
Abstract
The field of the biology of ageing has received increasing attention from a biomedical point of view over the past decades. The main reason has been the realisation that increases in human population life expectancy are accompanied by late onset diseases. Indeed, ageing is the most important risk factor for a number of neoplastic, neurodegenerative and metabolic pathologies. Advances in the knowledge of the genetics of ageing, mainly through research in model organisms, have implicated various cellular processes and the respective signalling pathways that regulate them in cellular and organismal ageing. Associated with ageing is a dysregulation of metabolic homeostasis usually manifested as age-related obesity, diminished insulin sensitivity and impaired glucose and lipid homeostasis. Metabolic deterioration contributes to the ageing phenotype and metabolic pathologies are thought to be one of the main factors limiting the potential for lifespan extension. Great efforts have been directed towards identifying pharmacological interventions with the potential to improve healthspan and a number of natural and synthetic compounds have shown promise in achieving beneficial metabolic effects.
Similar content being viewed by others


Introduction
Cell signalling pathways process cues from the extracellular environment and signals of cellular status to ensure cells respond appropriately to maintain their homeostasis. Metabolic homeostasis is a key component of cellular and organismal homeostasis. In multicellular organisms, metabolic homeostasis requires the coordinate response of distinct cell and tissue types. Cell signalling pathways that sense the availability of nutrients and the energy status of the cells communicate with hormonal and growth factor signalling pathways to co-ordinately regulate whole body metabolic homeostasis. Ageing results in gradual deterioration of various cellular functions including of metabolic regulation. The age-related decline in metabolic homeostasis is likely an important contributing factor to general organismal ageing, as a number of interventions, genetic and pharmacological, affecting the activity of metabolic pathways also affect the rate of ageing. Indeed, the Insulin/IGF-1 Signalling (IIS) pathway and the mechanistic Target Of Rapamycin (mTOR) pathway are the most extensively studied pathways shown to regulate lifespan and healthspan in a number of model organisms (Fontana et al. 2010; Kenyon 2010). Moreover, calorie restriction, the most potent environmental intervention shown to extend lifespan and healthspan in a number of species, is accompanied by alterations in the insulin/IGF-1 circulating levels, whereas inhibition of the mTOR pathway has been shown to have both common and distinct effects with calorie restriction (Kaeberlein and Kennedy 2009; Miller et al. 2014).
The relationship between ageing and metabolic regulation is bidirectional: Ageing impairs the activity of key metabolic signalling pathways and the ensuing metabolic dysregulation results in accelerated ageing. For example, age-related impairment in the activity of the insulin signalling pathway results in insulin resistance. The ensuing hyperglycemia, as a result of dysregulated glucose clearance, promotes formation of advanced glycation end products (AGEs), which in turn cause tissue damage further exacerbating metabolic dysregulation and accelerating the organismal ageing process (Semba et al. 2010).
The finding that loss-of-function mutations in genes encoding components of the IIS pathway can improve health span and in many cases the metabolic profile of aged mice is seemingly paradoxical (Barzilai et al. 2012). Various potential mechanisms have been proposed to explain this phenomenon. An important implication of these findings is that pharmacological targeting of metabolic signalling pathways can produce beneficial metabolic effects and ameliorate age-related metabolic pathologies. The activity of cell signalling pathways can readily be manipulated, as a large number of their component molecules are enzymes such as kinases and phosphatases, which can be inhibited or activated with the use of natural or synthetic compounds.
Here we summarise some key findings on the role of signalling pathways in metabolic homeostasis over the course of ageing, generated mainly through research in model organisms, and the evidence supporting pharmacological manipulation of these pathways as a means to improve metabolic health at old age.
Calorie restriction in lifespan and healthspan
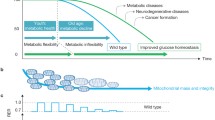
Calorie restriction is the most potent environmental intervention known to increase lifespan and healthspan in a number of species including primates (Colman et al. 2014). The molecular mechanisms underlying the beneficial effects of calorie restriction on lifespan and healthspan have been extensively studied and debated over the years (Masoro 2009). Various mechanisms have been proposed to explain the effects of calorie restriction ranging from enhanced stress resistance and improved proteostasis (Mitchell et al. 2016) to reduced inflammation (Chung et al. 2002). With regards to metabolic effects, calorie restricted animals are normally leaner and more insulin sensitive and glucose tolerant than ad libitum fed animals. It has recently been reported that calorie restriction induces browning of the adipose tissue with profound beneficial metabolic consequences (Fabbiano et al. 2016). Also calorie restriction was recently reported to protect from accelerated ageing induced by DNA-repair deficiency (Vermeij et al. 2016). Significant progress has been made in the identification of the signalling pathways mediating the effects of calorie restriction (Fig. 1). Growth factor, energy and nutrient sensing pathways likely have a prominent role in mediating the effects of calorie restriction (Anderson and Weindruch 2010; Lopez-Lluch and Navas 2016). The fact that circulating IGF-1 and insulin levels in calorie restricted animals are lower than those in ad libitum fed ones (Argentino et al. 2005; Huffman et al. 2008; Weiss et al. 2006) has pointed to a potential role for the somatotropic (Growth Hormone (GH)/IGF-1) axis and the IIS pathways in the mediation of the effects of calorie restriction. Genetic evidence has lent further support for a role of somatotropic signalling (Bonkowski et al. 2006). Also, the mTOR and AMP-activated protein kinase (AMPK) pathways together with sirtuins, a family of NAD-dependent deacetylases have also been implicated as effectors of the benefits associated with calorie restriction. In line with genetic studies, unbiased gene expression analyses in tissues of calorie restricted animals have revealed that genes encoding components of the somatotropic and the IIS pathways as well as genes involved in metabolic processes and energy metabolism are consistently part of the molecular signature of calorie restriction (Plank et al. 2012). Therefore, pharmacological agents targeting components of these pathways might have the potential to mimic the beneficial effects of calorie restriction. The field of calorie restriction mimetic compounds has been an area of intensive research, as it holds great promise for therapeutic applications in the combat against age-related diseases. Below, we briefly summarise the key evidence implicating these cell signalling pathways in lifespan and healthspan extension and we discuss the progress in pharmacological targeting of these pathways with a focus on improvements in metabolic homeostasis.

