ono K, Sowa N, Wada H, Takaya T, Horie T, et al. Lectin-like oxidized low-density lipoprotein receptor-1 is required for the adipose tissue expression of proinflammatory cytokines in high-fat diet-induced obese mice. Biochem Biophys Res Commun. 2010;398:576–80.
Fantuzzi G, Mazzone T. Adipose tissue and atherosclerosis: exploring the connection. Arterioscler Thromb Vasc Biol. 2007;27:996–1003.
Mehta JL, Hu B, Chen J, Li D. Pioglitazone inhibits LOX-1 expression in human coronary artery endothelial cells by reducing intracellular superoxide radical generation. Arterioscler Thromb Vasc Biol. 2003;23:2203–8.
Mehta JL, Chen J, Yu F, Li DY. Aspirin inhibits ox-LDL-mediated LOX-1 expression and metalloproteinase-1 in human coronary endothelial cells. Cardiovasc Res. 2004;64:243–9.
Chen J, Li D, Schaefer R, Mehta JL. Cross-talk between dyslipidemia and renin-angiotensin system and the role of LOX-1 and MAPK in atherogenesis studies with the combined use of rosuvastatin and candesartan. Atherosclerosis. 2006;184:295–301.
Hofnagel O, Luechtenborg B, Eschert H, Weissen-Plenz G, Severs NJ, Robenek H. Pravastatin inhibits expression of lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) in Watanabe heritable hyperlipidemic rabbits: a new pleiotropic effect of statins. Arterioscler Thromb Vasc Biol. 2006;26:604–10.
Kobayashi N, Honda T, Yoshida K, Nakano S, Ohno T, Tsubokou Y, et al. Critical role of bradykinin-eNOS and oxidative stress-LOX-1 pathway in cardiovascular remodeling under chronic angiotensin-converting enzyme inhibition. Atherosclerosis. 2006;187:92–100.
Takayama M, Matsubara M, Arakawa E, Takada C, Ina Y, Hasegawa K, et al. Comparison of the antiatherosclerotic effects of dihydropyridine calcium channel blocker and HMG-CoA reductase inhibitor on hypercholesterolemic rabbits. Vascul Pharmacol. 2007;46:302–8.
Ouslimani N, Mahrouf M, Peynet J, Bonnefont-Rousselot D, Cosson C, Legrand A, et al. Metformin reduces endothelial cell expression of both the receptor for advanced glycation end products and lectin-like oxidized receptor 1. Metabolism. 2007;56:308–13.
Li L, Renier G. The oral anti-diabetic agent, gliclazide, inhibits oxidized LDL-mediated LOX-1 expression, metalloproteinase-9 secretion and apoptosis in human aortic endothelial cells. Atherosclerosis. 2009;204:40–6.
Chui PC, Guan HP, Lehrke M, Lazar MA. PPARgamma regulates adipocyte cholesterol metabolism via oxidized LDL receptor 1. J Clin Invest. 2005;115:2244–56.
Oka K, Sawamura T, Kikuta K, Itokawa S, Kume N, Kita T, et al. Lectin-like oxidized low-density lipoprotein receptor 1 mediates phagocytosis of aged/apoptotic cells in endothelial cells. Proc Natl Acad Sci U S A. 1998;95:9535–40.
Marsche G, Levak-Frank S, Quehenberger O, Heller R, Sattler W, Malle E. Identification of the human analog of SR-BI and LOX-1 as receptors for hypochlorite-modified high density lipoprotein on human umbilical venous endothelial cells. FASEB J. 2001;15:1095–7.
Shi X, Niimi S, Ohtani T, Machida S. Characterization of residues and sequences of the carbohydrate recognition domain required for cell surface localization and ligand binding of human lectin-like oxidized LDL receptor. J Cell Science. 2001;114:1273–82.
Burkitt MJ. A critical overview of the chemistry of copper-dependent low density lipoprotein oxidation: roles of lipid hydroperoxides, [alpha]-tocopherol, thiols, and ceruloplasmin. Arch Biochem Biophysics. 2001;394:117–35.
Ylä-Herttuala S, Palinski W, Rosenfeld ME, Parthasarathy S, Carew TE, Butler S, et al. Evidence for the presence of oxidatively modified low density lipoprotein in atherosclerotic lesions of rabbit and man. J Clin Invest. 1989;84:1086–95.
