
Abstract
Checkpoint inhibitors have recently gained FDA approval for the treatment of cisplatin-resistant recurrent and metastatic head and neck squamous cell carcinoma (HNSCC) by outperforming standard of care chemotherapy and inducing durable responses in a subset of patients. These monoclonal antibodies unleash the patient’s own immune system to target cancer cells. HNSCC is a good target for these agents as there is ample evidence of active immunosurveillance in the head and neck and a number of immune evasion mechanisms by which HNSCCs form progressive disease including via the PD-1/PD-L1 axis. As HNSCCs typically possess a moderately high mutation burden, they should express numerous mutation-derived antigen targets for immune detection. However, with response rates less than 20% in clinical trials, there is a need for biomarkers to screen patients as well as clinical trials evaluating novel combinations to improve outcomes. The aim of this review is to provide historical and mechanistic context for the use of checkpoint inhibitors in head and neck cancer and provide a perspective on the role of novel checkpoints, biomarkers, and combination therapies that are evolving in the near term for patients with HNSCC.
Similar content being viewed by others

1 Introduction
Despite advances in chemotherapeutics, targeted radiation, and surgical techniques, patients with advanced head and neck squamous cell carcinoma (HNSCC) still suffer poor outcomes (50% 5 year survival) [1]. Therefore, there is a need for novel treatment options for patients with advanced stage or recurrent/metastatic disease. Immunotherapies, which recruit the patient’s own immune system to combat tumors, have experienced a surge in interest and enthusiasm since the cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) blocking antibody, ipilimumab, was found to improve overall survival in patients with metastatic melanoma [2]. Over the last decade, there has been an exponential growth in efforts to characterize other checkpoint pathways, identify targets of response, discover other mechanisms of inducing tumor-specific immune responses, and to uncover biomarkers of response. Already, two clinical trials have demonstrated that checkpoint blockade with anti-PD-1 antibodies, nivolumab or pembrolizumab, are efficacious in recurrent/metastatic HNSCC and these results led to the Food and Drug Administration (FDA) approval in the USA [3, 4]. However, the majority of patients do not respond to single-agent checkpoint blockade and more clarity is needed to optimally integrate immunotherapeutics into the HNSCC treatment paradigm. In this review, we provide a bench-to-bedside, mechanistic rationale for checkpoint immunotherapeutics in HNSCC and look forward at the possible directions of this approach.
2 Historical context
2.1 Cancer immunoediting hypothesis
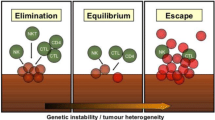
The foundation of modern immunotherapy approaches to head and neck malignancies dates back to William Coley’s successful treatment of a patient with a sarcoma of the head and neck using Coley’s toxins. Targeted approaches of HNSCCs with locoregional delivery of the pro-inflammatory cytokine IL-2 were attempted nearly 25 years ago [5,6,7]. The modern field of immunotherapies developed out of a convergence of two breakthroughs in cancer research. The first was the conceptualization of the “cancer immunoediting hypothesis” which articulated the role of the immune system in cancer development from initial elimination of highly immunogenic tumor cells to equilibrium and ultimately to an escape phase of progressive tumor growth [8,9,10]. Seminal studies, performed in mice, demonstrated that the immune system actively eliminates cancer cells that are immunogenic, forcing a selection pressure on the tumors and generating cancer cells that are less easily targeted and eliminated by the immune system. This hypothesis was validated by the finding that tumors derived in immunocompetent mice were less immunogenic than tumors of similar etiology derived in immunodeficient mice which did not face similar immunoediting [11,12,13,14]. These data extended the understanding of why immunosuppressed patients have a higher propensity to develop malignancies [15]. Implicit in these findings was the recognition that cancer cells may present antigens, mutated or modified proteins not expressed on normal tissue, that enable the immune system to selectively identify cancer cells as non-self [16, 17]. Early efforts to identify these tumor antigens in head and neck and other cancers focused mostly on tumor-associated antigens (TAAs), normal proteins overexpressed or modified on cancer cells, or cancer testis antigens (CTAs), proteins selectively expressed in germ cells that may also present in tumor cells [18,19,20]. We now know that tumor antigens generated as a result of nonsynonymous point mutations—termed neoantigens because they are uniquely expressed by tumor cells—may be a more appropriate target because of the lack of central tolerance [21].
2.2 Next-generation sequencing and a focus on neoantigens
The second breakthrough responsible for modern genomics-driven immunotherapies was the advent of next generation sequencing technologies that enabled the rapid and reliable detection of mutations within tumor cells. Pioneering work by Vogelstein and Allison used next-generation sequencing to establish a platform for high-throughput prediction of mutation-derived neoantigens [22]. Subsequent work by Robert Schreiber and colleagues used a similar approach based on cDNA capture-sequencing to identify and validate a neoantigen in a methylcholanthrene (MCA)-induced sarcoma cell line that was responsible for immune-mediated tumor rejection in immunocompetent mice [23]. This same group later demonstrated that neoantigen-specific CD8+ T cells are key mediators of checkpoint blockade-induced tumor rejection [24]. Next-generation sequencing technology has revolutionized the discovery of neoantigens within tumors and has opened the door for more personalized immunotherapeutic approaches such as mutation-guided vaccines and adoptive T cell therapies.
3 Basis for immunotherapy in HNSCC
3.1 Evidence of immunosurveillance in HNSCC
Head and neck squamous cell carcinoma is a logical target for immunotherapies. These tumors are characterized by a substantial immune infiltrate [25], frequent generation of neoantigens [21], and an array of mechanisms to enable immune evasion [26]. There is considerable evidence of active immunosurveillance in HNSCC. One particular study of patients who underwent bone marrow transplantation identified a 17.4-fold increased risk for oral cavity squamous cell carcinoma [27]. Another study reported a 22-fold increase in the risk of premalignant lip leukoplakia following renal transplantation (13% compared to 0.6%) [28]. Similar findings have been extensively reviewed elsewhere [29], demonstrating the critical role of the immune system in preventing clinically apparent head and neck malignancies.
3.2 Role of TIL in HNSCC
Infiltrating lymphocytes play a considerable role in the evolution of HNSCC tumors. A recent analysis of transcriptome data compiled by The Cancer Genome Atlas (TCGA) identified that HNSCC tumors contain high levels of immune infiltrates compared to other tumor types and that HPV-positive tumors were third only to lung adenocarcinoma and kidney clear cell carcinoma [25]. The limited literature on the prognostic significance of TIL in HNSCC mostly supports that lymphocyte infiltration is associated with better outcomes [29]. A study of 161 patients with a median follow up of 4 years found high CD8 expression on TILs to be an independent prognostic parameter of improved overall survival (OS) and local progression-free survival in a multivariate analysis although no staining was done for CD4 expression [30]. Another recent study of 278 patients found CD4 and CD8 expression associated with improved OS and relapse-free survival although only CD4 expression remained significant in multivariate analysis [31]. Looking specifically at markers of activation, one study of 64 patients with HNSCC identified the level of infiltrating PD-1-positive T cells to be significantly correlated with favorable clinical outcomes [32]. As PD-1 expression is a marker of antigen-experienced cells [33], this may indicate the importance of the presence of activated effector T cells rather than the total number of infiltrating lymphocytes. This is significant as the presence of neoantigen-specific T cells has been associated with responsiveness to checkpoint inhibitors [34,35,36].
3.3 Mutation load and neoantigens in HNSCC
Investigation of the frequency of mutations and predicted neoantigens and association with a clinical benefit was a logical extension of clinical trials with checkpoint inhibitors. Genomics-based discovery efforts in these clinical trials confirmed that a higher mutation burden was associated with a higher number of predicted neoantigens and improved responsiveness to checkpoint blockade [34,35,36]. Data from the TCGA and other datasets have demonstrated that HNSCCs generally possess a moderately high mutation rate [37] and are predicted to have an array of neoantigen targets [21]. A previous work to identify HNSCC antigens has been reviewed elsewhere [38] but to date only includes one mutation-derived neoantigen from the gene CASP8 [39]. Additionally, HNSCC is unique in that in addition to TAAs, CTAs, and mutation-derived neoantigens, they may also present viral antigens. HPV-associated oropharyngeal cancer cells may present viral components to the immune system offering a unique and potentially highly immunogenic target(s) for immune recognition and activation [40].
