
Abstract
Hemopexin (HPX) binds heme tightly, thus protecting cells from heme toxicity during hemolysis, trauma and ischemia-reperfusion injury. Heme uptake via endocytosis of heme-HPX followed by heme catabolism by heme oxygenase-1 (HMOX1) raises regulatory iron pools, thus linking heme metabolism with that of iron. Normal iron homeostasis requires copper-replete cells. When heme-HPX induces HMOX1, the copper-storing metallothioneins (MTs) are also induced whereas the copper-responsive copper chaperone that delivers copper to Cu, Zn superoxide dismutase, CCS1, is decreased; both are known responses when cellular copper levels rise. Endocytosis of heme-HPX is needed to regulate CCS1 since the signaling ligand cobalt-protoporphyrin (CoPP)-HPX, which does not induce HMOX1 but does co-localize with heme-HPX in endosomes, also decreased CCS1. These observations support that heme-HPX mobilizes copper in cells. The regulation of both hmox1 and mt1 is prevented by the copper-chelator, bathocuproinedisulfonate (BCDS), but not uptake of heme-AlexaFluor-labeled HPX into endosomes. Supporting a role for copper in HMOX1 regulation by heme-HPX, nutritional copper deficiency generated by tetraethylene pentamine or 232 tetraamine prevented HMOX1 induction. Using conditions that mimic maturing endosomes, we found that copper prevents rebinding of heme to apo-HPX. A model is presented in which copper endocytosis together with that of heme-HPX provides a means to facilitate heme export from HPX in the maturing endosomes: heme is needed for hmox1 transcription, while cytosolic copper and CCS1 provide a link for the known simultaneous regulation of hmox1 and mt1 by heme-HPX.
Similar content being viewed by others

Introduction
Heme is derived from methemoglobin and cellular heme-proteins during hemolysis, trauma and ischemia-reperfusion injury. The glycoprotein hemopexin (HPX) is the only well-characterized, extracellular chelator of this heme. By sequestering heme, HPX plays a vital role as an anti-oxidant extracellularly for all cells (Gutteridge and Smith 1988) and also uniquely delivers heme to cells in a safe, controlled manner via receptor-mediated endocytosis of heme-HPX complexes (Davies et al. 1979; Smith and Morgan 1979; Smith and Hunt 1990). Comparison of wild type and HPX-null mice reveals that HPX protects the kidney, liver and spleen during severe hemolysis (Tolosano et al. 1999). Albumin also binds heme but is not protective since it is present in normal amounts in the serum of HPX-null mice. Haptoglobin binds hemoglobin not heme, whereas HPX does not bind hemoglobin. After splenectomy, phenyl hydrazine-induced hemolysis produces hepatic fibrosis of HPX and haptoglobin double-knockout mice (Tolosano et al. 2002). Thus, neither albumin nor the liver could clear heme or hemoglobin to compensate for the absence of HPX and the hemoglobin-binding haptoglobin (Tolosano et al. 2002). These and other published data support that HPX is the first line of defense against heme-mediated oxidative damage to cells.
Several lines of evidence link HPX-mediated heme binding and heme uptake with the regulation not only of heme homeostasis but also that of iron. For example, heme transported by HPX is degraded by heme oxygenase-1 (HMOX1 Alam and Smith 1989) in a variety of cell types, including liver (Smith and Morgan 1979), lymphocytes (Smith et al. 1997), polymorphonuclear cells (Okazaki et al. 1989), monocytes and macrophages (Alam and Smith 1989) and mouse hepatoma cells (Smith and Ledford 1988). After intravenous injection of [55Fe]-heme-HPX (Davies et al. 1979) in rats, the radioactive iron is rapidly released from heme and stored in liver ferritin. Transferrin secretion (Smith 1999) and expression of the transferrin receptor one are decreased (Alam and Smith 1989) while ferritin is induced (Sung et al. 2000a, b) by heme-HPX. Iron from heme catabolism enters regulatory pools since the cell permeable iron chelator Desferal (DF) inhibits ferritin induction by heme-HPX (Sung et al. 2000a). The HMOX1 gene is transcriptionally activated by heme-HPX (Alam and Smith 1989). In our hands, heme uptake from HPX is similar to iron uptake by transferrin including recycling of intact HPX from liver in rats (Smith and Morgan 1978, 1979). Both hemopexin and transferrin were shown biochemically and morphologically to co-localize in vesicles of the endocytotic pathway in Hep G2 cells (Smith and Hunt 1990). Thus, endocytosis of heme-HPX increases cellular iron. We have defined two processes of heme uptake by HPX in hepatocytes: a high affinity, low capacity “specific” system and a low affinity, high capacity “selective” system that were both distinguished from the uptake of exogenous free heme, i.e., non-HPX bound heme (Smith and Morgan 1981).
Intracellular levels of non-protein bound iron, which is potentially toxic via the Fenton reaction (Uauy et al. 1998), need to be kept extremely low via storage on ferritin, export by ferroportin (FPN1) or utilization for biochemical processes including the synthesis of iron-enzymes. It is established that copper is needed to maintain iron homeostasis (Han and Wessling-Resnick 2002; Chung et al. 2004). For example, iron export by ferroportin requires copper and the multi-copper oxidase hephestin (Vulpe et al. 1999). Thus, heme uptake by HPX not only affects iron homeostasis but also that of copper. In addition, the HPX system is protective during infections and inflammation that are two oxidative stress conditions in which metals such as iron are sequestered within cells. Copper, like iron, is redox active; and cells minimize intracellular levels of non-protein bound copper, such that free copper is virtually undetectable (Rae et al. 1999). The cytosolic proteins that bind copper include the metallothioneins (e.g., MT-1 and MT-2) as well as the tripeptide glutathione and several copper chaperones that move copper for cupro-enzyme synthesis. MT-1 null mice have revealed that MTs protect cells against oxidative stress (Conrad et al. 2000) in part by binding redox-active copper. The transcriptional activation of hmox1 by heme-HPX is regulated simultaneously with that of mt1. This gene regulation by heme-HPX is linked with copper since induction of HMOX1 and MT-1 mRNAs is prevented when cells are incubated with the copper chelator, bathocuproine disulfonate (BCDS) (Sung et al. 2000a). Activation of both hmox1 and mt1 by heme-HPX is inhibited by BCDS, but not by the [BCDS]2–Cu(I) complex (Alam and Smith 1992; Sung et al. 2000b; Vanacore et al. 2000). In contrast, induction of HMOX1 and MT-1 mRNA levels by heme-HPX were not affected by the iron chelators, DF and α,α′-dipyridyl (Sung et al. 2000a).
A link between MTs and the copper chaperone CCS1 for Cu, Zn superoxide dismutase (SOD Schmidt et al. 1999) is clear from the work of Tapia and colleagues who showed that cytosolic MTs act to safely store copper under normal and supraphysiological concentrations (Tapia et al. 2004). Other cytosolic proteins that bind copper include glutathione and the copper chaperones, which move copper to sites for cuproenzyme synthesis, e.g., CCS1 for Cu, Zn superoxide dismutase. Both SOD and CCS1 have been shown to play a significant role in protecting cells against oxidative damage (Fridovich 1995). Intriguingly, MT also is needed for induction by copper of the genes for SOD and CCS1 (Tapia et al. 2004). Thus, CCS1 acts together with MTs to minimize levels of non-protein bound redox active copper in the cytosol.
As part of our ongoing investigations into the link between copper and heme uptake by HPX, we have here tested the hypothesis that endocytosis of heme-HPX increases intracellular copper and that this copper is needed for the cellular and regulatory effects of heme-HPX for hmox1 and mt1. To verify that copper is the transition element required for HMOX1 regulation by heme-HPX, we used two well-established models of nutritional copper deficiency using the cell permeable, Cu(II) chelators: 2, 3, 2 tetraamine (232tet) (Hopkins and Failla 1999) or tetraethylene pentamine (TEPA) (Narayanan et al. 2001). Regulation of hmox1 by heme-HPX requires heme uptake (Smith et al. 1993), as well as normal endosome acidification since this HMOX1 regulation is prevented by the V-type ATPase inhibitor, bafilomycin (Flaherty et al. 2007). In vitro, protonation of the heme-coordinating histidines prevents heme binding (Morgan and Muller-Eberhard 1972), and this is part of the process whereby heme is released from HPX within endosomes and travels to the nucleus to regulate hmox1 (Alam et al. 2003; Sun et al. 2004). Thus, our previous findings that hmox1 regulation requires heme passage through endosomes and that the copper chelator BCDS prevents the simultaneous regulation of hmox1 and mt1 provide a rationale to investigate the following hypotheses: first, that copper is needed for the endocytosis of heme-HPX complexes, which we tested by determining whether copper deficiency decreased or prevented endocytosis; and second, whether copper acts at the level of the endosome in the release of heme from heme-HPX, which we tested by determining whether copper prevented the re-association of heme with HPX over the pH range of endosomes. A recent report provided evidence that metals including copper reduce the thermal stability of heme-HPX (Rosell et al. 2005). Finally, we tested the hypothesis that endocytosis of heme-HPX mobilizes copper into cells by determining whether the copper chaperone CCS1, which responds to changes in intracellular copper (Bertinato and L’Abbe 2003; West and Prohaska 2004), is regulated by heme-HPX. Significantly, endocytosis of heme-HPX complexes decreases levels of the copper-responsive copper chaperone CCS1 consistent with an increase in cytosolic copper. A model is presented that describes specific and separate roles for copper potentially linked by CCS1 in two pathways for hmox1 and mt1 regulation by heme-HPX.