Boyd HC, Gown AM, Wolfbauer G, Chait A. Direct evidence for a protein recognized by a monoclonal antibody against oxidatively modified LDL in atherosclerotic lesions from a Watanabe heritable hyperlipidemic rabbit. Am J Pathol. 1989;135:815–25.
Itabe H, Takeshima E, Iwasaki H, Kimura J, Yoshida Y, Imanaka T, et al. A monoclonal antibody against oxidized lipoprotein recognizes foam cells in atherosclerotic lesions. Complex formation of oxidized phosphatidylcholines and polypeptides. J Biol Chem. 1994;269:15274–9.
Ehara S, Ueda M, Naruko T, Haze K, Itoh A, Otsuka M, et al. Elevated levels of oxidized low density lipoprotein show a positive relationship with the severity of acute coronary syndromes. Circulation. 2001;103:1955–60.
Nishi K, Itabe H, Uno M, Kitazato KT, Horiguchi H, Shinno K, et al. Oxidized LDL in carotid plaques and plasma associates With plaque instability. Arterioscler Thromb Vasc Biol. 2002;22:1649–54.
Palinski W, Hörkkö S, Miller E, Steinbrecher UP, Powell HC, Curtiss LK, et al. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J Clin Invest. 1996;98:800–14.
Kakutani M, Ueda M, Naruko T, Masaki T, Sawamura T. Accumulation of LOX-1 ligand in plasma and atherosclerotic lesions of Watanabe heritable hyperlipidemic rabbits: identification by a novel enzyme immunoassay. Biochem Biophys Res Commun. 2001;282:180–5.
Oka K, Yasuhara M, Suzumura K, Tanaka K, Sawamura T. Antioxidants suppress plasma levels of lectinlike oxidized low-density lipoprotein receptor-ligands and reduce atherosclerosis in watanabe heritable hyperlipidemic rabbits. J Cardiovasc Pharmacol. 2006;48:177–83.
Sato Y, Nishimichi N, Nakano A, Takikawa K, Inoue N, Matsuda H, et al. Determination of LOX-1-ligand activity in mouse plasma with a chicken monoclonal antibody for ApoB. Atherosclerosis. 2008;200:303–9.
Apostolov EO, Shah SV, Ray D, Basnakian AG. Scavenger receptors of endothelial cells mediate the uptake and cellular proatherogenic effects of carbamylated LDL. Arterioscler Thromb Vasc Biol. 2009;29:1622–30.
Lu J, Yang JH, Burns AR, Chen HH, Tang D, Walterscheid JP, et al. Mediation of electronegative low-density lipoprotein signaling by LOX-1: a possible mechanism of endothelial apoptosis. Circ Res. 2009;104:619–27.
Shin HK, Kim YK, Kim KY, Lee JH, Hong KW. Remnant lipoprotein particles induce apoptosis in endothelial cells by NAD(P)H oxidase-mediated production of superoxide and cytokines via lectin-like oxidized low-density lipoprotein receptor-1 activation: prevention by cilostazol. Circulation. 2004;109:1022–8.
Aramaki Y, Mitsuoka H, Toyohara M, Jinnai T, Kanatani K, Nakajima K, et al. Lectin-like oxidized LDL receptor-1 (LOX-1) acts as a receptor for remnant-like lipoprotein particles (RLPs) and mediates RLP-induced migration of vascular smooth muscle cells. Atherosclerosis. 2008;198:272–9.
Moriwaki H, Kume N, Sawamura T, Aoyama T, Hoshikawa H, Ochi H, et al. Ligand specificity of LOX-1, a novel endothelial receptor for oxidized low density lipoprotein. Arterioscler Thromb Vasc Biol. 1998;18:1541–7.
Kakutani M, Masaki T, Sawamura T. A platelet-endothelium interaction mediated by lectin-like oxidized low-density lipoprotein receptor-1. Proc Natl Acad Sci U S A. 2000;97:360–4.
Shimaoka T, Kume N, Minami M, Hayashida K, Sawamura T, Kita T, et al. LOX-1 supports adhesion of Gram-positive and Gram-negative bacteria. J Immunol. 2001;166:5108–14.
Cominacini L, Fratta Pasini A, Garbin U, Pastorino A, Rigoni A, Nava C, et al. The platelet-endothelium interaction mediated by lectin-like oxidized low-density lipoprotein receptor-1 reduces the intracellular concentration of nitric oxide in endothelial cells. J Am Coll Cardiol. 2003;41:499–507.