4 Immunological barriers in the HNSCC microenvironment
4.1 Immune evasion mechanisms
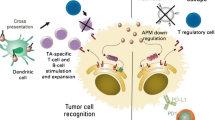
Despite the presence of infiltrating T cells and a potential plethora of antigen targets, HNSCCs evade the immune system via an equally diverse set of mechanisms. This can be separated into mechanisms of immune evasion intrinsic to the tumor cells and immunosuppressive forces in the microenvironment. Mechanisms of HNSCC immune evasion include downregulation of human leukocyte antigen (HLA) class I molecules [41,42,43,44,45], secretion of immunosuppressive and proapoptotic factors [46,47,48], and upregulation of inhibitory cell surface molecules [49,50,51,52]. In a recent study, head and neck cancer cells were shown to produce tumor-derived exosomes, small membrane-bound vesicles, containing an assortment of immunosuppressive molecules including inhibitory cytokines, death receptor ligands such as FASL, and checkpoint receptor ligands such as PD-L1 [53]. Additionally, tumor cells may upregulate enzymes such as indoleamine 2,3 dioxygenase (IDO1) depleting critical nutrients from the microenvironment and inducing T cell dysfunction [54].
4.2 Permissive microenvironmental factors
Beyond tumor-specific factors, the immune microenvironment of HNSCC is frequently infiltrated with pro-tumoral myeloid derived suppressor cells (MDSCs) [55]. Due to the rapid proliferation of tumor cells, abnormal vasculature and lymphatics may arise which can induce a hypoxic tumor microenvironment [56, 55]. In HNSCC, hypoxia has been shown to induce MDSC recruitment through increased production of macrophage migration inhibitory factory (MIF) and interleukin-6 [57]. Although counterintuitive, regulatory T cells that dampen tumor-specific responses are associated with good prognosis. Tregs have been shown to induce immune suppression and promote tumor progression through TGF-β and IL-10 [58]. However, consistent with previous studies in HNSCC [59] and as found in other cancer types [60], Treg infiltration has also been associated with improved survival in the aforementioned TCGA study [25, 59, 60].
5 Checkpoint inhibitors targeting the PD-1/PD-L1/PD-L2 axis
5.1 Background
Immunologic checkpoints are a complex homeostatic system of signaling pathways that mediate the targeted activation or selective tolerance of the immune system towards target cells. These pathways serve to establish an effector response to non-self antigens while preventing the induction of autoimmune activity. Tumor cells hijack these mechanisms to create an immunosuppressive and protumor microenvironment [61]. The first immune checkpoint to be clinically targeted was CTLA-4. T cell activation requires two signals, signal one being the binding of the T cell receptor (TCR) to its cognate antigen peptide in the context of the major histocompatibility complex (MHC) and signal two being the costimulatory signal through engagement of CD28 on the T cell with CD80 or CD86 on the APC. CTLA-4 competes with CD28 for the same ligands but with higher avidity binding and induces downregulation of T cell responses [62]. The critical role of CTLA-4 in suppressing immune function is best illustrated in CTLA-4 knockout mice which suffer fatal lymphoproliferative disorders [63, 64]. Based on encouraging preclinical data pioneered by Dr. James Allison, clinical testing of CTLA-4 blocking antibodies began in 2000 and based on a randomized control trial, the first CTLA-4 mAb, ipilimumab, gained FDA approval in melanoma in 2010 [65]. The trial was notable because it was the first to demonstrate a survival benefit in advanced melanoma, was the first illustration of the potential benefits of checkpoint blockade, and importantly, roughly 20% of the patients experienced durable responses 2 years after start of treatment [66]. Further trials have demonstrated that treatment with PD-1 blocking mAbs was more efficacious and better tolerated than ipilimumab [67, 68] and much of the checkpoint blockade trials in HNSCC have therefore focused on the PD-1/PD-L1 axis.
Programmed cell death 1 (PD-1) is an immune checkpoint whose purpose is to limit the effector function of T cells at the site of inflammation to protect against immune-mediated damage to normal tissue [69] (Fig. 1). Binding of PD-1 to its ligands, PD-L1 and PD-L2, induces an exhausted T cell phenotype characterized by impaired proliferation, migration, cytokine production, and reduced T cell-target cell contact [70, 61, 71]. PD-L1 expression is induced on tumor cells and immune cells via inflammatory signals, most notably IFNγ. Regulation of PD-L1 expression is mediated through signaling pathways related to proliferation and activation, most notably NFkB, MAPK, PI3K, mTOR, and JAK/STAT [72]. Additionally, independent of external signaling pathways, a subset of HNSCC has PD-L1 overexpression as a result of oncogene activation or genomic amplification [72]. Although the intrinsic pathways are less well defined, a recent study implicated overexpression of EGFR as being associated with PD-L1 expression in a JAK2/STAT1-dependent manner [73]. In this study, TCGA data were utilized to demonstrate a significant correlation between PD-L1, JAK2/STAT1, and EGFR in the transcriptome and this data was validated with IHC staining of HNSCC in vitro. To further validate this dependency, the authors demonstrate that silencing of STAT1 by shRNA, or inhibition of JAK2 and EGFR with BMS911345 and cetuximab, respectively, abrogates PD-L1 expression at the mRNA and protein level. These findings support further investigation of combination treatments involving anti-PD1 mAbs with cetuximab and JAK2 inhibitors in HNSCC.

T cell priming and effector function
5.2 Clinical trials of anti-PD-1 therapeutics in HNSCC
Monoclonal antibodies targeting the PD-1/PD-L1/PD-L2 axis have demonstrated remarkable success in a wide range of malignancies with many patients experiencing durable responses [2, 74,75,76, 67, 77, 78]. Two PD-1 inhibitors, pembrolizumab and nivolumab, have been evaluated in patients with recurrent/metastatic HNSCC who failed first-line therapy. Keynote 012 was a phase Ib trial of pembrolizumab in patients whose tumors had PD-L1 expression (> 1% tumor cells by IHC) followed by an expansion cohort without a PD-L1 selection. In the initial cohort of 60 patients, pembrolizumab was well tolerated with 17% of patients having a grade 3 or 4 drug-related adverse event [79]. The overall response rate as measured by Response Evaluation Criteria In Solid Tumors (RECIST) was 18%, but 25% in HPV-associated HNSCC and 14% in HPV-negative HNSCC [79]. In the expansion cohort of 132 patients, the ORR was unchanged, but patients with PD-L1-positive tumors (> 1% tumor cells by IHC) had an ORR of 22% compared to 14% in those with PD-L1-negative tumors [4]. Impressively, some responses were durable with a median duration of response (DOR) not reached and 83% of responses still ongoing with a median follow-up of 9 months. Additionally, 4/132 (3%) patients experienced a complete response [4]. A long-term follow up of the trial, presented at ASCO in 2016, demonstrated that of those patients who responded, 85% lasted greater than 6 months and 71% lasted at least 12 months (median DOR still not reached) [80].
Checkmate 141 was a phase III trial where 361 patients with platinum-resistant, recurrent HNSCC were randomized 2:1 to receive nivolumab (3 mg/kg every 2 weeks) or standard, single-agent systemic therapy. Patients who received nivolumab had a median overall survival of 7.5 months versus 5.1 months in the control arm and a 30% lower risk of death [3]. Furthermore, the 1-year survival with nivolumab was 36%, significantly higher than 16.6% in the control arm [3]. The nivolumab response rate was 13.3% with 6 patients (2.5%) experiencing a complete response and another 26 patients (11%) with a partial response [3]. No significant difference was noted in the rate of progression-free survival between the two groups [3]. Consistent with the Keynote 012 trial, the improvement in overall survival was greater in patients with HPV-associated tumors than non-HPV-associated tumors. Of those patients with p16-positive tumors, the median overall survival was 9.1 months with nivolumab versus 4.4 months in the control arm [3]. In patients with p16-negative tumors, the median overall survival was 7.5 months with nivolumab versus 5.8 months in the control arm [3]. Importantly, nivolumab was well tolerated with only 13.1% of patients reporting a grade 3 or 4 treatment-related adverse event compared to 35.1% with standard of care chemotherapy [3].
Together, these trials demonstrate that PD-1 blockade is efficacious in recurrent/metastatic HNSCC. In the single-arm Keynote 012 trial with pembrolizumab, the objective response rate was 18% and in the phase III randomized Checkmate 141 trial with nivolumab, the objective response rate was 13.3%. In both trials, patients with HPV-associated HNSCC or PD-L1 positive tumors had improved outcomes although it is clear that additional screening tools are needed to better select patients for anti-PD-1 therapy.