Materials and methods
Chemicals and reagents
Dulbecco’s Modified Eagle Medium (DMEM), fetal bovine serum (FBS), Gentamicin and Alexa Fluors 488 and 594 were purchased from Invitrogen Corporation (Carlsbad, CA). Metalloporphyrins were obtained from Frontier Scientific (Logan, UT) and their concentrations determined as described (Eskew et al. 1999). All other chemicals were purchased from either Fisher Scientific Company (Pittsburg, PA) or Sigma Chemical Company (St. Louis, MO) including TEPA and 232tet; and BCDS from Aldrich Chemical Co. (Milwaukee, WI). 232tet binds copper more tightly than TEPA (affinity constant of 23.9 vs. 9.4). As with BCDS, TEPA binds Cu(I) more tightly than Cu(II) with an affinity constant of 15.8 (R.C. Hider, personal communication, email July 17, 2007). There are no equivalent figures for the binding of Cu(I) by 232tet. The preference that BCDS has for Cu(I) is conferred by methyl groups (logK of 19 for Cu(I) but only 11.7 for Cu(II); based on values for 2,9 dimethyl-1,10 phenanthroline, R.C. Hider, personal communication).
HPX purification and formation of heme-HPX complexes
Stoichiometric heme-HPX and cobalt-protoporphryin IX (CoPP)-HPX complexes (>90–95% saturated) were made, characterized and quantitated following published procedures and extinction coefficients (Smith et al. 1993; Eskew et al. 1999). Deuteroheme, used for pH titration experiments, has a millimolar extinction coefficient of 1.7 in DMSO, λ max at 392 nm (Brown and Lantzke 1969).
Cell culture, lysis and immunoblot analyses
Minimal deviation hepatoma (Hepa) cells were cultured as previously (Smith and Ledford 1988) and their properties that include many liver-specific functions have been described (Ledford and Davis 1983). Nutritional copper deficiency was generated by incubating Hepa cells, growing in DMEM containing 2% FBS, with the cell permeable, Cu(II) chelators: tetraethylene pentamine (TEPA; 0–50 μM for 24 and 40 h (Narayanan et al. 2001)) or 2, 3, 2 tetraamine (232tet; at 5–50 μM for 24 and 40 h, based on published protocols).Whole cell extracts were prepared from PBS-rinsed Hepa cells scraped into cell lysis buffer (100 μl; Cell Signaling Technology Inc., Beverley, MA) and protein concentrations determined as previously (Rish et al. 2007). For protein detection, immunoblotting was carried out with primary antibodies: anti-HMOX-1 (raised against a synthetic peptide 1–30 amino acid residues: SPA 896, Assay Designs, Ann Arbor, MI; 1:5000); anti-CCS1 (1:1000 dilution; a generous gift from Dr. J. Prohaska); and anti-α-tubulin (mouse monoclonal anti-α-tubulin, Santa Cruz Biotechnology Inc., Santa Cruz CA;1:2000 dilution). Scanning with the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE) required the secondary antibodies: IRDye 800CW goat anti-rabbit IgG (Rockland Immunochemicals, Gilbertsville, PA) and Alexa Fluor 680-conjugated goat anti-mouse IgG (Invitrogen, CA). Quantitation of immunoblots was carried out using UN-SCAN-IT gel digitizing software (Silk Scientific, Utah, USA).
Preparation of Alexa Fluor-labeled proteins, cell surface binding and endocytosis
Heme-HPX and CoPP-HPX in PBS (1 mg/ml) were labeled with either Alexa Fluor 594 or 488 (Invitrogen, CA) essentially according to the manufacturer’s instructions. Hepa cells were cultured on poly-l-lysine coated eight-well chamber slides (seeding density of 1.5 × 104 cells/well), rinsed twice with pre-warmed gas-equilibrated HEPES.NaOH-buffered, serum-free medium, pH 7.4, then incubated for 30 min with 20 μM BCDS at 37°C. For surface binding, heme-AF594HPX (500 nM) was added for 1 h to cells cooled on ice or for endocytosis incubated at 37°C for 10 min. After rinsing, cells were fixed in 3.7% formaldehyde/PBS (10 min, room temperature) or permeabilized with 0.1% Triton-X 100/PBS (5 min, room temperature) before mounting in 50% glycerol (containing 1 μg/ml DAPI as nuclear stain) and visualized using a NIKON TE2000-U inverted microscope. Co-staining with a marker for early endosomes, early endosomal antigen-1 (EEA-1) was used to localize heme-AF594HPX intracellularly. After fixing and permeabilizing, cells were blocked for 1 h at room temperature in 20 Tris–HCl. pH 7.6 containing 137 mM NaCl, 0.1% v/v Tween 20 with 5% w/v BSA (5% BSA-TBS-T) prior to overnight incubation with rabbit polyclonal anti-EEA1 antibody (1:1000 dilution, Abcam, UK) and visualized using Alexa Fluor 488 goat anti-rabbit IgG (1:1000 dilution, Invitrogen). Confocal images of AF594CoPP-HPX were obtained with an Olympus Fluoview confocal microscope (FV300). XZ-stacks were composed of 40 slices taken at 250 nm steps.
Preparation of copper-nitrilotriacetate (Cu-NTA)
Cu-NTA was prepared essentially as for ferric-NTA (Johnson and Enns 2004). This was used to provide a known concentration of copper while minimizing potential problems associated with copper salts, which can form relatively insoluble copper hydroxide. Equimolar Cu-NTA or Cu(NTA)2 were made by mixing appropriate amounts of 20 mM NTA (38.23 mg/ml 1 M NaOH titrated to pH 7.4 with 1 M HEPES) with 10 mM CuCl2 in water and diluted for use at the concentrations and molar ratios indicated. Copper-TEPA and copper-232tet complexes, needed for certain control experiments, were prepared by adding equimolar amounts of Cu-NTA to the chelator.
pH titration of heme-HPX
Rabbit HPX (Morgan and Smith 1984; Eskew et al. 1999) and human HPX (Vretblad and Hjorth 1977) were isolated, stoichiometric deuteroheme-HPX complexes prepared and pH titrations carried out as published (Morgan and Muller-Eberhard 1972). The concentration and percent saturation of deuteroheme-HPX (0.95:1 equivalents, 10 ml) in 0.1 M KCl were determined as described (Morgan and Muller-Eberhard 1972) from the absorbance at 404 nm using a Hewlett Packard 8452A Diode Array Spectrophotometer. The pH of the deuteroheme-HPX complex (10 μM in 10 ml) was gradually titrated from pH ~7.3 to a pH of ~4.2 by the addition of 0.5 M HCl (1–3 μl) at room temperature and the pH was gradually restored to neutral (7.0–7.2) by the addition of 0.5 M KOH (1–3 μl). The sample pH was measured directly (Corning Pinnacle pH meter), and the OD 404 nm was recorded initially and after each addition of acid or alkali. The sample absorbance was then normalized with respect to the initial value and respective dilution factor. For metal competition experiments, at the pH of 4.2–4.5, Cu-NTA, Cu(NTA)2, or CaCl2 (500 mM stock in water) were added to the solution at the concentrations indicated in the figure legends and the pH was gradually restored to neutral (7.0–7.2). The final volume remained below 105% of the initial sample volume in all experiments.
Statistical analyses
PRISM 5.0 (GraphPad Software Incorporated, San Diego, CA) was used for statistical analyses with an unpaired two-tailed t-test and confidence interval of 95%. The sample number and number of independent experiments are described in the text and/or figure legends.