Murphy JE, Tacon D, Tedbury PR, Hadden JM, Knowling S, Sawamura T, et al. LOX-1 scavenger receptor mediates calcium-dependent recognition of phosphatidylserine and apoptotic cells. Biochem J. 2006;393:107–15.
Li L, Roumeliotis N, Sawamura T, Renier G. C-reactive protein enhances LOX-1 expression in human aortic endothelial cells: relevance of LOX-1 to C-reactive protein-induced endothelial dysfunction. Circ Res. 2004;95:877–83.
Sakamoto N, Ishibashi T, Sugimoto K, Sawamura T, Sakamoto T, Inoue N, et al. Role of LOX-1 in monocyte adhesion-triggered redox, Akt/eNOS and Ca2+ signaling pathways in endothelial cells. J Cell Physiol. 2009;220:706–15.
Honjo M, Nakamura K, Yamashiro K, Kiryu J, Tanihara H, McEvoy LM, et al. Lectin-like oxidized LDL receptor-1 is a cell-adhesion molecule involved in endotoxin-induced inflammation. Proc Natl Acad Sci U S A. 2003;100:1274–9.
Casciola-Rosen L, Rosen A, Petri M, Schlissel M. Surface blebs on apoptotic cells are sites of enhanced procoagulant activity: implications for coagulation events and antigenic spread in systemic lupus erythematosus. Proc Natl Acad Sci. 1996;93:1624–9.
Black S, Kushner I, Samols D. C-reactive Protein. J Biol Chem. 2004;279:48487–90.
Volanakis JE, Kaplan MH. Interaction of C-reactive protein complexes with the complement system. II. Consumption of guinea pig complement by CRP complexes: requirement for human C1q. J Immunol. 1974;113:9–17.
Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. New Engl J Med. 1999;340:448–54.
Verma S, Szmitko PE, Ridker PM. C-reactive protein comes of age. Nat Clin Pract Cardiovasc Med. 2005;2:29–36.
Fujita Y, Kakino A, Nishimichi N, Yamaguchi S, Sato Y, Machida S, et al. Oxidized LDL receptor LOX-1 binds to C-reactive protein and mediates its vascular effects. Clin Chem. 2009;55:285–94.
Shih HH, Zhang S, Cao W, Hahn A, Wang J, Paulsen JE, et al. CRP is a novel ligand for the oxidized LDL receptor LOX-1. Am J Physiol Heart Circ Physiol. 2009;296:H1643–50.
Ohki I, Ishigaki T, Oyama T, Matsunaga S, Xie Q, Ohnishi-Kameyama M, et al. Crystal structure of human lectin-like, oxidized low-density lipoprotein receptor 1 ligand binding domain and its ligand recognition mode to OxLDL. Structure. 2005;13:905–17.
Fujita Y, Kakino A, Harada-Shiba M, Sato Y, Otsui K, Yoshimoto R, et al. C-reactive protein uptake by macrophage cell line via class—A scavenger receptor. Clin Chem. 2010;56:478–81.
Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. New Engl J Med. 1997;336:973–9.
Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. New Engl J Med. 2000;342:836–43.
Ridker PM, Rifai N, Rose L, Buring JE, Cook NR. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. New Engl J Med. 2002;347:1557–65.
Ridker PM, Danielson E, Fonseca FAH, Genest J, Gotto AM, Kastelein JJP, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. New Engl J Med. 2008;359:2195–207.
Li D, Chen H, Romeo F, Sawamura T, Saldeen T, Mehta JL. Statins modulate oxidized low-density lipoprotein-mediated adhesion molecule expression in human coronary artery endothelial cells: role of LOX-1. J Pharmacol Exp Ther. 2002;302:601–5.
De Maio A. Extracellular heat shock proteins, cellular export vesicles, and the Stress Observation System: A form of communication during injury, infection, and cell damage. Cell Stress Chaperones. 2011;16:235–49.
Delneste Y, Magistrelli G, Gauchat J, Haeuw J, Aubry J, Nakamura K, et al. Involvement of LOX-1 in dendritic cell-mediated antigen cross-presentation. Immunity. 2002;17:353–62.
Xie J, Zhu H, Guo L, Ruan Y, Wang L, Sun L, et al. Lectin-like oxidized low-density lipoprotein receptor-1 delivers heat shock protein 60-fused antigen into the MHC class I presentation pathway. J Immunol. 2010;185:2306–13.