5.3 Biomarkers of response
Based on the findings from trials with pembrolizumab and nivolumab, the FDA approved these agents for treatment of cisplatin-resistant recurrent/metastatic disease. However, efficacy is limited to a subset of patients and defining biomarkers of response has been a major aim of many investigations. Studies of anti-PD-1 responders across cancers have broadly identified biomarkers based on immunologic, genetic, and virological criteria [69]. Looking at immunologic biomarkers, Taube and colleagues conceptualized four patterns of tumor-host interactions: PD-L1 expression geographically associated with infiltrating immune cells indicating an inflamed phenotype characterized by IFNγ release, PD-L1 expression in the absence of immune cells indicating intrinsic PD-L1 upregulation, low PD-L1 with infiltrating immune cells which are presumably antigen naive, and low PD-L1 expression and no infiltrating immune cells which many in the field refer to as “cold tumors” [81]. Many studies have reported that tumors with PD-L1 expression are more likely to respond to inhibitors of the PD-1/PD-L1/PD-L2 axis, although lack of PD-L1 expression is not an indicator of resistance [82]. Intuitively, evidence of pre-existing adaptive immune response would portend a more likely response to PD-1 blockade and indeed a study of melanoma patients identified the presence of pre-existing CD8+ T cells along the invasive front of the tumor to be associated with response to PD-1 blockade [83].
As mentioned above, a higher mutation rate is associated with more frequent neoantigen production and therefore a greater chance of presentation of immunogenic tumor antigens. This hypothesis was tested in a clinical trial of patients whose tumors possess mismatch repair deficiencies, a DNA-repair dysfunction that results in 10–100 times the number of somatic mutations as mismatch repair proficient tumors [84]. Patients with mismatch repair deficiencies had profound responses to checkpoint blockade and this has ultimately led to the accelerated FDA approval of pembrolizumab in tissue/site agnostic disease with mismatch repair deficiency [84]. This is relevant for two reasons: (1) Microsatellite instability has been associated with the carcinogenesis of a subset of HNSCCs which would presumably respond better to checkpoint blockade [85], and (2) there are a wide range of mutation burdens among HNSCCs, and this could be further explored as a biomarker of susceptibility to checkpoint blockade [21].
Human papillomavirus (HPV) positive HNSCC, with expression of viral proteins, presents a logical target for immune-mediated therapy for two reasons. All expressed viral gene products could potentially serve as tumor antigens and therefore increases the likelihood of T cell specificity and second, the HPV-16 and HPV-18 E6 and E7 oncogenes are critical for tumorigenesis and are therefore likely expressed in every cancer cell and are unlikely to be lost or downregulated [69]. In fact, in a study of HLA-A*0201+ patients with HPV-16 associated HNSCC, HPV-16 E711–20- or E786–93-specific T cells were isolated from the peripheral blood, demonstrating the antigenic potential of the viral components [86].
6 Future directions in checkpoint therapy
6.1 Natural kill cell targeted therapies
Natural kill (NK) cells are innate immune cells involved in detection and eradication of virally infected or transformed cells but, unlike T cells, do not possess clonally rearranged antigen receptors [87]. Instead, NK cells respond to a complex system of activating and inhibitory surface receptors [87]. Whereas T cells become activated through binding of their cognate antigen in the context of MHC I, NK cells are suppressed by the presence of MHC I molecules through binding of killing immunoglobulin-like receptors (KIRs) [88]. NK cell activation results in the release of pro-inflammatory cytokines, such as IFNγ and TNFα, and cytotoxic effectors, such as granzyme B and perforin [89]. There has been growing enthusiasm for immunotherapeutic approaches that capitalize on the pro-inflammatory and anti-tumor properties of NK cells. Lirilumab and monalizumab are blocking monoclonal antibodies that have been developed to interfere with MHC I–KIR binding, thereby leading to NK cell activation [89]. Lirilumab was evaluated in a trial in combination with nivolumab across multiple cancer types and results were presented at SITC 2016. In the subset of patients with HNSCC (n = 29), the combination was clinically effective with an ORR of 24.1% with 3 patients having a complete response [90]. Future clinical trials will better evaluate the effectiveness of this combination and other approaches to capitalize on the potential of NK cell-based immunotherapies.
6.2 Incorporating checkpoint therapy into multimodality treatment of HNSCC
Traditionally, the three pillars of HNSCC treatment have been surgery, chemotherapy, and radiation depending on tumor location, stage, and presence of high-risk pathology. Although response rates are low, cetuximab represents the only FDA-approved targeted therapeutic agent for HNSCC patients. Immunotherapy represents a potent and durable approach to be included in the treatment paradigm for HNSCC. However, much work is needed to optimize the timing of immunotherapeutics to maximize clinical response (Fig. 2). While chemotherapy can frequently be immunosuppressive, there is some evidence that certain chemotherapy agents may prove synergistic with checkpoint blockade by recruitment of CD8+ T cells to the tumor microenvironment [91, 92].

Future directions in personalized immuno-oncology
6.3 Incorporating radiotherapy into immunotherapeutic approaches
There is a significant body of evidence supporting the role of ionizing radiation therapy as an immunomodulatory agent. Studies in immunocompetent and immunocompromised mice demonstrate a critical role of the immune system in the success of radiation therapy through a type-I interferon and CD8+ T cell dependent response [93, 94]. Furthermore, ionizing radiation may induce MHC class I upregulation, improving T cell detection of neoantigens [95]. Importantly, ionizing radiation also incites the generation of novel peptides which become expressed/upregulated following treatment as a result of activation of the mTOR pathway [96]. However, a more detailed analysis at the effects of chemoradiation indicate a complex picture with variable changes in systemic CD8+ T cells but consistently reported increases in immunosuppressive Tregs and MDSCs [97,98,99].
Activation of a tumor-specific immune response following radiation has been shown to induce an abscopal effect in metastatic cancer. The abscopal effect results when local radiation of a tumor induces a systemic immune response leading to rejection of distant metastatic tumor sites. This notably occurred in a patient with metastatic melanoma with progressive disease despite treatment with the CTLA-4 inhibitor, ipilimumab, who received palliative radiation to a painful paraspinal lesion [100]. Following radiation, she had significant regression of the paraspinal lesion as well as multiple metastatic lesions outside the field of radiation [100]. Although cases like this are uncommon, it demonstrates the potential synergy of radiation and checkpoint blockade to produce both local and systemic effects.
6.4 The role of surgery in immunotherapy
The optimal integration of surgery in immunotherapeutic approaches remains to be fully defined. Surgery remains a common first-line treatment modality for locally advanced HNSCC when there is potential for curative management, although a significant subset of patients still develops locoregional recurrence or metastatic disease despite optimal management. Therefore, there is growing interest in incorporating checkpoint inhibitors in the neoadjuvant setting for primary locally advanced HNSCC with the goal of priming immunity prior to resection in order to eradicate recurrent disease before it becomes clinically apparent. A preclinical model of metastatic breast cancer found that neoadjuvant immunotherapy with regulatory T cell (Treg) depletion reduced recurrence, metastases, and improved overall survival—19/20 mice with long-term survival (> 250 days) in the neoadjuvant cohort compared to 5/20 mice surviving in the adjuvant treatment-only cohort [101]. With a neoadjuvant anti-PD1 regimen, again the overall survival was better in the neoadjuvant setting than the adjuvant setting (67% survival compared to 0% in the adjuvant setting) [101]. Additionally, there has been renewed interest in the role of “cytoreductive surgery” in the recurrent/metastatic setting with the goal of (1) decreasing the systemic immunosuppressive effects of the primary tumor, (2) palliate symptoms, and (3) investigate biomarkers for novel treatment regimens [102].
6.5 Towards combination treatment and novel approaches for non-responsive tumors
Despite the approaches outlined above, a significant number of patients with advanced disease fail or progress on single-modality checkpoint inhibitor therapy. There are many reasons why patients progress on checkpoint inhibitor therapy including lack of strong tumor antigens, impaired expression of tumor antigens, poor infiltration or activation of T cells in the tumor, and immunosuppressive forces in the tumor microenvironment [103]. Understanding the mechanism of immune evasion of a tumor will enable practitioners to more precisely design combination therapies or personalized therapies. One common approach combining anti-CTLA-4 and anti-PD1 antibodies was demonstrated in a preclinical model of melanoma to increase recruitment of effector T cells and improve tumor rejection (65% rejection vs. 10% with monotherapy) [104]. This led to clinical trials in patients with unresectable melanoma of combination nivolumab with ipilimumab versus either as monotherapy. The combination resulted in a progression-free survival of 11.5 vs. 2.9 months with ipilimumab alone or 6.9 months with nivolumab alone [105]. Despite the clinical benefit, the combination treatment was associated with significant morbidity with 55% of patients reporting grade 3 or 4 adverse events vs. 16 and 27% with monotherapy using nivolumab or ipilimumab, respectively [105].