Results
Nutritional copper deficiency prevents induction of HMOX1 by heme-HPX
To confirm and extend our previous observations that copper is the trace metal needed for hmox1 regulation by heme-HPX (Sung et al. 2000a, b), we first determined whether nutritional copper deficiency prevented HMOX1 induction by heme-HPX. Incubation of Hepa cells with either TEPA or 232tet (10–50 μM for 24 or 40 h) impaired or prevented the induction of HMOX1 protein by heme-HPX (Fig. 1a, b). The inhibitory effects of both chelators were abolished by incubating the cells with pre-formed copper-TEPA complexes (Fig. 1c) or copper-232tet complexes (data not shown). We used changes in the level of the copper chaperone, CCS1, to monitor the copper status of the cells (Cobine et al. 2006), since CCS1 responds to cellular copper (Bertinato and L’Abbe 2003; West and Prohaska, 2004). CCS1 protein levels were significantly increased 1.4- to 1.6-fold after 24 or 40 h treatment with either TEPA or 232tet (Fig. 1d), but not with copper-chelator complexes (Fig. 1e). We conclude that copper is needed for the induction of HMOX1 by heme-HPX in mouse Hepa cells.

HMOX1 induction by heme-HPX is impaired in copper-deficient Hepa cells. For HMOX1 induction, cultured Hepa cells that were growing in unsupplemented medium or medium supplemented with either a TEPA or b 232tet (at 5, 10, 20 μM) for 24 or 40 h as indicated) were rinsed and then incubated for 4 h in serum-free HEPES-buffered DMEM, pH 7.4, supplemented with heme-hemopexin (10 μM) or PBS and either TEPA or 232tet. Immunoblotting of cell extracts (20 μg) revealed that heme-HPX induced HMOX1 ~3 to 4-fold in copper-replete but not copper-deficient Hepa cells. Data shown (sample pixels normalized to α-tubulin pixels using the software, UNSCAN-IT) are columns, means; bars, SEM. from three independent experiments. When TEPA (10 μM) was first incubated with equimolar amounts of Cu-NTA before addition to the cell medium, it was no longer an effective inhibitor of HO1 induction (c). Data shown, normalized to α-tubulin, are from two independent experiments. Fluorescence was recorded using an Odyssey Imaging System scanner and colored images converted to grayscale with Adobe Photoshop 7.0. Increases in protein levels of the copper chaperone CCS1 confirm that copper deficiency was induced by these two cell permeable Cu(II) chelators TEPA or 232tet (d). Immuno-blot with the anti-CCS1 antibodies of Hepa cell extracts (20 μg) obtained after growing cells in DMEM containing 2% FBS in the absence or presence of either TEPA or 232tet (10, 20 or 50 μM) for 24 or 40 h, as indicated. Data shown (sample normalized to α-tubulin), are from two independent experiments. P values relative to the DMEM control are as follows * <0.05, ** <0.01 and *** <0.005. e CCS1 levels were not increased in cells after incubation with equimolar TEPA-Cu-NTA (5, 10, 20 μM) or Cu-NTA (5, 10, 20 μM) for 24 or 40 h. The P value = 0.0006 for TEPA versus TEPA/Cu-NTA, and P value = 0.0003 for TEPA versus Cu-NTA
The endosome is important for heme uptake from heme-HPX and HMOX1 induction by heme-HPX
Two properties of BCDS suggest that its inhibitory effect on the coordinated HMOX1 and MT-1 gene regulation by heme-HPX (Sung et al. 2000a, b) involve a role for copper at the cell surface. BCDS is not permeable to cells due to the presence of sulfonic groups that remain charged from pH 7.4 to 2.0 (R.C. Hider, personal communication); and BCDS has a preference for Cu(I), which is the form transported by the copper permease Ctr1 at the plasma membrane (Lee et al. 2002). We therefore, next investigated whether BCDS adversely affected receptor recognition and binding of heme-HPX since that would prevent or decrease the endocytosis of heme-HPX complexes. Surface binding and endocytosis of heme-Alexa-Fluor594 labeled HPX complexes in mouse Hepa cells takes place in the presence of BCDS (Fig. 2a). This was not unexpected since signaling to RelA/NFκB from heme-HPX is not affected by BCDS (Vanacore et al. 2000). Confirming our earlier published work (Smith and Hunt 1990), heme-HPX traffics through early endosomes, shown here by co-localization with the early endosomal antigen (EEA1; Fig. 2b). BCDS, also inhibits the induction of HMOX1 protein by heme-hemopexin (Fig. 2c), previously shown by us at the mRNA level (Sung et al. 2000a, b). Overall, these data support that copper at the cell surface available to BCDS is not needed for heme-HPX binding or endocytosis per se.

BCDS prevents the induction of HO1 protein by heme-HPX but without decreasing the binding and endocytosis of heme-HPX. To assess whether BCDS treatment impaired surface binding of heme-HPX (a), Hepa cells were incubated for 30 min with BCDS (20 μM) at 37°C, then cooled by placing on ice, followed by incubation for 1 h at 2°C (on ice) after addition of 500 nM heme-AF594HPX, prepared as described in the Sect. “Materials and Methods”. Nuclei were stained with DAPI (blue). Confirming our previous work in HepG2 cells (Smith and Hunt 1990), the diffuse red fluorescence is indicative of surface binding of heme-AF594HPX. After incubation with 500 nM heme-AF594HPX on ice for 1 h, cells were then warmed to 37°C for 10 min, to investigate endocytosis in the presence of the copper chelator, bathocuproine disulfonate, BCDS (b). In the presence of BCDS, heme-AF594HPX is rapidly taken up and produces a punctate fluorescence indicative of a location in endosomes as we have published (Smith and Hunt 1990), and co-localizes with EEA1 in early endosomes as shown here by the yellow overlay (RHS). Incubation of Hepa cells with BCDS (10 μM) for 30 min nevertheless prevents induction of HMOX1 protein by heme-HPX (10 μM for 4 h; c Shown by immunoblotting 20 μg of Hepa cell extracts as described in the Sect. “Materials and Methods” and quantitated as described in the legend to Fig. 1. The blot shown is a representative one from three independent experiments
Based on the data presented above, the lack of hmox1 regulation by CoPP-HPX is presumably not due to decreased surface binding without endocytosis. A more likely explanation is that CoPP is not released intracellularly from HPX as previously proposed (Smith et al. 1993). Alternatively, CoPP-HPX might bind at the cell surface but not undergo endocytosis. Although the signaling ligand CoPP-HPX competes with heme-HPX for surface binding sites (Smith et al. 1993), whether it is taken up by endocytosis like heme-HPX has not previously been investigated directly. We therefore next determined whether CoPP-HPX was internalized and present in endosomes using immunofluorescence microscopy. After incubation of Hepa cells with CoPP-Alexa-Fluor 594 HPX for 10 min at 37°C, the staining is punctate as seen with heme-HPX in endosomes (Fig. 3a). Evidence for the presence of CoPP-HPX within cells is provided by the Z-stack images from confocal microscopy (Fig. 3b). Furthermore, CoPP-Alexa-Fluor 594 HPX was shown to co-localize with heme-Alexa-Fluor 488 HPX (Fig. 3a). Therefore, CoPP-HPX is taken up into cells by endocytosis and traffics into the endosome, like heme-HPX.