Signalling pathways implicated in age-related metabolic decline. Calorie restriction is the most potent environmental intervention that improves the metabolic profile and extends healthspan and lifespan of various animal species. Calorie restriction is thought to be suppressing the GH/IGF-1, insulin/PI3K, and mTOR pathways and activating the AMPK pathway. These pathways provide potential targets for therapeutic intervention to improve metabolic homeostasis at old age. Pointed arrows indicate activatory and blunt arrows inhibitory actions
Growth factor, energy and nutrient sensing pathways in metabolic regulation and ageing
The main cell signalling pathways that have been implicated in the modulation of the rate of ageing have at the same time important roles in metabolic regulation. These are the somatotropic axis, insulin/IGF-1, mTOR and AMPK signalling pathways. These pathways are interlinked to ensure coordinate regulation and fine-tuning of cellular metabolic responses in line with cellular energy status, nutrient availability and hormonal/growth factor signalling input (Fig. 2). Feedback loops operate within the pathways to regulate signal intensity and duration. A key feedback mechanism for downregulation of IIS involves phosphorylation of the Insulin Receptor Substrates (IRS) by p70 ribosomal protein S6-kinase-1 (S6K1) following activation of mTOR (Harrington et al. 2004; Shah et al. 2004). In fact, sustained activation of S6K1 and various other stress-induced serine/threonine kinases is thought to be a major cellular mechanism in the development of insulin resistance (Tanti and Jager 2009). Key mediators of the metabolic effects of the IIS pathway are Phosphoinositide 3-Kinase (PI3K) and its downstream effectors, serine/threonine kinase Akt and FOXO transcription factors (Whiteman et al. 2002). FOXO transcription factors are essential mediators of the lifespan extending effects of IIS attenuation (Martins et al. 2016). Consistent with this, the gene encoding FOXO3 is one of few human genes consistently associated with longevity in a number of distinct populations (Morris et al. 2015). FOXO transcription factors have multiple metabolic effects. Notably, FOXO1 plays an essential role in the regulation of hepatic glucose production (Gross et al. 2008). mTOR’s most extensively studied role is in the regulation of protein translation. mTOR key effectors in this process are the above mentioned S6K1 and the translational repressor eIF4E-Binding Protein 1 (4EBP1). mTOR has also prominent roles in lipid biosynthesis (Caron et al. 2015).

Interrelationships between growth factor, nutrient availability and energy sensing pathways in metabolic regulation in health. The Ras/ERK and PI3K/Akt pathways are activated upon insulin/IGF1 stimulation. Akt, a key effector of insulin/PI3K signalling mediates most of the metabolic actions of insulin, notably stimulation of glucose uptake and glycogen synthesis and inhibition of lipolysis. The mTOR pathway integrates signals from growth factor stimulation (via Akt), aminoacid availability and energy status (via AMPK). mTOR-activated S6K1 is a key component of a feedback loop that downregulates insulin's signal. FOXO transcription factors, which upon phosphorylation by Akt are inhibited through nuclear exclusion, also have metabolic roles, prominently in the regulation of gluconeogenesis. AMPK is activated by low energy (high AMP/ATP and/or ADP/ATP ratio) stress via phosphorylation by LKB1 and promotes glucose uptake and fatty acid oxidation. The majority of the molecular components of these pathways have been omitted from the schematic for simplicity
Closely intertwined with the IIS and mTOR pathways is the Liver Kinase B (LKB) 1/AMPK pathway, which plays a role in energy status sensing (Garcia and Shaw 2017). Overexpression of one of the AMPK subunits has been shown to increase lifespan in C. elegans (Apfeld et al. 2004). AMPK activation brings about beneficial metabolic effects mainly by promoting glucose uptake and fatty acid oxidation. AMPK activation is thought to mediate the effects of the anti-diabetic biguanide drug metformin at least in part, as additional mechanisms have been shown to underlie metformin's effects in the liver; notably suppression of gluconeogenesis through inhibition of mitochondrial glycerophosphate dehydrogenase (Madiraju et al. 2014) and antagonism of glucagon action through accumulation of AMP and consequent inhibition of adenylyl cyclase (Miller et al. 2013).
The Ras/Extracellular Signal Regulated Kinase (ERK) pathway is an essential pathway in transmission of mitogenic signalling, which is also activated downstream the insulin/IGF-1 receptor via IRS. The Ras/ERK pathway has also been implicated in the modulation of lifespan (Slack 2017). It has recently been reported that administration of trametinib, an inhibitor of ERK activation, extends the lifespan of D. melanogaster (Slack et al. 2015). A key downstream effector of the pathway in lifespan extension is AOP, a transcriptional repressor of the ETS family. Whether a similar lifespan extending effect of ERK inhibition could be attainable in mammalian species remains to be seen. Evidence for metabolic effects of Ras/ERK pathway perturbations, at least under obesogenic conditions, has been presented before and it is summarised in (Slack 2017). However, the potential metabolic effects of long-term administration of this inhibitor in mammalian species also remain to be seen.
The somatotropic, insulin/IGF-1 and mTOR signalling pathways have been extensively studied in the context of ageing and age-related metabolic homeostasis over the years and they are discussed below in more detail, together with the role of sirtuins, a class of histone deacetylases, which have emerged as important regulators of cellular energy homeostasis.