Two other common checkpoint targets currently in clinical trials are the lymphocyte activation gene 3 (LAG-3) and T cell membrane protein 3 (TIM-3). LAG-3 acts synergistically with PD-L1 and its binding of MHC class II molecules induces an inhibitory signal to T cells and enhances Treg-mediated suppression [66, 106]. TIM-3 similarly incites a suppressive effect and has been implicated as a potential resistance mechanism for anti-PD1-treated HNSCC tumors [107, 108].
Other efforts have focused on agonists of the tumor necrosis factor (TNF) receptor family such as CD40, 4-1BB, OX40, and GITR in combination with anti-PD-1 to promote APC activation and enhance anti-tumor T cell response based on encouraging preclinical data [109,110,111]. These molecules exhibit distinct expression patterns with CD40 predominantly expressed on dendritic cells, 4-1BB on T cells, NK cells and monocytes, OX40 on T cells, NK cells and neutrophils, and GITR predominantly on Tregs [66]. Therefore, each represents a potential pathway to synergize with checkpoint blockade and overcome immune resistance. Additional combination therapy approaches have utilized agonists of the STING pathway based on encouraging preclinical data demonstrating that combination intratumoral STING agonist and systemic anti-PD1 can overcome adaptive resistance to checkpoint alone [112]. A full list of current clinical trials utilizing checkpoint inhibitors in HNSCC is listed in Table 1.
7 Adoptive T cell therapy
The ultimate goal of any immunotherapy approach is to increase the number and effectiveness of tumor-specific T cells within the tumor to induce clinical regression of disease. Priming, recruitment, and activity of T cells at the tumor site may be dysfunctional for many reasons as outlined above and therefore there is interest in combining checkpoint inhibitors with approaches that increase the frequency of tumor-specific T cells. As a result, there is enthusiasm for adoptive T cell therapy (ACT) as a means to cultivate tumor-specific T cells ex vivo and re-infuse them into the patient to mount a fulminant anti-tumor immune response. Briefly, tumors are excised from patients and fragments are cultured in high-dose IL-2 to expand the lymphocyte compartment [113]. Expanded T cell populations are then screened for tumor specificity and re-infused into a lymphodepleted tumor-bearing host [113]. This technique has been successful in many patients, independent of checkpoint blockade, and is capable of inducing durable tumor regression [114, 115].
In one dramatic example, a patient with metastatic colon cancer was identified to have T cells specific for a neoantigen encoded by a driver mutation in the KRAS gene [116]. These T cells were enriched ex vivo and reinfusion of 1 × 1011 cells induced objective regression of all the metastatic lesions [116]. After one lesion ultimately progressed, it was resected and found to have loss of the coding region of the HLA-C*08:02 class I molecule responsible for presenting the KRAS-encoded neoantigen, providing a mechanism of immune escape [116]. This case is pertinent to HNSCC because HPV-associated HNSCC similarly have potential tumor antigens in the HPV-16 and HPV-18 E6 and E7 genes, which are known oncogenic drivers. Therefore, targeted recognition of these antigens by adoptively transferred lymphocytes could be a potent modality against these tumor cells and this approach is currently being evaluated in a clinical trial at the National Cancer Institute (NCT02280811) [117]. A similar clinical trial of adoptive T cell therapy in metastatic cervical cancer enrolled nine patients and selected for T cell cultures based on reactivity to HPV-16 or HPV-18 E6 and E7 [118]. Three of the 9 patients experienced an objective tumor response with 2 of the 3 responders experiencing a durable response ongoing 15 and 22 months [118]. Interestingly, a deeper analysis of the T cell reactivity from two patients with durable responses indicated that the vast majority of infused T cells were actually specific for mutation-derived neoantigens rather than HPV-associated antigens E6 and E7, indicating that approaches combining both mutation-derived and viral antigens should be considered [119]. The lack of more consistent responses to ACT also provides support for the use of combination approaches with checkpoint inhibitors in order to both increase tumor-specific T cells and reduce immunosuppression in the tumor microenvironment.
8 Vaccine therapy in HNSCC
8.1 Personalized cancer vaccines
Another mechanism for increasing the frequency of tumor-specific T cells for monotherapy or in combination with checkpoint inhibitors would be via a therapeutic vaccine approach. There has been interest in cancer vaccine development for decades as an immunotherapeutic agent. Historically, cancer vaccine trials in a range of malignancies including HNSCC have been failures. There are several explanations why these vaccines have failed, but it is the likely that the vaccine target or adjuvant were ineffective at priming a tumor-specific response or that immunosuppression in the tumor microenvironment was insurmountable [120]. Most commonly, these trials have utilized TAAs or CTAs linked to an adjuvant to facilitate APC uptake and T cell activation [121,122,123]. Although individual phase I or phase II trials have shown promise, the most comprehensive evaluation were two phase III randomized, double-blinded clinical trials using MAGE-A3 in a vaccine for patients with non-small cell lung cancer in one trial and melanoma in the other [124]. Ultimately, both trials failed to demonstrate improvement in disease-free survival [124].
Therefore, focus has turned to neoantigens as promising vaccine targets given their tumor-restricted expression, lack of central tolerance, and the known tumor-rejection efficacy of neoantigen-specific T cells. In preclinical models, neoantigen vaccines have demonstrated success in inducing tumor control and rejection [24, 125]. Neoantigens are predicted based on whole exome sequencing or cDNA capture-sequencing, screened for RNA expression, prioritized based on MHC class I or II binding affinity and then validated by assaying tumor infiltrating lymphocytes against candidate epitopes using ELISPOT, intracellular cytokine staining or tetramer staining [126]. However, in the clinical setting, there is rarely time to validate candidate epitopes for pre-existing T cell reactivity. Therefore, current clinical trials apply filters to the predicted neoantigens based on variant allele frequency, RNA expression, and binding affinity and the top predicted neoantigens are included in the vaccine. A small clinical trial of three patients with melanoma treated with a neoantigen dendritic cell vaccine found the vaccine to be safe and increased neoantigen-specific immunity [127]. There are now several therapeutic neoantigen vaccine clinical trials active in a handful of malignancies including glioblastoma (NCT02510950, NCT02287428), renal cell carcinoma (NCT02950766), melanoma (NCT01970358), colon cancer (NCT01885702), breast cancer (NCT02348320, NCT02427581), and other assorted tumor types (NCT02897765), although no neoantigen vaccine trials are available in HNSCC to date.
8.2 Vaccine approaches in HPV-associated HNSCC
Preventative vaccines for HPV have had tremendous success in the prevention of persistent genital HPV infection (90–100%) and likely has similar efficacy in oropharyngeal infection although data is limited based on lack of routine screening [128]. In the therapeutic setting, there is interest in using known HPV antigens as vaccine targets to induce tumor-specific immune responses. A phase II clinical trial was conducted using E6 and E7 synthetic peptides in a vaccine for women with HPV-positive grade 3 vulvar intraepithelial neoplasia (VIN) [129]. At the 1-year mark, 15 of 19 patients had an objective clinical response with 9 complete responses [129]. A subsequent trial of E6 and E7 vaccine in advanced/recurrent HPV-associated gynecological carcinoma found a majority of patients with induction of immune response, although no clinically evident tumor regression was noted [130]. This may indicate that E6 and E7 vaccination is more effective in early-stage cancer and should be considered as part of a combination therapy regimen for more advanced tumors. Two clinical trials are currently evaluating therapeutic HPV vaccines in HPV-associated HNSCC. In one, ADXS11-001, a live attenuated Listeria monocytogenes bacterium encoding HPV 16 E7 is given in treatment naïve patients prior to transoral robotic surgery (NCT02002182). In the other, patients with advanced disease receive nivolumab in combination with ISA 101, which consists of 13 synthetic long peptides encoding the HPV 16 E6 and E7 proteins (NCT02426892).