Goh S-Y, Cooper ME. The role of advanced glycation end products in progression and complications of diabetes. J Clin Endocrinol Metab. 2008;93:1143–52.
Jono T, Miyazaki A, Nagai R, Sawamura T, Kitamura T, Horiuchi S. Lectin-like oxidized low density lipoprotein receptor-1 (LOX-1) serves as an endothelial receptor for advanced glycation end products (AGE). FEBS Lett. 2002;511:170–4.
Ishino K, Wakita C, Shibata T, Toyokuni S, Machida S, Matsuda S, et al. Lipid peroxidation generates body odor component trans-2-nonenal covalently bound to protein in vivo. J Biol Chem. 2010;285:15302–13.
Shibata T, Shimozu Y, Wakita C, Shibata N, Kobayashi M, Machida S, et al. Lipid peroxidation modification of protein generates Ne-(4-oxononanoyl)lysine as a pro-inflammatory ligand. J Biol Chem. 2011;286:19943–57.
Chen M, Kakutani M, Minami M, Kataoka H, Kume N, Narumiya S, et al. Increased expression of lectin-like oxidized low density lipoprotein receptor-1 in initial atherosclerotic lesions of Watanabe heritable hyperlipidemic rabbits. Arterioscler Thromb Vasc Biol. 2000;20:1107–15.
Kataoka H, Kume N, Miyamoto S, Minami M, Moriwaki H, Murase T, et al. Expression of lectinlike oxidized low-density lipoprotein receptor-1 in human atherosclerotic lesions. Circulation. 1999;99:3110–7.
Eichhorn B, Muller G, Leuner A, Sawamura T, Ravens U, Morawietz H. Impaired vascular function in small resistance arteries of LOX-1 overexpressing mice on high-fat diet. Cardiovasc Res. 2009;82:493–502.
Inoue K, Arai Y, Kurihara H, Kita T, Sawamura T. Overexpression of lectin-like oxidized low-density lipoprotein receptor-1 induces intramyocardial vasculopathy in apolipoprotein E-null mice. Circ Res. 2005;97:176–84.
Ishigaki Y, Katagiri H, Gao J, Yamada T, Imai J, Uno K, et al. Impact of plasma oxidized low-density lipoprotein removal on atherosclerosis. Circulation. 2008;118:75–83.
Finn AV, Nakano M, Narula J, Kolodgie FD, Virmani R. Concept of vulnerable/unstable plaque. Arterioscler Thromb Vasc Biol. 2010;30:1282–92.
Ishino S, Mukai T, Kume N, Asano D, Ogawa M, Kuge Y, et al. Lectin-like oxidized LDL receptor-1 (LOX-1) expression is associated with atherosclerotic plaque instability–analysis in hypercholesterolemic rabbits. Atherosclerosis. 2007;195:48–56.
Hearse DJ, Bolli R. Reperfusion induced injury: manifestations, mechanisms, and clinical relevance. Cardiovasc Res. 1992;26:101–8.
Fischman DL, Leon MB, Baim DS, Schatz RA, Savage MP, Penn I, et al. A randomized comparison of coronary-stent placement and balloon angioplasty in the treatment of coronary artery disease. N Engl J Med. 1994;331:496–501.
Serruys PW, de Jaegere P, Kiemeneij F, Macaya C, Rutsch W, Heyndrickx G, et al. A comparison of balloon-expandable-stent implantation with balloon angioplasty in patients with coronary artery disease. N Engl J Med. 1994;331:489–95.
Hinagata J, Kakutani M, Fujii T, Naruko T, Inoue N, Fujita Y, et al. Oxidized LDL receptor LOX-1 is involved in neointimal hyperplasia after balloon arterial injury in a rat model. Cardiovasc Res. 2006;69:263–71.
Yao EH, Fukuda N, Ueno T, Matsuda H, Matsumoto K, Nagase H, et al. Novel gene silencer pyrrole-imidazole polyamide targeting lectin-like oxidized low-density lipoprotein receptor-1 attenuates restenosis of the artery after injury. Hypertension. 2008;52:86–92.
Tatsuguchi M, Furutani M, Hinagata J, Tanaka T, Furutani Y, Imamura S, et al. Oxidized LDL receptor gene (OLR1) is associated with the risk of myocardial infarction. Biochem Biophys Res Commun. 2003;303:247–50.