CoPP-HPX is taken up into cells by endocytosis and co-localizes with heme-HPX in endosomes. Unlike heme-HPX, CoPP-HPX complexes are stable to the acid pH of maturing endosomes. To assess whether CoPP-HPX follows the same route of internalization as heme-HPX (a), Hepa cells were incubated for 30 min at 37°C in the presence of 500 nM heme-AF488HPX and CoPP-AF594HPX, prepared as described in the Sect. “Materials and Methods”. CoPP-HPX co-localizes with heme-HPX intracellularly as shown by the yellow overlay (a). Confocal microscopy with an Olympus Fluoview microscope (FV300) was used to demonstrate the intracellular location of CoPP-AF594HPX (b). The figure shown is an XZ image from a stock of 40 slices taken at 250 nm steps. The original magnification = 600×. To investigate whether CoPP remains bound to HPX at the acidic pH known to exist in maturing endosomes, a pH titration was carried out to determine whether there was a loss in absorbance in the Sôret region, indicative of heme bound to HPX, as the pH is decreased from neutral to 4.2–4.5 (c). On the LHS, the data show that heme binding by HPX is lost as the pH decreases but regained upon restoring the pH to neutral. In contrast to heme-HPX complexes, the absorbance of CoPP-HPX is unchanged as the pH is decreased, thus confirming the stability of this complex. Therefore, it is unlikely that CoPP is released from HPX in the endosome, consistent with the lack of HMOX1 induction by CoPP-HPX. The combined data from three independent titrations of rabbit HPX are shown. The last 3 data points in the titrations are significantly different with P values of 0.0004, 0.0007 and <0.0001, respectively
The next set of experiments show that CoPP-HPX, unlike heme-HPX, is stable at acidic pH. When heme-HPX undergoes endocytosis (Fig. 3; (Smith and Hunt 1990) within cells, these complexes are exposed to significant pH changes, e.g., from 7.4 in the plasma to ca. 6.5 in early endosomes, 5.5 in the multi-vesicular body and 6.8 in recycling endosomes (Tycko and Maxfield 1982; van Renswoude et al. 1982; Clague 1998). HPX does not bind heme at pH below five (see Fig. 4; (Morgan and Muller-Eberhard 1972) due to protonation of the heme iron-coordinating histidine residues of HPX. This has been proposed as part of the mechanism to release heme from HPX within maturing endosomes (Morgan and Smith 2001). Consistent with this, we have recently shown that acidification of the endosome is needed for HMOX1 regulation by heme-HPX (Flaherty et al. 2007) via heme translocation to the nucleus. Since, on a molar basis, CoPP induces HMOX1 mRNA more extensively than heme (Smith et al. 1993), if CoPP were released from HPX within cells, it would be a very effective inducer of HMOX1. Thus, the observed lack of HMOX1 induction by CoPP-HPX suggests that the CoPP-HPX complex is stable within endosomes. Therefore, we investigated the influence of pH on the stability of CoPP-HPX, anticipating that CoPP would not be released from HPX at acid pH. Protoheme and mesoheme are not water soluble (Margalit and Rotenberg 1984), so for these pH titration experiments the more water soluble heme analog deuteroheme was used, as published (Morgan and Muller-Eberhard 1972). Deuteroheme dissociates from rabbit HPX at low pH ~4.5 and, in our hands, is completely recovered as the pH is restored to neutral (Fig. 3c, LHS). In contrast to deuteroheme, CoPP remains bound to HPX at these acidic pHs (Fig. 3c, RHS). Taken together, our observations here support that CoPP-HPX is internalized into the same intracellular compartment as heme-HPX, and that CoPP-HPX is stable as the maturing endosomes acidify, as previously proposed (Smith et al. 1993). These data also provide additional strong evidence that heme uptake and release from HPX in endosomes is needed for HMOX1 induction.

Copper prevents the re-association of heme with apo-HPX over the pH range found in maturing endosomes. Similar pH titrations to those described in legend to Fig. 4 were carried out using rabbit (a) or human (b) HPX but when a pH of 4.2–4.5 was reached the Cu-NTA complexes were added Cu(NTA)2 at either 200:400 μM [A, top, Cu(NTA)2] or 600 μM:1.2 mM [A, middle, Cu(NTA)2] or 200 μM: 200 μM Cu-NTA [A, bottom panel and panel B, Cu-NTA] complexes were added as indicated. Only at the highest concentration of copper (~ 60 μM) was the rate at which heme rebound to HPX inhibited. The recovery of initial heme binding to rabbit or human HPX in the absence and presence of copper was 96 and 80%, respectively. Heme binding is recovered by human HPX, which is more sensitive to the inhibitory effect of copper (at 200 μM Cu-NTA). Data shown are representative of 2–3 independent titrations. For the statistical analyses, data from three independent titrations of rabbit HPX were combined and for human HPX from three independent titrations using three different protein isolates. The P values are <0.05 (*), <0.01 (**) and <0.001 (***). Small differences in the titration such as the decrease in absorbance reflecting the amount of heme bound at pH ~4.2–4.5 were apparent between rabbit and human HPX, and were consistently observed and may reflect subtle differences in the kinetics of heme binding by the two congeners. c Sites that can accommodate calcium have been identified in HPX and calcium is positively charged as is copper. However, calcium chloride (3 mM) did not inhibit heme rebinding to rabbit HPX or to human HPX (data not shown)
Evidence that copper prevents the re-association of heme with HPX over the pH range that exists in endosomes
We next addressed which intracellular sites of copper might have an impact on the cellular effects of heme-HPX endocytosis. After surface binding and internalization via coated pits, heme-HPX is found in endosomes (Smith and Hunt 1990; Fig. 2b here). Copper levels within endosomes are unknown. However, endocytosis of copper is likely for the following reasons. The iron transporter DMT1 (isoform 1A) can also transport copper across the plasma membrane, but whether as Cu(I) or Cu(II) is currently unresolved (Arredondo et al. 2003). Furthermore, a protein copper permease, Ctr2, and a reductase have been identified and proposed to function in copper export from endosomes (Rees and Thiele 2007). This would be analogous to iron export from endosomes via the divalent metal transporter DMT1 and the reductase, Steap3 (Ohgami et al. 2005). The sequestration of redox-active copper by intracellular copper-binding proteins including MTs protect cells from oxidative stress (Conrad et al. 2000). Both MT-1 and MT-2 are induced by heme-HPX (Ren and Smith 1995; Escriba et al. 2002).
Low affinity binding sites for metals, including copper, have been reported for hemopexin at pH 7.0 (Rosell et al. 2005). If the endocytosis of heme-HPX stimulates copper uptake into endosomes, then this copper may bind to receptor-associated apo-HPX at acidic pH, and prevent the re-association of heme with HPX. This would facilitate heme release from HPX and subsequent heme export from the endosome to the cytosol. To mimic conditions within the endosome, we exposed heme-HPX complexes to decreasing pH and then, at pH 4.2–4.5, added copper (as preformed nitrilotriacetate complexes, Cu-NTA, which are kinetically labile at acid pH (Bolton et al. 1996), R.C. Hider, personal communication, email July 10, 2007). The Cu-NTA complexes used generate a range of free copper concentrations (see Table 1). The highest concentration of free copper generated was ~60 μM at pH 4.0, which is 3.5-fold higher than plasma levels of ~17 μM, where copper is predominantly bound to albumin and present in ceruloplasmin (Linder 1991, 2002). No extensive inhibitory effects of copper were apparent at the lower concentrations of copper (~20 μM) generated from Cu(NTA)2 (200 μM:400 μM) or Cu(NTA)2 (600 μM:1.2 mM; Fig. 4a, top and middle panels, respectively). However, when 200 μM:200 μM Cu:NTA was added, which generates 57.5 μM “free” copper, the rate of deuteroheme rebinding to rabbit HPX was significantly slower (Fig. 4a, lower panel). In the absence of copper, 96% of the original binding was recovered by rabbit HPX and ~80% in the presence of 57.5 μM copper (Fig. 4a). About 92% of the initial heme binding was recovered with human-HPX at pH 7.0 (Fig. 4b) but was essentially prevented by this high concentration of copper (Fig. 4b). Lower amounts of copper had little effect on human HPX (data not shown). Importantly, there was no detectable change in the human HPX protein due to the exposure to copper as shown by SDS–PAGE analysis under both reducing and non-reducing conditions (data not shown). Calcium, which like copper is positively charged, has also been proposed to bind to HPX (Paoli et al. 1999). However, there was no detectable inhibitory effect of calcium (3 mM final conc.) on heme rebinding using either rabbit (Fig. 4c) or human HPX (data not shown). Thus, these data support that within the environment of mature endosomes, copper could facilitate heme release from HPX.
Evidence that heme-HPX regulates the levels of the copper chaperone CCS1
We next address what the copper targets might be for these three copper chelators. Presumably, only copper residing at the cell surface poised for uptake into cells is potentially available to all of them. It is established that copper is moved across the plasma membrane via Ctr1 (Lee et al. 2002), but passively down a concentration gradient. This copper is then transferred directly to copper chaperones in the cytosol (Rae et al. 2001) and see model depicted in Fig. 6). If, on the other hand, endocytosis of heme-HPX is accompanied by or stimulates copper endocytosis, then cytosolic copper levels would rise when copper is exported from endosomes. A copper transporter, Ctr2, has been proposed to export copper from endosomes to cytosol (Rees and Thiele 2007). Significantly, any cytosolic targets following uptake of heme-HPX are predicted to also be affected by the endocytosis of CoPP-HPX. What might this target be? MTs bind redox active copper, are induced by copper and also by heme-HPX. Proteins known to respond rapidly to changes in copper levels are copper chaperones. CCS1 is a chaperone that delivers copper to pools of nascent apo-Cu, Zn superoxide dismutase (SOD, Rae et al. 2001). Cu, ZnSOD is a cytosolic enzyme that protects cells from oxidative stress (Hunt et al. 1996). Since the HPX system is protective, if endocytosis of heme-HPX raises cell copper levels, CCS1 is a likely chaperone target. In addition, CCS1 is known to respond to changes in intracellular copper and decreases when copper levels rise (Bertinato and L’Abbe 2003). Danzeisen and coworkers (Danzeisen et al. 2007) have suggested that CCS1 is the best protein to monitor changes in copper status.