9 Conclusion
Immunotherapies hold tremendous promise for patients with HNSCC. HNSCC is an excellent target for immunotherapeutic approaches because of the high mutation burden, frequent infiltration with T cells, upgregulation of PD-L1, and the potential for targeted therapy against shared HPV antigens. We are still in the early stages of understanding the potential of immunotherapies with much unknown about the optimal way to combine surgery, chemotherapy, and radiation with immunotherapeutics. Additionally, next-generation sequencing and neoantigen prediction pipelines now afford the potential for personalized therapeutic neoantigen vaccines. A substantial number of clinical trials are ongoing that address many of these avenues and the next decade promises to fundamentally alter our approach and options for patients with HNSCC.
References
Warnakulasuriya, S. (2009). Global epidemiology of oral and oropharyngeal cancer. Oral Oncology, 45(4–5), 309–316. doi:10.1016/j.oraloncology.2008.06.002.
Hodi, F. S., O’Day, S. J., McDermott, D. F., Weber, R. W., Sosman, J. A., Haanen, J. B., et al. (2010). Improved survival with ipilimumab in patients with metastatic melanoma. The New England Journal of Medicine, 363(8), 711–723. doi:10.1056/NEJMoa1003466.
Ferris, R. L., Blumenschein, G., Jr., Fayette, J., Guigay, J., Colevas, A. D., Licitra, L., et al. (2016). Nivolumab for recurrent squamous-cell carcinoma of the head and neck. The New England Journal of Medicine, 375(19), 1856–1867. doi:10.1056/NEJMoa1602252.
Chow, L. Q., Haddad, R., Gupta, S., Mahipal, A., Mehra, R., Tahara, M., et al. (2016). Antitumor activity of pembrolizumab in biomarker-unselected patients with recurrent and/or metastatic head and neck squamous cell carcinoma: results from the phase Ib KEYNOTE-012 expansion cohort. Journal of Clinical Oncology, 34(32), 3838–3845.
Whiteside, T. L., Letessier, E., Hirabayashi, H., Vitolo, D., Bryant, J., Barnes, L., et al. (1993). Evidence for local and systemic activation of immune cells by peritumoral injections of interleukin 2 in patients with advanced squamous cell carcinoma of the head and neck. Cancer Research, 53(23), 5654–5662.
Dadian, G., Riches, P., Henderson, D., MacLennan, K., Lorentzos, A., Moore, J., et al. (1993). Immune changes in peripheral blood resulting from locally directed interleukin-2 therapy in squamous cell carcinoma of the head and neck. European Journal of Cancer Part B: Oral Oncology, 29(1), 29–34.
De Stefani, A., Forni, G., Ragona, R., Cavallo, G., Bussi, M., Usai, A., et al. (2002). Improved survival with perilymphatic interleukin 2 in patients with resectable squamous cell carcinoma of the oral cavity and oropharynx. Cancer, 95(1), 90–97.
Dunn, G. P., Bruce, A. T., Ikeda, H., Old, L. J., & Schreiber, R. D. (2002). Cancer immunoediting: from immunosurveillance to tumor escape. Nature Immunology, 3(11), 991–998. doi:10.1038/ni1102-991.
Dunn, G. P., Old, L. J., & Schreiber, R. D. (2004). The immunobiology of cancer immunosurveillance and immunoediting. Immunity, 21(2), 137–148. doi:10.1016/j.immuni.2004.07.017.
Dunn, G. P., Old, L. J., & Schreiber, R. D. (2004). The three Es of cancer immunoediting. Annual Review of Immunology, 22, 329–360. doi:10.1146/annurev.immunol.22.012703.104803.
Shankaran, V., Ikeda, H., Bruce, A. T., White, J. M., Swanson, P. E., Old, L. J., et al. (2001). IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature, 410(6832), 1107–1111. doi:10.1038/35074122.
Smyth, M. J., Thia, K. Y., Street, S. E., MacGregor, D., Godfrey, D. I., & Trapani, J. A. (2000). Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. The Journal of Experimental Medicine, 192(5), 755–760.
Street, S. E., Cretney, E., & Smyth, M. J. (2001). Perforin and interferon-γ activities independently control tumor initiation, growth, and metastasis. Blood, 97(1), 192–197.
Street, S. E., Trapani, J. A., MacGregor, D., & Smyth, M. J. (2002). Suppression of lymphoma and epithelial malignancies effected by interferon γ. Journal of Experimental Medicine, 196(1), 129–134.
Agraharkar, M. L., Cinclair, R. D., Kuo, Y.-F., Daller, J. A., & Shahinian, V. B. (2004). Risk of malignancy with long-term immunosuppression in renal transplant recipients. Kidney International, 66(1), 383–389.
Klein, G., Sjogren, H. O., Klein, E., & Hellstrom, K. E. (1960). Demonstration of resistance against methylcholanthrene-induced sarcomas in the primary autochthonous host. Cancer Research, 20, 1561–1572.
Herin, M., Lemoine, C., Weynants, P., Vessiere, F., Van Pel, A., Knuth, A., et al. (1987). Production of stable cytolytic T-cell clones directed against autologous human melanoma. International Journal of Cancer, 39(3), 390–396.
Old, L. J., & Chen, Y. T. (1998). New paths in human cancer serology. The Journal of Experimental Medicine, 187(8), 1163–1167.
Sahin, U., Tureci, O., Schmitt, H., Cochlovius, B., Johannes, T., Schmits, R., et al. (1995). Human neoplasms elicit multiple specific immune responses in the autologous host. Proceedings of the National Academy of Sciences of the United States of America, 92(25), 11810–11813.
van der Bruggen, P., Traversari, C., Chomez, P., Lurquin, C., De Plaen, E., Van den Eynde, B., et al. (1991). A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science, 254(5038), 1643–1647.
Schumacher, T. N., & Schreiber, R. D. (2015). Neoantigens in cancer immunotherapy. Science, 348(6230), 69–74. doi:10.1126/science.aaa4971.
Segal, N. H., Parsons, D. W., Peggs, K. S., Velculescu, V., Kinzler, K. W., Vogelstein, B., et al. (2008). Epitope landscape in breast and colorectal cancer. Cancer Research, 68(3), 889–892. doi:10.1158/0008-5472.CAN-07-3095.
Matsushita, H., Vesely, M. D., Koboldt, D. C., Rickert, C. G., Uppaluri, R., Magrini, V. J., et al. (2012). Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature, 482(7385), 400–404. doi:10.1038/nature10755.
Gubin, M. M., Zhang, X., Schuster, H., Caron, E., Ward, J. P., Noguchi, T., et al. (2014). Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature, 515(7528), 577–581. doi:10.1038/nature13988.
Mandal, R., Şenbabaoğlu, Y., Desrichard, A., Havel, J. J., Dalin, M. G., Riaz, N., et al. (2016). The head and neck cancer immune landscape and its immunotherapeutic implications. JCI insight, 1(17).
Ferris, R. L. (2015). Immunology and immunotherapy of head and neck cancer. Journal of Clinical Oncology, 33(29), 3293–3304.
Bhatia, S., Louie, A. D., Bhatia, R., O’donnell, M. R., Fung, H., Kashyap, A., et al. (2001). Solid cancers after bone marrow transplantation. Journal of Clinical Oncology, 19(2), 464–471.
King, G. N., Healy, C. M., Glover, M. T., Kwan, J. T., Williams, D. M., Leigh, I. M., et al. (1995). Increased prevalence of dysplastic and malignant lip lesions in renal-transplant recipients. New England Journal of Medicine, 332(16), 1052–1057.
Uppaluri, R., Dunn, G. P., & Lewis, J. S., Jr. (2008). Focus on TILs: prognostic significance of tumor infiltrating lymphocytes in head and neck cancers. Cancer Immunity, 8, 16.
Balermpas, P., Rödel, F., Weiss, C., Rödel, C., & Fokas, E. (2014). Tumor-infiltrating lymphocytes favor the response to chemoradiotherapy of head and neck cancer. Oncoimmunology, 3(1), e27403.
Nguyen, N., Bellile, E., Thomas, D., McHugh, J., Rozek, L., Virani, S., et al. (2016). Tumor infiltrating lymphocytes and survival in patients with head and neck squamous cell carcinoma. Head & neck.
Badoual, C., Hans, S., Merillon, N., Van Ryswick, C., Ravel, P., Benhamouda, N., et al. (2013). PD-1–expressing tumor-infiltrating T cells are a favorable prognostic biomarker in HPV-associated head and neck cancer. Cancer Research, 73(1), 128–138.
Gros, A., Parkhurst, M. R., Tran, E., Pasetto, A., Robbins, P. F., Ilyas, S., et al. (2016). Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nature Medicine, 22(4), 433–438.