Somatotropic signalling in ageing and metabolism
Signalling via the growth hormone/insulin-like growth factor-1 (GH/IGF-1) axis, known as the somatotropic axis, is essential for body growth. IGF-1 is produced by the liver upon stimulation by GH released from the anterior pituitary gland. Loss-of-function mutations in components of the somatotropic axis have been shown to affect longevity in mammals (Junnila et al. 2013). A number of mouse mutants with spontaneous or targeted loss-of-function mutations that diminish production or impair sensing of GH have been described (Brown-Borg 2015). All these mutants have smaller body size and substantially extended lifespan. The first group of this type of mutants to be studied were hypopituitary mice, such as Snell and Ames dwarf mice which bear the Pit-1 dw and the Prop-1 df (Prophet of Pit-1) gene mutations, respectively. Both mutations affect the activity of the Pit-1 transcription factor, which is required for proper development of the anterior pituitary gland. Both Snell and Ames mice exhibit a deficiency in release of pituitary hormones (growth hormone, thyroid stimulating hormone and prolactin), greatly reduced body size, reduced fertility, increased adiposity, undetectable circulating IGF-1, and low insulin and glucose levels. The other group of somatotropic axis mutants consists of GH-resistant mice, specifically GH receptor and GH-binding protein (GHR/BP) knockout mouse (Laron mouse) (Zhou et al. 1997) as well as GH-deficient mice, due to a spontaneous mutation (little mice) or targeted disruption of the Ghrhr gene (GHRH-KO), which encodes the GH-releasing hormone receptor (Alba and Salvatori 2004; Godfrey et al. 1993). Hypopituitary dwarf mice display a 40–70% increase in mean lifespan depending on the specific mutation and sex (Brown-Borg et al. 1996; Flurkey et al. 2001). Even though these mutant mice are deficient in other hormones in addition to GH, their lifespan extension seems to be primarily due to GH deficiency, as the lifespan of Ames dwarf mice treated with GH reverts back to that of control animals (Panici et al. 2010).
Hypopituitary, GH deficient and GH resistant mice have been extensively characterised with respect to their metabolic phenotypes, over the years (Bartke and Westbrook 2012). Ames mice, as wells as GHR-KO mice, display increased adiposity, hypoinsulinemia and hypoglycaemia, increased adiponectin levels and reduced serum lipids. Interestingly, visceral fat from GHR-KO mice has been shown to produce higher levels of adiponectin, which likely explains the improved insulin sensitivity and glucose tolerance in the face of increased adiposity (List et al. 2011). Indeed removal of visceral fat from GHR-KO mice resulted in reduced insulin sensitivity in contrast to wild type mice, in which the same intervention improved insulin sensitivity (Masternak et al. 2012). Furthermore, indirect calorimetry experiments revealed increased oxygen consumption and reduced respiratory quotient indicating that these mutants have elevated oxidative metabolism and preferentially utilise lipids as fuel (Westbrook et al. 2009). Such properties can explain the beneficial metabolic parameters, i.e. improved insulin sensitivity and glucose homeostasis of these dwarf mutants. Interestingly, increased respiration was evident only under standard vivarium temperature (commonly 23 °C) whereas under mouse thermoneutral conditions (30 °C) there was no difference (Bartke and Westbrook 2012). This suggests that the small body size of these mutants could be the key determinant of their metabolic phenotype, as it necessitates an increased metabolic rate in order to maintain body temperature.
The extent to which reduction in the IGF-1 accounts for the life extending effects of GH-deficient and GH-resistant mice is not fully clear, but both GH-deficient and GH-resistant mice consistently show more robust lifespan extension than IGF-1-deficient mice (Bartke 2009). Gene inactivation of the IGF-1 receptor in homozygosity is perinatally lethal, but haploinsufficiency of the IGF-1 receptor (IGF-1R+/−) resulted in enhanced stress resistance and lifespan extension by 33% in female, but not male, mice (Holzenberger et al. 2003). Another study conducted independently, found a lifespan extension of only 4.7% in female IGF-1R+/− mice, though it confirmed the previously reported stress resistance (Bokov et al. 2011). In terms of metabolic phenotypes, the latter study demonstrated that over ageing the mice developed insulin resistance and the males glucose intolerance as well. A follow up study from the Holzenberger lab, demonstrated that the magnitude of the lifespan extension effect, but not stress resistance, was depended on genetic background (Xu et al. 2014). In terms of metabolic phenotypes, the study corroborated the development of insulin resistance in male mice. Thus, downregulation of IGF-1 signalling does not seem to offer the same beneficial effects with GH deficiency/resistance. Therefore, GH deficiency/resistance likely mediates its life extending effects through mechanisms distinct from IGF-1 deficiency to a large extent.
The neuroendocrine axis of growth control has been implicated in human ageing as well. GH levels drop with age, a process known as somatopause. Certain manifestations of tsomatopause, such as reduction of muscle mass, causing age-related sarcopenia, and increase in visceral adiposity are partly reversible by GH treatment (Rudman et al. 1990). Hence, GH replacement therapy has been proposed as an anti-ageing intervention. However, the findings from research in GH deficient/resistant mouse mutants do not support such an effect of GH and on the contrary suggest that such an intervention could have a negative impact on human ageing (Bartke 2008). Also, in humans there is an apparent negative correlation between height and longevity, thus suggesting a potential role of GH and IGF-1 regulated growth in promoting ageing (Samaras and Storms 1992). There is also genetic evidence implicating the activity of the somatotropic axis in human ageing. Allele frequency studies have reported that polymorphic variants of genes related to GH synthesis, IGF-1 signalling and insulin action change in frequency with age. Bonafè et al. found that allele A of IGF-1R is associated with low plasma IGF-1 and is more frequent among long-lived people (Bonafe et al. 2003). Moreover, van Heemst et al. found that women carrying a SNP variant of the GH1 gene for human growth hormone are 2 cm shorter and exhibit a 0.80-fold reduced mortality (van Heemst et al. 2005). Furthermore, female offspring of centenarian Ashkenazi Jews were found to be heterozygous for loss-of-function mutations in the gene encoding IGF-1R (Suh et al. 2008). However, such evidence is essentially correlative and it does not reveal the underlying mechanisms of lifespan extension. Body size per se is not necessarily a determinant of lifespan extension. Reduced susceptibility to pathologies such as cancer could underlie the lifespan extending effect in this context (de Magalhaes and Faragher 2008). This notion is further supported by the recent report that male carriers of a common GHR allele lacking exon 3 (d3-GHR) live longer despite being taller than carriers of the wild-type allele (Ben-Avraham et al. 2017). Interestingly, a study that monitored 99 Ecuadorian GH receptor deficient dwarfs for 22 years, has shown that they were less susceptible to cancer and obesity, but not long-lived (Guevara-Aguirre et al. 2011). Therefore, diminished somatotropic signalling can have beneficial metabolic effects, even in the absence of lifespan effects in humans.