To provide evidence that endocytosis of heme-HPX increases intracellular copper, we therefore next investigated whether heme-HPX decreased CCS1 levels. To confirm that this was associated with endocytosis of HPX complexes, we also investigated whether CoPP-HPX acted analogously to heme-HPX. In order to more readily detect and monitor changes in CCS1 levels, Hepa cells were incubated with TEPA for 24 h to raise CCS1 protein as in previous studies on the copper regulation of CCS1 using rat hepatoma H4IIE cells (Bertinato and L’Abbe 2003). Incubation of TEPA-treated Hepa cells with either heme-HPX or CoPP-HPX caused a significant decrease of ~25% in CCS1 levels within 4–6 h and a decrease of 43% by 10 h (Fig. 5a, b, respectively). This is similar to the 30% decrease over 3 h when copper-deficient H4IIE cells (generated by incubation with 20 μM 232tet) were incubated with ~30 μM CuSO4 (i.e., 50 μM CuSO4 in the presence of 20 μM 232tet (Bertinato and L’Abbe 2003). We also monitored the response of Hepa cells to the addition of copper sulfate added to the medium. CCS1 levels decreased but only by 10% at 6 h and 29% at 10 h (Fig. 5b). The specificity of this effect of the HPX complexes is shown by the fact that exogenous heme (2–10 μM), which extensively induces HMOX1, does not affect CCS1 levels (Fig. 5c).

CCS1 levels are rapidly decreased by heme-HPX. a A time course study reveals that following incubation with TEPA (20 μM) for 24 h, addition of 10 μM heme-HPX (open squares) or equivalent volume of PBS (open circles) to the medium of Hepa cells caused a rapid decrease in CCS1 levels (detected by immunoblotting as described in the legend to Fig. 1). Data shown are mean ± SEM. of 10–14 data points from 3 to 4 independent experiments. CCS1 is degraded by the proteosome (Bertinato and L’Abbe 2003) and consistent with this lactacystin prevents the decrease in CCS1 by heme-HPX (filled squares). The P values shown are <0.05 (*), <0.01 (**) and <0.001 (***) for heme-HPX in the presence and absence of lactacystin (b). Incubating TEPA-treated Hepa cells with CoPP-HPX (diamonds) or PBS (circles) caused a decrease in CCS1 levels similar to heme-HPX. Data shown are mean ± SEM. of 6–12 data points from three independent experiments. As a positive control, Hepa cells are incubated with about 30 μM CuSO4 (i.e., 50 μM CuSO4 in the presence of 20 μM TEPA; triangles) following published procedures with rat hepatoma (H4IIE) cells (Bertinato and L’Abbe 2003). CCS1 levels declined but slightly more slowly than in response to the HPX complexes, Data shown are mean ± SEM. of 6–10 data points from three independent experiments. The P values shown are <0.05 (*), <0.01 (**) and <0.001 (***) versus PBS control. In contrast to the HPX complexes, exogenous heme, which is an extensive inducer of HMOX1, did not alter CCS1 levels (c). Data shown are the mean ± SEM. of 2–4 data points from two independent experiments with DMSO (open circles) or 2, 5 and 10 μM heme (open diamonds, squares and triangles, respectively)
In response to elevated copper CCS1 is degraded by the proteosome in rat IIE cells (Bertinato and L’Abbe 2003). Consistent with this, CCS1 degradation by heme-HPX in Hepa cells was prevented by the proteosome inhibitor, lactacystin (Fig. 5a). Lactacystin also prevented the decreases in CCS1 in response to CoPPHPX (data not shown). Thus, the regulation of CCS1 by both heme-HPX and CoPP-HPX support that cytosolic copper levels increase in response to the endocytosis of these two HPX complexes.
Discussion
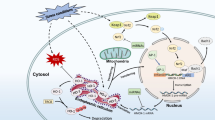
We have here tested the hypotheses that heme-HPX endocytosis mobilizes copper, alters its distribution intracellularly, and that copper plays a key role for HMOX1 regulation by heme-HPX. The data show that endocytosis of heme-HPX decreases levels of the copper chaperone CCS1, consistent with an increase in cellular copper. We have also provided evidence that copper in the endosome could prevent heme rebinding at acidic pH and thus facilitate heme release from HPX within the cell. CoPP-HPX, like heme-HPX, stimulates the nuclear translocation of MTF1, a transcriptional activator needed for MT gene regulation; and both MTF1 activation and MT-1 transcription by both HPX complexes is prevented by BCDS (Sung et al. 2000a, b; Vanacore et al. 2000). Regulation by heme-HPX of the copper chaperone CCS1 via mobilization of copper provides a means for the simultaneous regulation by heme-HPX of hmox1 with that of mt1 (see model, Fig. 6).

A model for sites for copper in the coordinate regulation of HMOX1 and MT-1 by heme-HPX. Published data support that after receptor-mediated endocytosis of heme-HPX(HHPX), heme is released from HPX within endosomes and exported (step c) for transfer to HMOX1 in the SER or into the nucleus (step d) for hmox1 regulation (overall steps a–e). The iron from heme catabolism via HMOX1 is stored on ferritin, iron uptake via transferrin and its receptor is decreased by heme-HPX (not shown). An established route for copper uptake into cytosol is via the copper chaperone, CCS1, which accepts copper from Ctr1 (step f) and delivers it to apo-superoxide dismutase (SOD). CCS1 responds to elevated copper concentrations by being targeted to the proteosome for degradation, which is prevented by lactacystin. Data presented here show that heme-HPX significantly decreases CCS1 levels within a few hours, and that this decrease is prevented by lactacystin. CCS1 also decreases when cells are incubated with a source of copper, or when cells are incubated with CoPP-HPX, which co-localizes with heme-HPX in endosomes. Overall, these observations strongly support that endocytosis of heme-HPX increases cytosolic copper. If endocytosis of copper occurs (see step a) perhaps via DMT1 and together with heme-HPX, our data support that under conditions that mimic maturing endosomes, copper prevents the rebinding of heme to HPX. Thus, copper availability potentially affects release of heme from HPX within endosomes making heme available for export from endosomes for hmox1 regulation. Copper has been proposed to be exported from endosomes to cytosol (Rees and Thiele 2007) as Cu(I) via Ctr2, a Ctr1-like protein with an associated reductase, Fre6. Alternatively, DMT1 may play a role. The coordinate regulation of hmox1 with mt1 (f–i) by heme-HPX via MTF1 (Ren and Smith 1995; Vanacore et al. 2000) is proposed to take place in response to a rise in cytosolic copper (f), displacing zinc from MTs due to its higher affinity (step g). This zinc then binds to a regulatory zinc-finger on the transcription factor, MTF1, stimulating its nuclear translocation (step h) and leading to activation of the mt1 gene (i). Overall, our data with TEPA, 232tet and BCDS, support that in addition to the cell surface where copper is accessible to all three chelators, a dynamic intracellular pool of copper affects both hmox1 and mt1 regulation by heme-HPX, which is proposed here to be linked by CCS1
First, using two models of nutritional copper deficiency, we confirmed and extended our previous work with the copper chelator, BCDS, and show that copper is the trace metal needed for induction of HMOX1 by heme-HPX in mouse Hepa cells. The ~1.5 to 1.6-fold rise in CCS1 protein levels in response to copper starvation created by TEPA or 232tet is similar to the doubling seen in response to 48 h treatment of rat hepatoma H4IIE cells with 20 μM 232tet (Bertinato and L’Abbe 2003). CCS1 levels are known to increase to varying degrees in different cell types and cell lines in response to low copper (Bertinato and L’Abbe 2003). In response to copper-deficiency induced by short term, mild copper chelation (e.g., 6–10 μM BCDS Sung et al. 2000b) and two nutritional copper-deficiency models (e.g., TEPA or 232tet at 5–10 μM), which were neither toxic nor chronic, there was no longer any induction of HMOX1 protein by heme-HPX in copper-deficient Hepa cells.