Rizvi, N. A., Hellmann, M. D., Snyder, A., Kvistborg, P., Makarov, V., Havel, J. J., et al. (2015). Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science, 348(6230), 124–128. doi:10.1126/science.aaa1348.
Snyder, A., Makarov, V., Merghoub, T., Yuan, J., Zaretsky, J. M., Desrichard, A., et al. (2014). Genetic basis for clinical response to CTLA-4 blockade in melanoma. The New England Journal of Medicine, 371(23), 2189–2199. doi:10.1056/NEJMoa1406498.
Van Allen, E. M., Miao, D., Schilling, B., Shukla, S. A., Blank, C., Zimmer, L., et al. (2015). Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science, 350(6257), 207–211. doi:10.1126/science.aad0095.
Alexandrov, L. B., Nik-Zainal, S., Wedge, D. C., Aparicio, S. A., Behjati, S., Biankin, A. V., et al. (2013). Signatures of mutational processes in human cancer. Nature, 500(7463), 415–421.
Allen, C. T., Judd, N. P., Bui, J. D., & Uppaluri, R. (2012). The clinical implications of antitumor immunity in head and neck cancer. Laryngoscope, 122(1), 144–157. doi:10.1002/lary.21913.
Mandruzzato, S., Brasseur, F., Andry, G., Boon, T., & van der Bruggen, P. (1997). A CASP-8 mutation recognized by cytolytic T lymphocytes on a human head and neck carcinoma. The Journal of Experimental Medicine, 186(5), 785–793.
Hoffmann, T. K., Arsov, C., Schirlau, K., Bas, M., Friebe-Hoffmann, U., Klussmann, J. P., et al. (2006). T cells specific for HPV16 E7 epitopes in patients with squamous cell carcinoma of the oropharynx. International Journal of Cancer, 118(8), 1984–1991.
Mandic, R., Lieder, A., Sadowski, M., Peldszus, R., & Werner, J. A. (2004). Comparison of surface HLA class I levels in squamous cell carcinoma cell lines of the head and neck. Anticancer Research, 24(2b), 973–979.
Grandis, J. R., Falkner, D. M., Melhem, M. F., Gooding, W. E., Drenning, S. D., & Morel, P. A. (2000). Human leukocyte antigen class I allelic and haplotype loss in squamous cell carcinoma of the head and neck: clinical and immunogenetic consequences. Clinical Cancer Research, 6(7), 2794–2802.
Bandoh, N., Ogino, T., Katayama, A., Takahara, M., Katada, A., Hayashi, T., et al. (2010). HLA class I antigen and transporter associated with antigen processing downregulation in metastatic lesions of head and neck squamous cell carcinoma as a marker of poor prognosis. Oncology Reports, 23(4), 933–939.
Ogino, T., Shigyo, H., Ishii, H., Katayama, A., Miyokawa, N., Harabuchi, Y., et al. (2006). HLA class I antigen down-regulation in primary laryngeal squamous cell carcinoma lesions as a poor prognostic marker. Cancer Research, 66(18), 9281–9289. doi:10.1158/0008-5472.can-06-0488.
Ferris, R. L., Whiteside, T. L., & Ferrone, S. (2006). Immune escape associated with functional defects in antigen-processing machinery in head and neck cancer. Clinical Cancer Research, 12(13), 3890–3895. doi:10.1158/1078-0432.ccr-05-2750.
Cross, D. S., Platt, J. L., Juhn, S. K., Bach, F. H., & Adams, G. L. (1992). Administration of a prostaglandin synthetase inhibitor associated with an increased immune cell infiltrate in squamous cell carcinoma of the head and neck. Archives of Otolaryngology–Head & Neck Surgery, 118(5), 526–528.
Gabrilovich, D. I., Chen, H. L., Girgis, K. R., Cunningham, H. T., Meny, G. M., Nadaf, S., et al. (1996). Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nature Medicine, 2(10), 1096–1103.
Young, M. R. (2006). Protective mechanisms of head and neck squamous cell carcinomas from immune assault. Head & Neck, 28(5), 462–470. doi:10.1002/hed.20331.
Lyford-Pike, S., Peng, S., Young, G. D., Taube, J. M., Westra, W. H., Akpeng, B., et al. (2013). Evidence for a role of the PD-1:PD-L1 pathway in immune resistance of HPV-associated head and neck squamous cell carcinoma. Cancer Research, 73(6), 1733–1741. doi:10.1158/0008-5472.can-12-2384.
Strome, S. E., Dong, H., Tamura, H., Voss, S. G., Flies, D. B., Tamada, K., et al. (2003). B7-H1 blockade augments adoptive T-cell immunotherapy for squamous cell carcinoma. Cancer Research, 63(19), 6501–6505.
Cho, Y. A., Yoon, H. J., Lee, J. I., Hong, S. P., & Hong, S. D. (2011). Relationship between the expressions of PD-L1 and tumor-infiltrating lymphocytes in oral squamous cell carcinoma. Oral Oncology, 47(12), 1148–1153. doi:10.1016/j.oraloncology.2011.08.007.
Lu, S. L., Reh, D., Li, A. G., Woods, J., Corless, C. L., Kulesz-Martin, M., et al. (2004). Overexpression of transforming growth factor beta1 in head and neck epithelia results in inflammation, angiogenesis, and epithelial hyperproliferation. Cancer Research, 64(13), 4405–4410. doi:10.1158/0008-5472.can-04-1032.
Whiteside, T. L. (2016). Exosomes and tumor-mediated immune suppression. The Journal of Clinical Investigation, 126(4), 1216–1223. doi:10.1172/jci81136.
Uyttenhove, C., Pilotte, L., Theate, I., Stroobant, V., Colau, D., Parmentier, N., et al. (2003). Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nature Medicine, 9(10), 1269–1274. doi:10.1038/nm934.
Davis, R. J., Van Waes, C., & Allen, C. T. (2016). Overcoming barriers to effective immunotherapy: MDSCs, TAMs, and Tregs as mediators of the immunosuppressive microenvironment in head and neck cancer. Oral Oncology, 58, 59–70. doi:10.1016/j.oraloncology.2016.05.002.
Le, Q.-T., Kong, C., Lavori, P. W., O’byrne, K., Erler, J. T., Huang, X., et al. (2007). Expression and prognostic significance of a panel of tissue hypoxia markers in head-and-neck squamous cell carcinomas. International Journal of Radiation Oncology* Biology* Physics, 69(1), 167–175.
Zhu, G., Tang, Y., Geng, N., Zheng, M., Jiang, J., Li, L., et al. (2014). HIF-alpha/MIF and NF-kappaB/IL-6 axes contribute to the recruitment of CD11b+Gr-1+ myeloid cells in hypoxic microenvironment of HNSCC. Neoplasia, 16(2), 168–179. doi:10.1593/neo.132034.
Bergmann, C., Strauss, L., Wang, Y., Szczepanski, M. J., Lang, S., Johnson, J. T., et al. (2008). T regulatory type 1 cells in squamous cell carcinoma of the head and neck: mechanisms of suppression and expansion in advanced disease. Clinical Cancer Research, 14(12), 3706–3715.
Nizard, M., Sandoval, F., Badoual, C., Pere, H., Terme, M., Hans, S., et al. (2013). Immunotherapy of HPV-associated head and neck cancer: critical parameters. Oncoimmunology, 2(6), e24534.
Badoual, C., Hans, S., Fridman, W. H., Brasnu, D., Erdman, S., & Tartour, E. (2009). Revisiting the prognostic value of regulatory T cells in patients with cancer. Journal of Clinical Oncology, 27(19), e5–e6.
Dong, H., Strome, S. E., Salomao, D. R., Tamura, H., Hirano, F., Flies, D. B., et al. (2002). Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nature Medicine, 8(8), 793–800. doi:10.1038/nm730.
Egen, J. G., Kuhns, M. S., & Allison, J. P. (2002). CTLA-4: new insights into its biological function and use in tumor immunotherapy. Nature Immunology, 3(7), 611–618.
Waterhouse, P., Penninger, J. M., Timms, E., & Wakeham, A. (1995). Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science, 270(5238), 985.
Chambers, C. A., Sullivan, T. J., & Allison, J. P. (1997). Lymphoproliferation in CTLA-4–deficient mice is mediated by costimulation-dependent activation of CD4+ T cells. Immunity, 7(6), 885–895.
Pardoll, D. M. (2012). The blockade of immune checkpoints in cancer immunotherapy. Nature Reviews Cancer, 12(4), 252–264.