Insulin/IGF-1 signalling pathway and metabolic ageing
During the 1980s and 1990s, mutagenic screen studies in C. elegans identified the first long-lived mutants, where single mutations produced large effects on lifespan. Worms with loss-of-function mutations in genes in the IIS pathway showed remarkable increases in mean and maximum lifespan and maintained a youthful morphology for longer (Kenyon 2011). Therefore, the IIS pathway was the first signalling pathway shown to play a pivotal role in the ageing process. Indeed, loss-of-function mutation of daf-2, the worm ortholog of the insulin/IGF-1 receptor, resulted in large increases in the lifespan of worms. In contrast, ubiquitous knock out of the Insulin Receptor (IR) gene in mice resulted in neonatal lethality (Accili et al. 1996). Moreover, a severely diminished IR gene function in humans causes Dohonue syndrome (leprechaunism) (Kitamura et al. 2003). Hence, it was initially thought that the role of IIS in the regulation of the ageing process could not be evolutionarily conserved and that the activity of the IIS pathway was unlikely to promote ageing in mammals. However, there is now ample evidence that reduction of the IIS pathway can delay ageing and improve the metabolic profile of mammalian species in late life. The first evidence was provided from the study of mice with adipose tissue-specific knockout of the insulin receptor (FIRKO mice, Fat-specific Insulin Receptor KnockOut). FIRKO mice were protected against age-related obesity and exhibited an 18% increase of mean lifespan in both sexes (Bluher et al. 2003). FIRKO mice displayed reduced insulin levels and resistance to age-related glucose intolerance (Bluher et al. 2002). More recently, extension of the maximal lifespan of heterozygous mice for a ubiquitous null mutation of the insulin receptor (IR-KO+/−) has been reported for male, but not female, mice (Nelson et al. 2012). Conversely, reduction of circulating insulin levels by targeted disruption of insulin genes has recently been reported to extend lifespan in female, but not male mice (Templeman et al. 2017). This is an example of how sex can differentially influence the effects of genetic interventions even on closely positioned components of signalling pathways.
Ubiquitous knock-out of the Insulin Receptor Substrate-1 (IRS1) has also been shown to extend lifespan (by 32% in females, weaker effect in males) and improve a wide range of markers of ageing such as immune and motor system dysfunction and bone and skin deterioration (Selman et al. 2008, 2011). IRS1 KO mice, although insulin resistant, display improved glucose tolerance at old age. Although Selman et al. found no lifespan effects in ubiquitous IRS2 heterozygous knockout mice (IRS2+/−), another study reported that both ubiquitous IRS2+/− and brain-specific IRS2 KO mice exhibited lifespan extension (Taguchi et al. 2007). Interestingly, ubiquitous IRS2+/− mice were found to be more insulin sensitive than wild-type littermates, but brain-specific IRS2 KO were insulin resistant and glucose intolerant.
A key effector molecule in IR is PI3K. The principal IIS-responsive mammalian isoform of PI3K is p110α (Foukas et al. 2006). Hence, mice heterozygous for a kinase-dead knock-in mutation in the gene encoding p110α (p110α KI) displayed insulin resistance and moderate glucose intolerance at young age. However, this partial inactivation of PI3K p110α exerted a protective effect in the long-term so that aged p110α KI mice were leaner and manifested an improved metabolic profile compared to their wild-type littermates (Foukas et al. 2013). These effects were more prominent in male mice. Consistent with this, male p110α KI mice showed a modest (approx. 7%) extension in their median lifespan. These findings are in line with the phenotypes reported for mice with systemic overexpression of the tumour suppressor phosphatase and tensin homolog (PTEN), which counteracts the activity of PI3K. PTEN transgenic (PTEN-Tg) mice exhibited increased energy expenditure, decreased adiposity, improved insulin sensitivity upon high-fat feeding or with aging, and extended lifespan (Ortega-Molina et al. 2012). High levels of expression of the uncoupling protein 1 (UCP1) in the brown adipose tissue (BAT) of PTEN transgenic mice resulted in enhanced nutrient burning capacity and reduced adiposity and associated pathologies. All the above evidence supports the notion that downregulation of the insulin signalling pathway can have important beneficial metabolic effects in mammals.