Levels of the copper chaperone CCS1 are shown here to rapidly decline in response to heme-HPX. Such a decline has previously been linked to increases in cellular copper upon incubation of cells with copper sulfate, followed by CCS1 degradation via the proteosome (Bertinato and L’Abbe 2003). Consistent with this, lactacystin prevented the decrease in CCS1 following incubation of cells with heme-HPX. Since CCS1 levels also decrease in response to CoPP-HPX, which was shown here to co-localize with heme-HPX in endosomes, our data support that endocytosis of HPX complexes is linked with an increase in cellular copper likely via copper uptake. In contrast, non-HPX bound heme had no effect on CCS1.
How might copper get across the plasma membrane into the cell? Cu(II) might be endocytosed via the divalent metal transporter, DMT1, that transports iron (Picard et al. 2000) and other metals including copper (Arredondo et al. 2003). DMT1 acts both at the plasma membrane for metal uptake and to export metal out of the endosome (Andrews 1999; Garrick et al. 1999). Normal endosome maturation and function is needed for HMOX1 induction by heme-HPX since when endosome acidification is prevented by bafilomycin, HMOX1 is no longer induced (Flaherty et al. 2007). Here, we have provided evidence for a role for copper in the endosome, which would facilitate heme export from this organelle by preventing heme rebinding to apo-HPX. This occurs over a pH range known to exist within endosomes. Furthermore, it is consistent with copper-binding sites on apo-HPX (Mauk et al. 2007) and that copper-binding interferes with the stability of heme-HPX complexes (Mauk et al. 2005). The reported relative affinities for metal-HPX interactions (e.g., Kds of ~200 μM and 2 mM for Mg characterized at pH 7.0 using metal affinity chromatography and potentiometry; (Mauk et al. 2005; Rosell et al. 2005) are potentially too low for HPX to play a role in the circulation at normal plasma copper levels (i.e., ~17 μM). However, higher metal concentrations are likely generated within the lumen of small intracellular vesicles, such as endosomes. The acidification of endosomes is accompanied by increases in chloride entry (Sonawane et al. 2002). Physiologically relevant endosomal chloride concentrations range from ~20 to ~75 mM upon acidification from pH 6.95 to 5.3 and vary slightly with cell type (Sonawane et al. 2002). Our titrations were carried out in this range with 100 mM KCl, and potassium is the physiologically relevant intracellular ion rather than sodium. Heme rebinding to rabbit or human HPX using conditions in vitro that mimic the endosome during ligand uptake and receptor recycling is significantly inhibited by copper. HPX has several sites that might bind positively charged calcium (Paoli et al. 1999). Calcium plays an important role in intracellular trafficking including endosomes (Sandvig et al. 2002) The specificity of these effects of copper is supported by our observation that calcium did not significantly decrease heme rebinding to HPX. In intact cells, due to the insolubility of protoheme at acid pH, close interactions between receptor-bound HPX and the putative endosomal heme exporter are anticipated. Heme exported from the endosome is then transported by unknown carrier(s) to the SER for catabolism by HMOX1 and also to the nucleus for HMOX1 gene regulation via Bach1 de-repression (Alam et al. 2003; Sun et al. 2004).
An increase in cytosolic copper in response to heme-HPX provides an explanation for mt1 activation by heme-HPX (Ren and Smith 1995; Sung et al. 2000a) via the pathway shown in Fig. 6. Copper endocytosis and endosomal export of copper (e.g., Cu(I) via Ctr2) to the cytosol more readily explains the simultaneous regulation of HMOX1 with MT-1 by heme-HPX than Cu(I) uptake to CCS1 via the copper permease Ctr1 in the plasma membrane (see Fig. 6). One possible candidate for copper endocytosis is the divalent metal transporter, DMT1, which transports iron across the plasma membrane (e.g., the 1A/IRE + isoform in enterocytes) and exports Fe(II) from transferrin in endosomes (e.g., the 1B/IRE- isoform in reticulocytes MacKenzie et al. 2007). As mentioned above, DMT1 has been proposed to transport copper and several other metals as well; although, direct evidence is lacking. In addition, there is some controversy as to whether Cu(I) or Cu(II) is transported by DMT1 (Arredondo et al. 2003). Although we have described a role for DMT1 in copper endocytosis, we cannot rule out a contribution from Ctr1 situated in the plasma membrane. When human Ctr1 is overexpressed in human embryonic kidney cells it undergoes endocytosis but only in response to high levels of copper. Ctr1 is subsequently degraded thus potentially minimizing further copper uptake (Petris et al. 2003).
Both heme-HPX and CoPP-HPX activate mt1 transcription (Ren and Smith 1995) and both complexes stimulate the nuclear translocation of the metal transcription factor-1 (MTF-1) needed for mt1 regulation (Heuchel et al. 1994). Significantly, MT-1mRNA induction and MTF1 activation by CoPP-HPX, as well as by heme-HPX, are prevented by BCDS (Vanacore et al. 2000). CoPP from CoPP-HPX does not accumulate intracellularly (Smith et al. 1993), as does heme from heme-HPX, thus endogenous HMOX1 is not induced. Nevertheless, CoPP-HPX activates the JNK and RelA/NFκB signaling cascades as does heme-HPX (Smith et al. 1993). Overall, these published data indicate that there is a copper-dependent step in the pathway for mt1 gene regulation via HPX receptors, which can be distinguished from that in heme uptake. We show here that BCDS does not prevent surface binding and uptake of heme-HPX by Hepa cells. In our model (Fig. 6), the path to MT-1 regulation via MTF1 is activated by increasing cytosolic copper because this copper will displace zinc from its storage sites on MT, since it is bound more tightly (Waalkes et al. 1984). Zinc then binds to a “zinc sensor” Zn finger on MTF1, so called since the zinc is interchangeable (Dalton et al. 1997; Bittel et al. 1998) which activates this transcription factor needed for mt1 transcription (Jiang et al. 2003). This zinc displacement has also been previously proposed (Tapia et al. 2004). We favor that copper enters cells when heme-HPX is taken up by endocytosis since this helps explain why both hmox1 and mt1 regulation by heme-HPX is prevented by copper chelators.
To summarize, we have found that three structurally distinct copper chelators BCDS, TEPA and 232tet all consistently prevent HMOX1 induction by heme-HPX. Heme-HPX increases cytosolic copper since CCS1 levels decrease when Hepa cells are incubated with heme-HPX. Thus, in addition to an extracellular source of copper at the plasma membrane (either Cu(II) potentially available to all of the chelators, or Cu(I) for TEPA or BCDS), our data support that a dynamic intracellular pool of cytosolic copper affects HMOX1 and MT-1 gene regulation by heme-HPX. Copper in the maturing endosome would facilitate heme release from HPX. We propose that CCS1 is one direct link between endosomal copper and the pathway for mt1 regulation by heme-HPX. This is currently under further investigation.
In conclusion, since heme catabolism via HMOX1 raises intracellular regulatory iron pools, these data show for the first time using the HPX system that heme and iron homeostasis in mammalian cells are linked with that of copper at the HPX-HMOX1 axis. Cells replete with HMOX1, Cu, ZnSOD and MTs would be able to survive the oxidative stress in situations where HPX is protective such as the inflammation that accompanies hemolysis, trauma, reperfusion injury as well as infection. Our research here points to the potential importance of assessing copper status in patients with hematological and neurological disorders since copper is needed for the simultaneous regulation of hmox1 and mt1 by the HPX system.