Khalil, D. N., Smith, E. L., Brentjens, R. J., & Wolchok, J. D. (2016). The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nature Reviews Clinical Oncology, 13(5), 273–290.
Robert, C., Schachter, J., Long, G. V., Arance, A., Grob, J. J., Mortier, L., et al. (2015). Pembrolizumab versus ipilimumab in advanced melanoma. New England Journal of Medicine, 372(26), 2521–2532.
Wolchok, J. D., Kluger, H., Callahan, M. K., Postow, M. A., Rizvi, N. A., Lesokhin, A. M., et al. (2013). Nivolumab plus ipilimumab in advanced melanoma. New England Journal of Medicine, 369(2), 122–133.
Topalian, S. L., Taube, J. M., Anders, R. A., & Pardoll, D. M. (2016). Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nature Reviews. Cancer, 16(5), 275–287. doi:10.1038/nrc.2016.36.
Barber, D. L., Wherry, E. J., Masopust, D., Zhu, B., Allison, J. P., Sharpe, A. H., et al. (2006). Restoring function in exhausted CD8 T cells during chronic viral infection. Nature, 439(7077), 682–687.
Fife, B. T., Pauken, K. E., Eagar, T. N., Obu, T., Wu, J., Tang, Q., et al. (2009). Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR–induced stop signal. Nature Immunology, 10(11), 1185–1192.
Ritprajak, P., & Azuma, M. (2015). Intrinsic and extrinsic control of expression of the immunoregulatory molecule PD-L1 in epithelial cells and squamous cell carcinoma. Oral Oncology, 51(3), 221–228. doi:10.1016/j.oraloncology.2014.11.014.
Concha-Benavente, F., Srivastava, R. M., Trivedi, S., Lei, Y., Chandran, U., Seethala, R. R., et al. (2016). Identification of the cell-intrinsic and -extrinsic pathways downstream of EGFR and IFNgamma that induce PD-L1 expression in head and neck cancer. Cancer Research, 76(5), 1031–1043. doi:10.1158/0008-5472.can-15-2001.
Topalian, S. L., Sznol, M., McDermott, D. F., Kluger, H. M., Carvajal, R. D., Sharfman, W. H., et al. (2014). Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. Journal of Clinical Oncology, 32(10), 1020–1030. doi:10.1200/jco.2013.53.0105.
Garon, E. B., Rizvi, N. A., Hui, R., Leighl, N., Balmanoukian, A. S., Eder, J. P., et al. (2015). Pembrolizumab for the treatment of non-small-cell lung cancer. The New England Journal of Medicine, 372(21), 2018–2028. doi:10.1056/NEJMoa1501824.
Motzer, R. J., Escudier, B., McDermott, D. F., George, S., Hammers, H. J., Srinivas, S., et al. (2015). Nivolumab versus everolimus in advanced renal-cell carcinoma. The New England Journal of Medicine, 373(19), 1803–1813. doi:10.1056/NEJMoa1510665.
Powles, T., Eder, J. P., Fine, G. D., Braiteh, F. S., Loriot, Y., Cruz, C., et al. (2014). MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature, 515(7528), 558–562.
Ansell, S. M., Lesokhin, A. M., Borrello, I., Halwani, A., Scott, E. C., Gutierrez, M., et al. (2015). PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. The New England Journal of Medicine, 372(4), 311–319. doi:10.1056/NEJMoa1411087.
Seiwert, T. Y., Burtness, B., Mehra, R., Weiss, J., Berger, R., Eder, J. P., et al. (2016). Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. The Lancet Oncology, 17(7), 956–965.
Chow, L., Mehra, R., Haddad, R., Mahipal, A., Weiss, J., Berger, R., et al. Biomarkers and response to pembrolizumab (pembro) in recurrent/metastatic head and neck squamous cell carcinoma (R/M HNSCC). In ASCO Meeting Abstracts, 2016 (Vol. 34, pp. 6010).
Taube, J. M., Anders, R. A., Young, G. D., Xu, H., Sharma, R., McMiller, T. L., et al. (2012). Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Science Translational Medicine, 4(127), 127ra137.
Zou, W., Wolchok, J. D., & Chen, L. (2016). PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Science Translational Medicine, 8(328), 328rv324.
Tumeh, P. C., Harview, C. L., Yearley, J. H., Shintaku, I. P., Taylor, E. J., Robert, L., et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature, 515(7528), 568–571. doi:10.1038/nature13954.
Le, D. T., Uram, J. N., Wang, H., Bartlett, B. R., Kemberling, H., Eyring, A. D., et al. (2015). PD-1 blockade in tumors with mismatch-repair deficiency. The New England Journal of Medicine, 372(26), 2509–2520. doi:10.1056/NEJMoa1500596.
Field, J., Kiaris, H., Howard, P., Vaughan, E., Spandidos, D., & Jones, A. (1995). Microsatellite instability in squamous cell carcinoma of the head and neck. British Journal of Cancer, 71(5), 1065–1069.
Albers, A., Abe, K., Hunt, J., Wang, J., Lopez-Albaitero, A., Schaefer, C., et al. (2005). Antitumor activity of human papillomavirus type 16 E7–specific T cells against virally infected squamous cell carcinoma of the head and neck. Cancer Research, 65(23), 11146–11155.
Moretta, A., Bottino, C., Mingari, M. C., Biassoni, R., & Moretta, L. (2002). What is a natural killer cell? Nature Immunology, 3(1), 6–8.
Moretta, A., Bottino, C., Vitale, M., Pende, D., Biassoni, R., Mingari, M. C., et al. (1996). Receptors for HLA class-I molecules in human natural killer cells. Annual Review of Immunology, 14(1), 619–648.
Muntasell, A., Ochoa, M. C., Cordeiro, L., Berraondo, P., de Cerio, A. L.-D., Cabo, M., et al. (2017). Targeting NK-cell checkpoints for cancer immunotherapy. Current Opinion in Immunology, 45, 73–81.
Leidner, R., Kang, H., Haddad, R., Segal, N. H., Wirth, L. J., Ferris, R. L., et al. (2016). Preliminary efficacy from a phase 1/2 study of the natural killer cell–targeted antibody, lirilumab in combination with nivolumab in squamous cell carcinoma of the head and neck. Abstract, 456, 11–13.
Soliman, H. H. (2017). nab-paclitaxel as a potential partner with checkpoint inhibitors in solid tumors. OncoTargets and therapy, 10, 101.
Pfirschke, C., Engblom, C., Rickelt, S., Cortez-Retamozo, V., Garris, C., Pucci, F., et al. (2016). Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity, 44(2), 343–354.
Burnette, B. C., Liang, H., Lee, Y., Chlewicki, L., Khodarev, N. N., Weichselbaum, R. R., et al. (2011). The efficacy of radiotherapy relies upon induction of type I interferon–dependent innate and adaptive immunity. Cancer Research, 71(7), 2488–2496.
Lee, Y., Auh, S. L., Wang, Y., Burnette, B., Wang, Y., Meng, Y., et al. (2009). Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood, 114(3), 589–595.
Lugade, A. A., Sorensen, E. W., Gerber, S. A., Moran, J. P., Frelinger, J. G., & Lord, E. M. (2008). Radiation-induced IFN-γ production within the tumor microenvironment influences antitumor immunity. The Journal of Immunology, 180(5), 3132–3139.
Reits, E. A., Hodge, J. W., Herberts, C. A., Groothuis, T. A., Chakraborty, M., Wansley, E. K., et al. (2006). Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. Journal of Experimental Medicine, 203(5), 1259–1271.
Parikh, F., Duluc, D., Imai, N., Clark, A., Misiukiewicz, K., Bonomi, M., et al. (2014). Chemoradiotherapy-induced upregulation of PD-1 antagonizes immunity to HPV-related oropharyngeal cancer. Cancer Research, 74(24), 7205–7216.
Schuler, P. J., Harasymczuk, M., Schilling, B., Saze, Z., Strauss, L., Lang, S., et al. (2013). Effects of adjuvant chemoradiotherapy on the frequency and function of regulatory T cells in patients with head and neck cancer. Clinical Cancer Research, 19(23), 6585–6596. doi:10.1158/1078-0432.ccr-13-0900.
Sridharan, V., Margalit, D. N., Lynch, S. A., Severgnini, M., Zhou, J., Chau, N. G., et al. (2016). Definitive chemoradiation alters the immunologic landscape and immune checkpoints in head and neck cancer. Br J Cancer.