mechanistic Target of Rapamycin (mTOR) pathway and metabolism
The mTOR signalling pathway is evolutionarily conserved and integrates nutrient availability, energy status and growth factor signalling in the control of cell growth and proliferation. It regulates a multitude of cellular processes such as mRNA translation, metabolism, autophagy and stress resistance (Cornu et al. 2013; Kapahi et al. 2010; Laplante and Sabatini 2012). The mTOR protein kinase is distributed in two distinct complexes, mTOR Complex 1 and 2 (mTORC1 and 2), each with distinct functions and substrates. mTORC1 consists of mTOR, mammalian lethal with sec-13 protein 8 (mLST8, also known as GβL), and regulatory-associated protein of TOR (Raptor). Additional components include DEP-domain-containing mTOR-interacting protein (DEPTOR) and proline-rich Akt substrate 40 kDa (PRAS40). mTORC1 is acutely sensitive to rapamycin and regulates ribosomal protein biogenesis, protein translation and autophagy. mTORC2 is composed of mTOR, rapamycin-insensitive companion of mTOR (Rictor), a G protein beta subunit-like associated to mTOR (mLST8), and stress-activated protein kinase-interacting protein 1 (mSIN1) (Cornu et al. 2013; Laplante and Sabatini 2012). S6K1 and 4E-BP1 are two well-characterised substrates of mTORC1 (Ma and Blenis 2009), whereas the hydrophobic motif phosphorylation site (S473) of Akt is a key substrate for mTORC2 (Sarbassov et al. 2005). Both mTORC1 and mTORC2 are activated by growth factors. mTORC1 activity is also modulated by availability of aminoacids and by energy levels through input from AMPK. Hence, mTORC1 is a central signalling node that integrates multiple inputs to regulate biological responses such as protein and lipid synthesis, autophagy and cell cycle progression. mTORC2 has been implicated in cytoskeletal organisation, cell survival and metabolism; in the latter to a large extent via its Akt phosphorylating activity.
Genetic or pharmacological inhibition of mTOR signalling has been found to extend the lifespan of invertebrate species including yeast, nematodes, fruit flies and mice (Kapahi et al. 2010; Lamming et al. 2013). Indeed, deletion of S6K1 protects against diet-induced obesity, enhances insulin sensitivity and increases lifespan in mice (Selman et al. 2009; Um et al. 2004). Moreover mice expressing a hypomorphic allele of mTOR and mice heterozygous for both mTOR and mLST8, are also long-lived (Lamming et al. 2012; Wu et al. 2013). Furthermore, it has recently been shown that long-lived heterozygous Akt1 mutants exhibit decreased mTORC1 activity (Nojima et al. 2013). A role for mTORC2 in the regulation of lifespan is still largely uncertain, but it could potentially have an influence through its role as a modulator of mTORC1 signalling.
The mTOR and IIS pathways are closely linked in the regulation of energy metabolism and glucose homeostasis (Zoncu et al. 2011). Early genetic approaches attempting ubiquitous inactivation of mTOR in mice resulted in early embryonic lethality and therefore did not contribute to the study of mTOR in metabolic regulation (Gangloff et al. 2004; Murakami et al. 2004). Hence, tissue-specific mutagenesis has been applied in order to study the roles of the two mTOR complexes in metabolic tissues, which often resulted in disparate metabolic effects depending on the targeted tissue (Table 1). Tissue-specific disruption of mTOR signalling pathway components has been shown to induce differential effects on the metabolic profile in rodents, summarised in (Polak and Hall 2009). Deletion of Raptor, a component of mTOR C1, specifically in the adipose tissue protected against diet-induced obesity (Polak et al. 2008), whereas deletion of Raptor in skeletal muscle was deleterious, causing severe muscular dystrophy (Bentzinger et al. 2008). Overexpression of a dominant-negative version of Raptor in the liver improved insulin sensitivity (Koketsu et al. 2008), whereas inhibition of mTOR by rapamycin reduced the production and release of insulin by pancreatic islets, leading to hypoinsulinemia and glucose intolerance (Zahr et al. 2008). Also, aminoacid-induced activation of mTOR or activation of S6K in the hypothalamus decreased food intake and body weight (Blouet et al. 2008; Cota et al. 2006). Similar approaches targeting mTORC2 in the adipose tissue by disruption of the Rictor gene resulted in larger body mass due to organ hypertrophy and hyperinsulinemia, but normal glucose tolerance, under standard chow feeding (Cybulski et al. 2009). These findings have been largely corroborated by another study utilising a similar targeting approach, with the exception of the glucose tolerance, which in the later study was found to be impaired (Kumar et al. 2010). From the above data, it becomes evident that the effect of downregulation of mTOR signalling on metabolism is difficult to predict. Pharmacological inhibition of mTOR with rapamycin apparently results in negative metabolic effects though these largely depend on the dosing regimen (see below). And hypomorphic mTOR expression has no metabolic phenotypes despite lifespan extension (Wu et al. 2013). Therefore, in the case of the mTOR pathway, a relation between body metabolic homeostasis and longevity is not obvious.
Insulin sensitivity and longevity
Ageing is associated with a reduction in insulin sensitivity both in humans and rodents (Basu et al. 2003; Escriva et al. 2007). Age-related insulin resistance is thought to be a result of increased visceral adiposity with age progression. Consistent with this notion, removal of visceral adipose tissue improves insulin sensitivity in mice (Gabriely et al. 2002). Adipose tissue development and function is highly dependent on sex steroids (White and Tchoukalova 2014) and therefore it is thought that differences in adipose tissue distribution between sexes would produce differences in development of insulin resistance, however there is no clear consensus about this (Kim and Reaven 2013).
The potential effect of insulin sensitivity on lifespan is still unclear (Barzilai and Ferrucci 2012). In humans, insulin resistance is accompanied by compensatory hyperinsulinemia and has clearly been implicated as a risk factor for multiple age-related diseases. Consistent with this, calorically restricted rodents and a number of long-lived mice (e.g. GH deficient/resistant mutants, FIRKO mice, S6K KO mice) display enhanced insulin sensitivity. However, as mentioned above, attenuation of the insulin signalling cascade via genetic inactivation of key IIS pathway intermediates has been associated with lifespan extension in model organisms (Kenyon 2010). Moreover, lifespan extension has been reported in mouse mutants for the IIS or mTOR pathways with normal (Lamming et al. 2012; Nojima et al. 2013; Wu et al. 2013) or reduced (Selman et al. 2008; Taguchi et al. 2007) insulin sensitivity. This implies that enhanced insulin sensitivity is not a requisite for extended longevity. However, insulin resistant mutants, such as IRS1−/− and PI3K p110α KI mice, display improved glucose tolerance compared to wild-type littermates at old age (Foukas et al. 2013; Selman et al. 2008). As mentioned in the Introduction, hyperglycemia contributes to a large extent to tissue damage and old age frailty. Therefore, improved glucose tolerance at old age might be a better indicator of a metabolic effect, which might translate to enhanced longevity, than insulin sensitivity.