Mango R, Clementi F, Borgiani P, Forleo GB, Federici M, Contino G, et al. Association of single nucleotide polymorphisms in the oxidised LDL receptor 1 (OLR1) gene in patients with acute myocardial infarction. J Med Genet. 2003;40:933–6.
Ohmori R, Momiyama Y, Nagano M, Taniguchi H, Egashira T, Yonemura A, et al. An oxidized low-density lipoprotein receptor gene variant is inversely associated with the severity of coronary artery disease. Clin Cardiol. 2004;27:641–4.
Morgan TM, Krumholz HM, Lifton RP, Spertus JA. Nonvalidation of reported genetic risk factors for acute coronary syndrome in a large-scale replication study. JAMA. 2007;297:1551–61.
Knowles JW, Assimes TL, Boerwinkle E, Fortmann SP, Go A, Grove ML, et al. Failure to replicate an association of SNPs in the oxidized LDL receptor gene (OLR1) with CAD. BMC Med Genet. 2008;9:23.
Biocca S, Falconi M, Filesi I, Baldini F, Vecchione L, Mango R, et al. Functional analysis and molecular dynamics simulation of LOX-1 K167N polymorphism reveal alteration of receptor activity. PLoS One. 2009;4:e4648.
Biocca S, Filesi I, Mango R, Maggiore L, Baldini F, Vecchione L, et al. The splice variant LOXIN inhibits LOX-1 receptor function through hetero-oligomerization. J Mol Cell Cardiol. 2008;44:561–70.
Mango R, Biocca S, del Vecchio F, Clementi F, Sangiuolo F, Amati F, et al. In vivo and in vitro studies support that a new splicing isoform of OLR1 gene is protective against acute myocardial infarction. Circ Res. 2005;97:152–8.
Rose-John S, Heinrich PC. Soluble receptors for cytokines and growth factors: generation and biological function. Biochem J. 1994;300:281–90.
Newman W, Beall L, Carson C, Hunder G, Graben N, Randhawa Z, et al. Soluble E-selectin is found in supernatants of activated endothelial cells and is elevated in the serum of patients with septic shock. J Immun. 1993;150:644–54.
De Caterina R, Basta G, Lazzerini G, Dell’Omo G, Petrucci R, Morale M, et al. Soluble Vascular Cell Adhesion Molecule-1 as a Biohumoral Correlate of Atherosclerosis. Arterioscler Thromb Vasc Biol. 1997;17:2646–54.
Honda M, Yamamoto S, Cheng M, Yasukawa K, Suzuki H, Saito T, et al. Human soluble IL-6 receptor: its detection and enhanced release by HIV infection. J Immun. 1992;148:2175–80.
Murase T, Kume N, Kataoka H, Minami M, Sawamura T, Masaki T, et al. Identification of soluble forms of lectin-like oxidized LDL receptor-1. Arterioscler Thromb Vasc Biol. 2000;20:715–20.
Mitsuoka H, Kume N, Hayashida K, Inui-Hayashiada A, Aramaki Y, Toyohara M, et al. Interleukin 18 stimulates release of soluble lectin-like oxidized LDL receptor-1 (sLOX-1). Atherosclerosis. 2009;202:176–82.
Hayashida K, Kume N, Murase T, Minami M, Nakagawa D, Inada T, et al. Serum soluble lectin-like oxidized low-density lipoprotein receptor-1 levels are elevated in acute coronary syndrome: a novel marker for early diagnosis. Circulation. 2005;112:812–8.
Kume N, Mitsuoka H, Hayashida K, Tanaka M, Kominami G, Kita T. Soluble lectin-like oxidized LDL receptor-1 (sLOX-1) as a sensitive and specific biomarker for acute coronary syndrome–Comparison with other biomarkers. J Cardiol. 2010;56:159–65.
Kume N, Mitsuoka H, Hayashida K, Tanaka M, Kita T. Soluble lectin-like oxidized low-density lipoprotein receptor-1 predicts prognosis after acute coronary syndrome—a pilot study. Circ J. 2010;74:1399–404.
Inoue N, Okamura T, Kokubo Y, Fujita Y, Sato Y, Nakanishi M, et al. LOX index, a novel predictive biochemical marker for coronary heart disease and stroke. Clin Chem. 2010;56:550–8.