Postow, M. A., Callahan, M. K., Barker, C. A., Yamada, Y., Yuan, J., Kitano, S., et al. (2012). Immunologic correlates of the abscopal effect in a patient with melanoma. New England Journal of Medicine, 366(10), 925–931.
Liu, J., Blake, S. J., Yong, M. C., Harjunpaa, H., Ngiow, S. F., Takeda, K., et al. (2016). Improved efficacy of neoadjuvant compared to adjuvant immunotherapy to eradicate metastatic disease. Cancer Discovery, 6(12), 1382–1399. doi:10.1158/2159-8290.cd-16-0577.
Bryan Bell, R., Gough, M. J., Seung, S. K., Jutric, Z., Weinberg, A. D., Fox, B. A., et al. (2016). Cytoreductive surgery for head and neck squamous cell carcinoma in the new age of immunotherapy. Oral Oncology, 61, 166–176. doi:10.1016/j.oraloncology.2016.08.020.
Kim, J., & Chen, D. (2016). Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure). Annals of Oncology, 27(8), 1492–1504.
Curran, M. A., Montalvo, W., Yagita, H., & Allison, J. P. (2010). PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proceedings of the National Academy of Sciences of the United States of America, 107(9), 4275–4280. doi:10.1073/pnas.0915174107.
Larkin, J., Hodi, F. S., & Wolchok, J. D. (2015). Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. The New England Journal of Medicine, 373(13), 1270–1271. doi:10.1056/NEJMc1509660.
Woo, S.-R., Turnis, M. E., Goldberg, M. V., Bankoti, J., Selby, M., Nirschl, C. J., et al. (2012). Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Research, 72(4), 917–927.
Li, J., Shayan, G., Avery, L., Jie, H.-B., Gildener-Leapman, N., Schmitt, N., et al. (2016). Tumor-infiltrating Tim-3+ T cells proliferate avidly except when PD-1 is co-expressed: evidence for intracellular cross talk. Oncoimmunology, 5(10), e1200778.
Shayan, G., Srivastava, R., Li, J., Schmitt, N., Kane, L. P., & Ferris, R. L. (2016). Adaptive resistance to anti-PD1 therapy by Tim-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer. Oncoimmunology(just-accepted), 00–00.
Vonderheide, R. H., & Glennie, M. J. (2013). Agonistic CD40 antibodies and cancer therapy. Clinical Cancer Research, 19(5), 1035–1043. doi:10.1158/1078-0432.ccr-12-2064.
Aspeslagh, S., Postel-Vinay, S., Rusakiewicz, S., Soria, J. C., Zitvogel, L., & Marabelle, A. (2016). Rationale for anti-OX40 cancer immunotherapy. European Journal of Cancer, 52, 50–66. doi:10.1016/j.ejca.2015.08.021.
Moran, A. E., Kovacsovics-Bankowski, M., & Weinberg, A. D. (2013). The TNFRs OX40, 4-1BB, and CD40 as targets for cancer immunotherapy. Current Opinion in Immunology, 25(2), 230–237. doi:10.1016/j.coi.2013.01.004.
Moore, E. C., Clavijo, P. E., Davis, R., Cash, H. A., Van Waes, C., Kim, Y. J., et al. (2016). Established T-cell inflamed tumors rejected after adaptive resistance was reversed by combination STING activation and PD-1-pathway blockade. Cancer Immunol Res, canimm. 0104.2016.
Rosenberg, S. A., & Restifo, N. P. (2015). Adoptive cell transfer as personalized immunotherapy for human cancer. Science, 348(6230), 62–68. doi:10.1126/science.aaa4967.
Robbins, P. F., Lu, Y. C., El-Gamil, M., Li, Y. F., Gross, C., Gartner, J., et al. (2013). Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nature Medicine, 19(6), 747–752. doi:10.1038/nm.3161.
Cohen, C. J., Gartner, J. J., Horovitz-Fried, M., Shamalov, K., Trebska-McGowan, K., Bliskovsky, V. V., et al. (2015). Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. The Journal of Clinical Investigation, 125(10), 3981–3991. doi:10.1172/jci82416.
Tran, E., Robbins, P. F., Lu, Y. C., Prickett, T. D., Gartner, J. J., Jia, L., et al. (2016). T-cell transfer therapy targeting mutant KRAS in cancer. The New England Journal of Medicine, 375(23), 2255–2262. doi:10.1056/NEJMoa1609279.
Hinrichs, C. S., & Rosenberg, S. A. (2014). Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunological Reviews, 257(1), 56–71. doi:10.1111/imr.12132.
Stevanovic, S., Draper, L. M., Langhan, M. M., Campbell, T. E., Kwong, M. L., Wunderlich, J. R., et al. (2015). Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. Journal of Clinical Oncology, 33(14), 1543–1550. doi:10.1200/jco.2014.58.9093.
Stevanović, S., Pasetto, A., Helman, S. R., Gartner, J. J., Prickett, T. D., Howie, B., et al. (2017). Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science, 356(6334), 200–205.
Hacohen, N., Fritsch, E. F., Carter, T. A., Lander, E. S., & Wu, C. J. (2013). Getting personal with neoantigen-based therapeutic cancer vaccines. Cancer Immunology Research, 1(1), 11–15. doi:10.1158/2326-6066.cir-13-0022.
Melero, I., Gaudernack, G., Gerritsen, W., Huber, C., Parmiani, G., Scholl, S., et al. (2014). Therapeutic vaccines for cancer: an overview of clinical trials. Nature Reviews. Clinical Oncology, 11(9), 509–524. doi:10.1038/nrclinonc.2014.111.
Yoshitake, Y., Fukuma, D., Yuno, A., Hirayama, M., Nakayama, H., Tanaka, T., et al. (2015). Phase II clinical trial of multiple peptide vaccination for advanced head and neck cancer patients revealed induction of immune responses and improved OS. Clinical Cancer Research, 21(2), 312–321.
DeLeo, A. B., & Whiteside, T. L. (2008). Development of multi-epitope vaccines targeting wild-type sequence p53 peptides. Expert Review of Vaccines, 7(7), 1031–1040. doi:10.1586/14760584.7.7.1031.
Gjerstorff, M. F., Andersen, M. H., & Ditzel, H. J. (2015). Oncogenic cancer/testis antigens: prime candidates for immunotherapy. Oncotarget, 6(18), 15772–15787. doi:10.18632/oncotarget.4694.
Castle, J. C., Kreiter, S., Diekmann, J., Lower, M., van de Roemer, N., de Graaf, J., et al. (2012). Exploiting the mutanome for tumor vaccination. Cancer Research, 72(5), 1081–1091. doi:10.1158/0008-5472.CAN-11-3722.
Gubin, M. M., Artyomov, M. N., Mardis, E. R., & Schreiber, R. D. (2015). Tumor neoantigens: building a framework for personalized cancer immunotherapy. The Journal of Clinical Investigation, 125(9), 3413–3421. doi:10.1172/JCI80008.
Carreno, B. M., Magrini, V., Becker-Hapak, M., Kaabinejadian, S., Hundal, J., Petti, A. A., et al. (2015). Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science, 348(6236), 803–808. doi:10.1126/science.aaa3828.
Kreimer, A. R. (2014). Prospects for prevention of HPV-driven oropharynx cancer. Oral Oncology, 50(6), 555–559. doi:10.1016/j.oraloncology.2013.06.007.
Kenter, G. G., Welters, M. J., Valentijn, A. R., Lowik, M. J., Berends-van der Meer, D. M., Vloon, A. P., et al. (2009). Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. The New England Journal of Medicine, 361(19), 1838–1847. doi:10.1056/NEJMoa0810097.
van Poelgeest, M. I., Welters, M. J., van Esch, E. M., Stynenbosch, L. F., Kerpershoek, G., van Meerten, E. L. V. P., et al. (2013). HPV16 synthetic long peptide (HPV16-SLP) vaccination therapy of patients with advanced or recurrent HPV16-induced gynecological carcinoma, a phase II trial. Journal of Translational Medicine, 11(1), 88.
Acknowledgements
Work in the Uppaluri lab is supported by the V Foundation, Merck Incorporated and NIH/NIDCR. PZ was supported by the “Development of Clinician/Researchers in Academic ENT” T32DC00022 from the National Institutes of Deafness and Other Communication Disorders.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zolkind, P., Uppaluri, R. Checkpoint immunotherapy in head and neck cancers. Cancer Metastasis Rev 36, 475–489 (2017). https://doi.org/10.1007/s10555-017-9694-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-017-9694-9