Sirtuins and metabolic effects of sirtuin activation
Sirtuins are a family of nicotinamide adenine dinucleotide (NAD+)-dependent histone deacetylases (Chang and Guarente 2014; Houtkooper et al. 2012). There are seven mammalian sirtuins (SIRT1-7) that differ in their tissue distribution, subcellular localisation, enzymatic activity and substrate specificity. In addition to histones, they also deacetylate transcription factors and other cellular proteins affecting gene expression activity. Their implication in lifespan regulation emerged when increasing the dosage of the sirtuin Sir2 was found to extend replicative lifespan in yeast (Kaeberlein et al. 1999). Various lines of evidence support the notion that sirtuins mediate the effects of calorie restriction to a large extent (Guarente 2011). They have also been shown to extend the lifespan of worms, flies and mice. The lifespan extending effects of sirtuin activation have been disputed, but the improvements in healthspan seem to be robust (Houtkooper et al. 2012). Sirtuins mediate various beneficial effects on metabolic tissues, such as reduced glycolysis and increased fatty acid oxidation in liver and muscle, reduced hepatic lipogenesis, adipose tissue browning and fat mobilisation (Chang and Guarente 2014). SIRT1, the principal mammalian sirtuin exerting metabolic effects, has been shown to be activated via the AMPK energy sensing pathway in skeletal muscle (Canto et al. 2010). Therefore, the sirtuin family provides promising therapeutic targets in metabolic diseases, such as age-related obesity and diabetes, and a number of sirtuin activators have been identified or developed. The natural polyphenol compound resveratrol has received great attention as a sirtuin activator with significant beneficial metabolic effects in mice fed a high-fat diet and in obese humans (Baur et al. 2006; Lagouge et al. 2006; Timmers et al. 2011). Resveratrol’s mechanism of action as a sirtuin activator has been reported to be indirect, through activation of energy sensing pathways and modulation of cAMP and NAD+ levels, according to some studies (Park et al. 2012; Um et al. 2010). However, later studies demonstrated that the substrate specificity of SIRT1 is sequence specific thus explaining the lack of activation by resveratrol against a number of substrates (Hubbard et al. 2013; Lakshminarasimhan et al. 2013). A number of synthetic SIRT1 activators have been developed and tested in preclinical trials (Carafa et al. 2016; Hubbard and Sinclair 2014; Sinclair and Guarente 2014). Moreover, some positive results have been reported from early stage clinical trials in inflammatory and metabolic disorders (Bonkowski and Sinclair 2016). However, a more detailed evaluation of their potential for clinical application remains to be seen.
Pharmacological interventions in healthspan extension
The demonstration that administration of the mTOR inhibitor rapamycin late in life can extend the lifespan of mice was a ground-breaking development in the biology of ageing, as it provided solid proof-of-principle that pharmacological treatment of ageing is possible (Harrison et al. 2009). It has boosted efforts to produce pharmacological agents to improve healthspan by combating age-related disease. This section summarises evidence supporting a potential effect of pharmacological agents, known to modulate the activity of signalling pathways implicated in regulation of healthspan and lifespan, on metabolic homeostasis. Specifically, the mTOR pathway inhibitor rapamycin, PI3K inhibitors (IIS pathway), somatoropic axis inhibitors and metformin (indirect activator of AMPK) are discussed in more detail. Sirtuin activators were briefly discussed above and in more detail in the literature cited therein.
Rapamycin
Rapamycin acts as an allosteric inhibitor of mTORC1 by forming a gain-of-function complex with the 12 kDa FK506-binding protein (FKBP12). Rapamycin has immunosuppressive and anti-proliferative properties in mammalian cells. It was approved as an immuno-suppressant in 1999. In recent years, interest has focused on the potential of rapamycin and its analogues (rapalogues) as anticancer drugs (Wander et al. 2011). Rapamycin has been reported to have both positive and negative effects on mammal metabolism. In obesity, the mTOR pathway is hyper-activated in the adipose tissue thus leading to increased lipogenesis, reduced lipolysis and fat accumulation. To limit its over-activation, mTORC1 blocks insulin signalling through a S6K-mediated feedback loop destabilising IRS (Harrington et al. 2004; Shah et al. 2004) and through direct phosphorylation and stabilisation of Grb10 (Hsu et al. 2011), provoking an insulin resistance state. Acute mTOR inhibition through rapamycin treatment improves insulin sensitivity in vitro and in vivo by disrupting this negative feedback loop of insulin signalling (Krebs et al. 2007; Tremblay and Marette 2001). Also rapamycin protects from diet-induced obesity and prevents weight gain in mice (Chang et al. 2009a; Makki et al. 2014). In rats and humans, it reduces age-related body weight gain when administered 3 times per week (Rovira et al. 2008). However, glucose intolerance and insulin resistance have been observed in a few strains of rodents treated daily with high doses of rapamycin. In fact, rapamycin increases lipolysis in the adipose tissue in rats, causing pronounced hyperlipidemia (Houde et al. 2010). Also, a 2-week rapamycin treatment aggravates hyperglycemia in a diabetic mouse model (Fraenkel et al. 2008) and similarly, rapamycin administration (6 weeks) exacerbates glucose intolerance in diet-induced obese KK/HIJ mice (Chang et al. 2009b). These controversial findings on the insurgence of insulin resistance might be explained by the duration and the doses of rapamycin treatment (Fang et al. 2013). Rapamycin could therefore still be a viable pharmacological option to promote beneficial metabolic effects in humans pending definition of appropriate dosing regimens.