Hamakawa Y, Omori N, Ouchida M, Nagase M, Sato K, Nagano I, et al. Severity dependent up-regulations of LOX-1 and MCP-1 in early sclerotic changes of common carotid arteries in spontaneously hypertensive rats. Neurol Res. 2004;26:767–73.
Blood cholesterol and vascular mortality by age, sex, and blood pressure: a meta-analysis of individual data from 61 prospective studies with 55?000 vascular deaths. Lancet. 2007;370:1829–39.
Tanizaki Y, Kiyohara Y, Kato I, Iwamoto H, Nakayama K, Shinohara N, et al. Incidence and risk factors for subtypes of cerebral infarction in a general population: the Hisayama study. Stroke. 2000;31:2616–22.
Okamura T, Tanaka H, Miyamatsu N, Hayakawa T, Kadowaki T, Kita Y, et al. The relationship between serum total cholesterol and all-cause or cause-specific mortality in a 17.3-year study of a Japanese cohort. Atherosclerosis. 2007;190:216–23.
Cui R, Iso H, Toyoshima H, Date C, Yamamoto A, Kikuchi S, et al. Serum total cholesterol levels and risk of mortality from stroke and coronary heart disease in Japanese: The JACC study. Atherosclerosis. 2007;194:415–20.
Amarenco P, Steg PG. The paradox of cholesterol and stroke. Lancet. 2007;370:1803–4.
Okamura T, Kokubo Y, Watanabe M, Higashiyama A, Miyamoto Y, Yoshimasa Y, et al. Low-density lipoprotein cholesterol and non-high-density lipoprotein cholesterol and the incidence of cardiovascular disease in an urban Japanese cohort study: the Suita study. Atherosclerosis. 2009;203:587–92.
Amarenco P, Labreuche J, Lavallee P, Touboul P-J. Statins in stroke prevention and carotid atherosclerosis: systematic review and up-to-date meta-analysis. Stroke. 2004;35:2902–9.
Matsumoto T, Fujita M, Sawamura T, Kakino A, Sato Y, Fujita Y, et al. Pitavastatin reduces lectin-like oxidized low-density lipoprotein receptor-1 ligands in hypercholesterolemic humans. Lipids. 2010;45:329–35.
Tang D, Lu J, Walterscheid JP, Chen HH, Engler DA, Sawamura T, et al. Electronegative LDL circulating in smokers impairs endothelial progenitor cell differentiation by inhibiting Akt phosphorylation via LOX-1. J Lipid Res. 2008;49:33–47.
Kanata S, Akagi M, Nishimura S, Hayakawa S, Yoshida K, Sawamura T, et al. Oxidized LDL binding to LOX-1 upregulates VEGF expression in cultured bovine chondrocytes through activation of PPAR-gamma. Biochem Biophys Res Commun. 2006;348:1003–10.
Akagi M, Ueda A, Teramura T, Kanata S, Sawamura T, Hamanishi C. Oxidized LDL binding to LOX-1 enhances MCP-1 expression in cultured human articular chondrocytes. Osteoarthr Cartil. 2009;17:271–5.
Zheng CJ, Han LY, Yap CW, Ji ZL, Cao ZW, Chen YZ. Therapeutic targets: progress of their exploration and investigation of their characteristics. Pharmacol Rev. 2006;58:259–79.
Lysko PG, Weinstock J, Webb CL, Brawner ME, Elshourbagy NA. Identification of a small-molecule, nonpeptide macrophage scavenger receptor antagonist. J Pharmacol Exp Ther. 1999;289:1277–85.
Syder AJ, Lee H, Zeisel MB, Grove J, Soulier E, Macdonald J, et al. Small molecule scavenger receptor BI antagonists are potent HCV entry inhibitors. J Hepatol. 2011;54:48–55.
Nishizuka T, Fujita Y, Sato Y, Nakano A, Kakino A, Ohshima S, et al. Procyanidins are potent inhibitors of LOX-1: a new player in the French Paradox. Proc Jpn Acad Ser B Phys Biol Sci. 2011;87:104–13.
Whitty A, Kumaravel G. Between a rock and a hard place? Nat Chem Biol. 2006;2:112–8.
Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov. 2004;3:301–17.
Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–9.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Yoshimoto, R., Fujita, Y., Kakino, A. et al. The Discovery of LOX-1, its Ligands and Clinical Significance. Cardiovasc Drugs Ther 25, 379–391 (2011). https://doi.org/10.1007/s10557-011-6324-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-011-6324-6