References
Alam J, Smith A (1989) Receptor-mediated transport of heme by hemopexin regulates gene expression in mammalian cells. J Biol Chem 264:17637–17640
Alam J, Smith A (1992) Heme-hemopexin-mediated induction of metallothionein gene expression. J Biol Chem 267(23):16379–16384
Alam J, Killeen E, Gong P, Naquin R, Hu B, Stewart D, Ingelfinger JR, Nath KA (2003) Heme activates the heme oxygenase-1 gene in renal epithelial cells by stabilizing Nrf2. Am J Physiol Renal Physiol 284(4):F743–F752
Andrews NC (1999) The iron transporter DMT1. Int J Biochem Cell Biol 31(10):991–994. doi:10.1016/S1357-2725(99)00065-5
Arredondo M, Munoz P, Mura CV, Nunez MT (2003) DMT1, a physiologically relevant apical Cu1+ transporter of intestinal cells. Am J Physiol Cell Physiol 284(6):C1525–C1530
Bertinato J, L’Abbe MR (2003) Copper modulates the degradation of copper chaperone for Cu, Zn superoxide dismutase by the 26 S proteosome. J Biol Chem 278(37):35071–35078. doi:10.1074/jbc.M302242200
Bittel D, Dalton T, Samson SL, Gedamu L, Andrews GK (1998) The DNA binding activity of metal response element-binding transcription factor-1 is activated in vivo and in vitro by zinc, but not by other transition metals. J Biol Chem 273(12):7127–7133. doi:10.1074/jbc.273.12.7127
Bolton H, Girvin DC, Plymale AE, Harvey SD, Workman DJ (1996) Degradation of metal-nitrilotriacetate complexes by Chelatobacter heintzii. Environ Sci Technol 30:931–938. doi:10.1021/es950397k
Brown SB, Lantzke IR (1969) Solution structures of ferrihaem in some dipolar aprotic solvents and their binary aqueous mixtures. Biochem J 115(2):279–285
Chung J, Prohaska JR, Wessling-Resnick M (2004) Ferroportin-1 is not upregulated in copper-deficient mice. J Nutr 134(3):517–521
Clague MJ (1998) Molecular aspects of the endocytotic pathway. Biochem J 336:271–282
Cobine PA, Pierrel F, Winge DR (2006) Copper trafficking to the mitochondrion and assembly of copper metalloenzymes. Biochim Biophys Acta 1763(7):759–772. doi:10.1016/j.bbamcr.2006.03.002
Conrad CC, Grabowski DT, Walter CA, Sabia M, Richardson A (2000) Using MT (−/−) mice to study metallothionein and oxidative stress. Free Radic Biol Med 28(3):447–462. doi:10.1016/S0891-5849(99)00263-4
Dalton TP, Bittel D, Andrews GK (1997) Reversible activation of mouse metal response element-binding transcription factor 1 DNA binding involves zinc interaction with the zinc finger domain. Mol Cell Biol 17(5):2781–2789
Danzeisen R, Araya M, Harrison B, Keen C, Solioz M, Thiele D, McArdle HJ (2007) How reliable and robust are current biomarkers for copper status? Br J Nutr 98:1–8
Davies DM, Smith A, Muller-Eberhard U, Morgan WT (1979) Hepatic subcellular metabolism of heme from heme-hemopexin: incorporation of iron into ferritin. Biochem Biophys Res Commun 91:1504–1511. doi:10.1016/0006-291X(79)91235-X
Escriba PV, Morales P, Smith A (2002) Membrane phospholipid reorganization differentially regulates metallothionein and heme oxygenase by heme-hemopexin. DNA Cell Biol 21:355–364. doi:10.1089/104454902753759762
Eskew JD, Vanacore RM, Sung L, Morales PJ, Smith A (1999) Cellular protection mechanisms against extracellular heme: heme-hemopexin, but not free heme, activates the N-terminal c-Jun kinase. J Biol Chem 274(2):638–648. doi:10.1074/jbc.274.2.638
Flaherty MM, Rish KR, Smith A, Crumbliss AL (2007) An investigation of hemopexin redox properties by spectroelectrochemistry: biological relevance for heme uptake. Biometals 21(3):259–271
Fridovich I (1995) Superoxide radical and superoxide dismutases. Annu Rev Biochem 64:97–112. doi:10.1146/annurev.bi.64.070195.000525
Garrick LM, Dolan KG, Romano MA, Garrick MD (1999) Non-transferrin-bound iron uptake in Belgrade and normal rat erythroid cells. J Cell Physiol 178(3):349–358. doi:10.1002/(SICI)1097-4652(199903)178:3<349::AID-JCP9>3.0.CO;2-R
Gutteridge JM, Smith A (1988) Antioxidant protection by haemopexin of haem-stimulated lipid peroxidation. Biochem J 256:861–865
Han O, Wessling-Resnick M (2002) Copper repletion enhances apical iron uptake and transepithelial iron transport by Caco-2 cells. Am J Physiol Gastrointest Liver Physiol 282(3):G527–G533
Heuchel R, Radtke F, Georgiev O, Stark G, Aguet M, Schaffner W (1994) The transcription factor MTF–1 is essential for basal and heavy metal- induced metallothionein gene expression. EMBO J 13(12):2870–2875
Hopkins RG, Failla ML (1999) Transcriptional regulation of interleukin-2 gene expression is impaired by copper deficiency in Jurkat human T lymphocytes. J Nutr 129(3):596–601
Hunt RC, Handy I, Smith A (1996) Heme-mediated reactive oxygen species toxicity to retinal pigment epithelial cells is reduced by hemopexin. J Cell Physiol 168(1):81–86. doi:10.1002/(SICI)1097-4652(199607)168:1<81::AID-JCP10>3.0.CO;2-S
Jiang H, Daniels PJ, Andrews GK (2003) Putative zinc-sensing zinc fingers of metal-response element-binding transcription factor-1 stabilize a metal-dependent chromatin complex on the endogenous metallothionein-I promoter. J Biol Chem 278(32):30394–30402. doi:10.1074/jbc.M303598200
Johnson MB, Enns CA (2004) Diferric transferrin regulates transferrin receptor 2 protein stability. Blood 104(13):4287–4293. doi:10.1182/blood-2004-06-2477
Ledford BE, Davis DF (1983) Kinetics of serum protein secretion by cultured hepatoma cells evidence for multiple secretory pathways. J Biol Chem 258:3304–3308
Lee J, Pena MM, Nose Y, Thiele DJ (2002) Biochemical characterization of the human copper transporter Ctr1. J Biol Chem 277(6):4380–4387. doi:10.1074/jbc.M104728200
Linder MC (1991) Biochemistry of copper. Plenum Press, NewYork
Linder MC (2002) Biochemistry and molecular biology of copper in mammals. Humana Press Inc., Totowa
MacKenzie B, Takanaga H, Hubert N, Rolfs A, Hediger MA (2007) Functional properties of multiple isoforms of human divalent metal-ion transporter 1 (DMT-1). Biochem J 403(1):59–69. doi:10.1042/BJ20061290
Margalit R, Rotenberg M (1984) Thermodynamics of porphyrin dimerization in aqueous solutions. Biochem J 219(2):445–450
Mauk MR, Rosell FI, Lelj-Garolla B, Moore GR, Mauk AG (2005) Metal ion binding to human hemopexin. Biochemistry 44(6):1864–1871. doi:10.1021/bi0481747
Mauk MR, Rosell FI, Mauk AG (2007) Structural modelling of metal ion binding to human haemopexin. Nat Prod Rep 24(3):523–532. doi:10.1039/b604184c
Morgan WT, Muller-Eberhard U (1972) Interactions of porphyrins with rabbit hemopexin. J Biol Chem 247:7181–7187
Morgan WT, Smith A (1984) Domain structure of rabbit hemopexin. Isolation and characterization of a heme-binding glycopeptide. J Biol Chem 259:12001–12006
Morgan W, Smith A (2001) Binding and transport of iron-porphyrins by hemopexin. In: Mauk G, Sykes AG (eds) Advances in inorganic chemistry, vol 51. Academic Press, New York
Narayanan VS, Fitch CA, Levenson CW (2001) Tumor suppressor protein p53 mRNA and subcellular localization are altered by changes in cellular copper in human HepG2 cells. J Nutr 131(5):1427–1432
Ohgami RS, Campagna DR, Greer EL, Antiochos B, McDonald A, Chen J, Sharp JJ, Fujiwara Y, Barker JE, Fleming MD (2005) Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat Genet 37(11):1264–1269. doi:10.1038/ng1658
Okazaki H, Taketani S, Kohno H, Tokunaga R, Kobayashi Y (1989) The hemopexin receptor on the cell surface of human polymorphonuclear leukocytes. Cell Struct Funct 14:129–140
Paoli M, Anderson BF, Baker HM, Morgan WT, Smith A, Baker EN (1999) Crystal structure of hemopexin reveals a novel high-affinity heme site formed between two beta-propeller domains. Nat Struct Biol 6(10):926–931. doi:10.1038/13294
Petris MJ, Smith K, Lee J, Thiele DJ (2003) Copper-stimulated endocytosis and degradation of the human copper transporter, hCtr1. J Biol Chem 278(11):9639–9646. doi:10.1074/jbc.M209455200
Picard V, Govoni G, Jabado N, Gros P (2000) Nramp 2 (DCT1/DMT1) expressed at the plasma membrane transports iron and other divalent cations into a calcein-accessible cytoplasmic pool. J Biol Chem 275(46):35738–35745. doi:10.1074/jbc.M005387200
Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O’Halloran TV (1999) Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science 284(5415):805–808. doi:10.1126/science.284.5415.805
Rae TD, Torres AS, Pufahl RA, O’Halloran TV (2001) Mechanism of Cu, Zn-superoxide dismutase activation by the human metallochaperone hCCS. J Biol Chem 276(7):5166–5176. doi:10.1074/jbc.M008005200
Rees EM, Thiele DJ (2007) Identification of a vacuole-associated metalloreductase and its role in CTR2-mediated intracellular copper mobilization. J Biol Chem 282:21629–21638
Ren Y, Smith A (1995) Mechanism of metallothionein gene regulation by heme-hemopexin. Roles of protein kinase C, reactive oxygen species, and cis-acting elements. J Biol Chem 270(41):23988–23995. doi:10.1074/jbc.270.41.23988
Rish KR, Swartzlander R, Sadikot TN, Berridge MV, Smith A (2007) Evidence that heme and heme-hemopexin interact with cell growth-associated plasma membrane electron transport. Biochem Biophys Acta Bioenerg 1767(9):1107–1117. doi:10.1016/j.bbabio.2007.06.003
Rosell FI, Mauk MR, Mauk AG (2005) pH- and metal ion-linked stability of the hemopexin-heme complex. Biochemistry 44(6):1872–1879. doi:10.1021/bi0480077
Sandvig K, Grimmer S, Lauvrak SU, Torgersen ML, Skretting G, van Deurs B, Iversen TG (2002) Pathways followed by ricin and Shiga toxin into cells. Histochem Cell Biol 117(2):131–141. doi:10.1007/s00418-001-0346-2
Schmidt PJ, Ramos-Gomez M, Culotta VC (1999) A gain of superoxide dismutase (SOD) activity obtained with CCS, the copper metallochaperone for SOD1. J Biol Chem 274(52):36952–36956. doi:10.1074/jbc.274.52.36952
Smith A (1999) Assessment of roles of redox-active iron and copper in heme uptake and gene regulation by heme-hemopexin. In: Ferreira GC, Moura JJG, Franco R (eds) Inorganic biochemistry and regulatory mechanisms of iron metabolism, chap 5. Wiley-VCH Publishing Co, Germany
Smith A, Hunt RC (1990) Hemopexin joins transferrin as representative members of a distinct class of receptor-mediated endocytic transport systems. Eur J Cell Biol 53:234–245
Smith A, Ledford BE (1988) Expression of the haemopexin-transport system in cultured mouse hepatoma cells. Links between haemopexin and iron metabolism. Biochem J 256:941–950
Smith A, Morgan WT (1978) Transport of heme by hemopexin to the liver: evidence for receptor-mediated uptake. Biochem Biophys Res Commun 84:151–157. doi:10.1016/0006-291X(78)90276-0
Smith A, Morgan WT (1979) Haem transport to the liver by haemopexin. Receptor-mediated uptake with recycling of the protein. Biochem J 182:47–54
Smith A, Morgan WT (1981) Hemopexin-mediated transport of heme into isolated rat hepatocytes. J Biol Chem 256:10902–10909
Smith A, Alam J, Escriba PV, Morgan WT (1993) Regulation of heme oxygenase and metallothionein gene expression by the heme analogs, cobalt-, and tin-protoporphyrin. J Biol Chem 268(10):7365–7371
Smith A, Eskew JD, Borza CM, Pendrak M, Hunt RC (1997) Role of heme-hemopexin in human T-lymphocyte proliferation. Exp Cell Res 232(2):246–254. doi:10.1006/excr.1997.3526
Sonawane ND, Thiagarajah JR, Verkman AS (2002) Chloride concentration in endosomes measured using a ratioable fluorescent Cl- indicator: evidence for chloride accumulation during acidification. J Biol Chem 277(7):5506–5513. doi:10.1074/jbc.M110818200
Sun J, Brand M, Zenke Y, Tashiro S, Groudine M, Igarashi K (2004) Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc Natl Acad Sci USA 101(6):1461–1466. doi:10.1073/pnas.0308083100
Sung L, Morales P, Shibata M, Shipulina N, Smith A (2000a) Defenses against extracellular heme-mediated oxidative damage: use of iron and copper chelators to investigate the role of redox active iron, copper and heme in the hemopexin heme transport system. In: Badman DG, Bergeron RJ, Brittenham GM (eds) Iron chelators: new developmental strategies. Saratoga Publishing Group, Saratoga, Florida, USA, pp 67–86
Sung L, Womack M, Shipulina N, Morales P, Smith A (2000b) Cell surface events for metallothionein-1 and heme oxygenase-1 regulation by the hemopexin heme transport system. Antioxid Redox Signal 2(4):753–765. doi:10.1089/ars.2000.2.4-753
Tapia L, Gonzalez-Aguero M, Cisternas MF, Suazo M, Cambiazo V, Uauy R, Gonzalez M (2004) Metallothionein is crucial for safe intracellular copper storage and cell survival at normal and supra-physiological exposure levels. Biochem J 378(Pt 2):617–624. doi:10.1042/BJ20031174
Tolosano E, Hirsch E, Patrucco E, Camaschella C, Navone R, Silengo L, Altruda F (1999) Defective recovery and severe renal damage after acute hemolysis in hemopexin-deficient mice. Blood 94(11):3906–3914
Tolosano E, Fagoonee S, Hirsch E, Berger FG, Baumann H, Silengo L, Altruda F (2002) Enhanced splenomegaly and severe liver inflammation in haptoglobin/hemopexin double-null mice after acute hemolysis. Blood 100(12):4201–4208. doi:10.1182/blood-2002-04-1270
Tycko B, Maxfield FR (1982) Rapid acidification of endocytic vesicles containing alpha 2-macroglobulin. Cell 28(3):643–651. doi:10.1016/0092-8674(82)90219-7
Uauy R, Olivares M, Gonzalez M (1998) Essentiality of copper in humans. Am J Clin Nutr 67(5 Suppl):952S–959S
van Renswoude J, Bridges KR, Harford JB, Klausner RD (1982) Receptor-mediated endocytosis of transferrin and the uptake of fe in K562 cells: identification of a nonlysosomal acidic compartment. Proc Natl Acad Sci USA 79(20):6186–6190. doi:10.1073/pnas.79.20.6186
Vanacore R, Eskew J, Morales P, Sung L, Smith A (2000) Role for copper in transient oxidation and nuclear translocation of MTF-1, but not of NFκB, by the hemopexin heme transport system. Antioxid Redox Signal 2(4):739–752. doi:10.1089/ars.2000.2.4-739
Vretblad P, Hjorth R (1977) The use of wheat-germ lectin-Sepharose for the purification of human haemopexin. Biochem J 167:759–764
Vulpe CD, Kuo YM, Murphy TL, Cowley L, Askwith C, Libina N, Gitschier J, Anderson GJ (1999) Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet 21(2):195–199. doi:10.1038/5979
Waalkes MP, Harvey MJ, Klaassen CD (1984) Relative in vitro affinity of hepatic metallothionein for metals. Toxicol Lett 20(1):33–39. doi:10.1016/0378-4274(84)90179-6
West EC, Prohaska JR (2004) Cu, Zn-superoxide dismutase is lower and copper chaperone CCS is higher in erythrocytes of copper-deficient rats and mice. Exp Biol Med (Maywood) 229(8):756–764
Acknowledgments
The authors wish to thank Dr. M. Ferrari (University of Missouri-K.C.) for his help with confocal microscopy; and are greatly indebted to Dr. J. Prohaska (University of Minnesota) for affinity purified antibodies to CCS1 and to Dr. R.C. Hider (Kings College, London, UK.) for the physical constants for the metal chelators, for the characterization of Cu-NTA complexes (speciation plots and the calculations of free copper from Cu-NTA complexes at different pHs). Drs. G. Anderson (QMIR, Brisbane, Australia), R.C. Hider and J. Price (University of Missouri-K.C.) are also thanked for reading the manuscript and for their helpful comments. This research was supported by the National Institutes of Health (R21DK 64363 to A.S.) and by a grant from the University of Missouri Research Board (A.S.). The authors declare no competing financial interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authorship contributions
R.L. and K.R. carried out experiments, participated in the data statistical analyses and helped set up the figures; R.H. and J.H. carried out the fluorescence microscopy experiments and R.H. made the microscopy figures. A.S. defined the experimental strategy, designed and supervised the research, interpreted data and wrote the manuscript.
Rights and permissions
About this article
Cite this article
Smith, A., Rish, K.R., Lovelace, R. et al. Role for copper in the cellular and regulatory effects of heme-hemopexin. Biometals 22, 421–437 (2009). https://doi.org/10.1007/s10534-008-9178-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10534-008-9178-z