GH/IGF1 axis inhibitors
Research in a number of GH deficient/resistant mice as well as in an Ecuadorian human GH receptor-deficient population, has clearly demonstrated that downregulation of this pathway exerts beneficial metabolic effects. Compounds targeting the activity of the somatotropic axis have been used in the treatment of acromegaly. Somatostatin analogues have been used to supress GH secretion, but with limited efficacy and substantial side-effects (Parkinson et al. 2002). Somatostatin inhibits secretion of insulin and it is therefore unlikely to have beneficial metabolic and healthspan effects. However, pegvisomat, a GH receptor antagonist, has demonstrated efficacy and an acceptable safety profile (van der Lely et al. 2012). Beneficial metabolic effects of short- to medium- term administration of pegvisomat in type-1 diabetes and acromegaly patients, respectively, have been reported (Lindberg-Larsen et al. 2007; Thankamony et al. 2014). Pegvisomat, is therefore a compound that could potentially be tested for healthspan effects upon long-term administration in humans, although logistic constraints related to high costs, make it an unlikely candidate as a widely available therapeutic modality.
PI3K inhibitors
As discussed above, long-lived IIS mutant mice, such as IRS1 KO (Selman et al. 2008), PTEN-Tg mice (Ortega-Molina et al. 2012) and PI3K p110α KI mice (Foukas et al. 2013) display a metabolic improvement at old age. Therefore it is reasonable to hypothesise that use of inhibitors against ‘druggable’ components of this pathway could represent a strategy in prevention or treatment of metabolic diseases associated with old age (such as obesity and type-2 diabetes) and in healthspan extension. A number of inhibitors against PI3Ks are currently available, mainly through efforts to target these enzymes in oncology. Proof of principle that PI3K inhibitors can also be useful in metabolic disease has been provided by a recent study (Ortega-Molina et al. 2015). Long-term administration of low doses of two pharmacological inhibitors of PI3K, CNIO-PI3Ki and GDC-0941, has been reported to reduce the adiposity and body weight of obese mice and rhesus monkeys. The same treatment did not affect adiposity of mice fed a standard chow. CNIO-PI3Ki is a small molecule ATP competitive dual inhibitor of the PI3K isoforms p110α and p110δ. Further testing for isoform specificity, using p110α and p110δ discriminating compounds, revealed that the anti-obesity effect was particularly prominent upon inhibition of p110α, the principal insulin activated isoform, although inhibition of p110δ did contribute to the overall effect (Lopez-Guadamillas et al. 2016). p110δ is highly expressed in leukocytes and its involvement was possibly due to an anti-inflammatory effect of p110δ inhibition, as inflammation is a well-established contributing factor in metabolic pathology. These findings suggest that pharmacological inhibition of PI3K could be an effective and safe anti-obesity intervention to prevent or reverse metabolic syndrome in humans.
Metformin
Metformin is a biguanide that has extensively been used in the treatment of type 2 diabetes. It works by decreasing glucose production in the liver, augmenting glucose utilisation by body tissues and increasing sensitivity to insulin. Metformin has been used for more than 60 years, it is safe and has also been reported to slow aging in C. elegans (Pryor and Cabreiro 2015). Metformin-treated worms not only live longer, but also stay healthier for longer (Onken and Driscoll 2010). Metformin administration increases the lifespan of mice by nearly 6% and improves various markers of healthspan (Martin-Montalvo et al. 2013). Also diabetic patients treated with metformin live longer than non-treated non-diabetic control subjects (Bannister et al. 2014). Therefore, metformin might have substantial beneficial effects on human healthspan. To test this idea, metformin is set to enter a ground breaking human trial as a potential anti-aging drug (Barzilai et al. 2016). A clinical trial called Targeting Aging with Metformin, or TAME will involve the administration of metformin to 3,000 people aged 70–80 years (at roughly 15 centres around the United States), who already have one or two of three conditions (cancer, heart disease or dementia) or are at risk of them. The participants will be monitored to test whether the medication prevents or delays development of diseases they do not already have, as well as of diabetes.
Conclusion and perspective
Recent advances in the elucidation of signalling pathways that modulate the rate of ageing have made it possible to study the effects of their manipulation on various pathologies associated with advanced age in model organisms. However, a number of confounding factors complicate the predictions for translational potential of the findings from model organism to humans (de Magalhaes 2014). Most of the research in the genetics of ageing and in testing interventions has used inbred animals with identical genetic backgrounds. Such models do not reflect the situation of human populations that are genetically heterogeneous. In many cases, the responses under investigation have been extremely variable between different strains. A prominent example is calory restriction in mice, where the outcome can vary from life extension to life shortening depending on the genetic background (Liao et al. 2010). Furthermore, as alluded at various places above, in many cases, genetic interventions have stronger or even restricted effects on one sex. It is conceivable that, as is the case in other diseases, notably in cancer, in age-related diseases there will not be a cure suitable for everyone, but treatments should be personalised based on individual genetics. Pharmacologic interventions might have to match the genetic and epigenetic make-up as well as the sex of individuals. To this end, continuous advances in next generation sequencing and human genetics, together with development of advanced bioinformatic methods, similar to those used to correlate longevity genes to age-related disease genes and to drugs (Fernandes et al. 2016), could make it possible to predict the likelihood of specific interventions to be effective in combating age-related diseases in particular individuals.
There is now substantial evidence that pharmacological interventions in the IIS, mTOR and AMPK pathways can have beneficial metabolic effects in mammalian organisms. As discussed above, interventions within these signalling pathways affect metabolic homeostasis, which appears to be a key determinant of healthspan. A number of compounds targeting these pathways have demonstrated good tolerability and substantial beneficial metabolic effects through preclinical testing in mice (Table 2). Whether such compounds will only have beneficial metabolic effects or more generalised healthspan effects will be of great interest to determine, but it will certainly be more challenging and it will require development of suitable biomarkers. Nevertheless, the application of signalling pathway inhibitors for prevention and treatment of age-related obesity and insulin resistance seems within grasp based on very promising results from preclinical stage testing. Exciting trials are under way to assess the effects of such compounds, such as the rapamycin trial in pet dogs (Kaeberlein et al. 2016) and the metformin trial in humans (Barzilai et al. 2016). Ongoing research in independent academic laboratories along with larger scale programmes such as the National Institute of Aging Intervention Testing Programme (NIA ITP) (https://www.nia.nih.gov/research/dab/interventions-testing-program-itp) have made great contributions to this effort and will likely identify additional compounds with potency to improve healthspan and lifespan to be subsequently tested in human trials. These trials hold great promise and a positive outcome would be a great return for the extensive efforts invested in research in the field of biology of ageing. Such developments are eagerly awaited by the respective scientific community and they are certain to be welcome by the general public.
References
Accili D et al (1996) Early neonatal death in mice homozygous for a null allele of the insulin receptor gene. Nat Genet 12:106–109. doi:10.1038/ng0196-106
Alba M, Salvatori R (2004) A mouse with targeted ablation of the growth hormone-releasing hormone gene: a new model of isolated growth hormone deficiency. Endocrinology 145:4134–4143. doi:10.1210/en.2004-0119
Anderson RM, Weindruch R (2010) Metabolic reprogramming, caloric restriction and aging. Trends Endocrinol Metab 21:134–141. doi:10.1016/j.tem.2009.11.005
Apfeld J, O’Connor G, McDonagh T, DiStefano PS, Curtis R (2004) The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev 18:3004–3009. doi:10.1101/gad.1255404
Argentino DP, Dominici FP, Al-Regaiey K, Bonkowski MS, Bartke A, Turyn D (2005) Effects of long-term caloric restriction on early steps of the insulin-signaling system in mouse skeletal muscle. J Gerontol A 60:28–34
Bannister CA et al (2014) Can people with type 2 diabetes live longer than those without? A comparison of mortality in people initiated with metformin or sulphonylurea monotherapy and matched, non-diabetic controls. Diabetes Obes Metab 16:1165–1173. doi:10.1111/dom.12354
Bartke A (2008) Growth hormone and aging: a challenging controversy. Clin Interv Aging 3:659–665
Bartke A (2009) The somatotropic axis and aging: mechanisms and persistent questions about practical implications. Exp Gerontol 44:372–374. doi:10.1016/j.exger.2009.04.001
Bartke A, Westbrook R (2012) Metabolic characteristics of long-lived mice. Front Genet 3:288. doi:10.3389/fgene.2012.00288
Barzilai N, Ferrucci L (2012) Insulin resistance and aging: a cause or a protective response? J Gerontol A 67:1329–1331. doi:10.1093/gerona/gls145
Barzilai N, Huffman DM, Muzumdar RH, Bartke A (2012) The critical role of metabolic pathways in aging. Diabetes 61:1315–1322. doi:10.2337/db11-1300
Barzilai N, Crandall JP, Kritchevsky SB, Espeland MA (2016) Metformin as a tool to target aging. Cell Metab 23:1060–1065. doi:10.1016/j.cmet.2016.05.011
Basu R et al (2003) Mechanisms of the age-associated deterioration in glucose tolerance: contribution of alterations in insulin secretion, action, and clearance. Diabetes 52:1738–1748
Baur JA et al (2006) Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444:337–342. doi:10.1038/nature05354
Ben-Avraham D et al (2017) The GH receptor exon 3 deletion is a marker of male-specific exceptional longevity associated with increased GH sensitivity and taller stature. Sci Adv 3:25. doi:10.1126/sciadv.1602025
Bentzinger CF et al (2008) Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab 8:411–424. doi:10.1016/j.cmet.2008.10.002
Blouet C, Ono H, Schwartz GJ (2008) Mediobasal hypothalamic p70 S6 kinase 1 modulates the control of energy homeostasis. Cell Metab 8:459–467. doi:10.1016/j.cmet.2008.10.004
Bluher M, Michael MD, Peroni OD, Ueki K, Carter N, Kahn BB, Kahn CR (2002) Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Dev Cell 3:25–38
Bluher M, Kahn BB, Kahn CR (2003) Extended longevity in mice lacking the insulin receptor in adipose tissue. Science 299:572–574. doi:10.1126/science.1078223
Bokov AF et al (2011) Does reduced IGF-1R signaling in Igf1r ± mice alter aging? PLoS ONE 6:e26891. doi:10.1371/journal.pone.0026891
Bonafe M et al (2003) Polymorphic variants of insulin-like growth factor I (IGF-I) receptor and phosphoinositide 3-kinase genes affect IGF-I plasma levels and human longevity: cues for an evolutionarily conserved mechanism of life span control. J Clin Endocrinol Metab 88:3299–3304. doi:10.1210/jc.2002-021810
Bonkowski MS, Sinclair DA (2016) Slowing ageing by design: the rise of NAD+ and sirtuin-activating compounds. Nat Rev Mol Cell Biol 17:679–690. doi:10.1038/nrm.2016.93
Bonkowski MS, Rocha JS, Masternak MM, Al Regaiey KA, Bartke A (2006) Targeted disruption of growth hormone receptor interferes with the beneficial actions of calorie restriction. Proc Natl Acad Sci USA 103:7901–7905. doi:10.1073/pnas.0600161103
Brown-Borg HM (2015) The somatotropic axis and longevity in mice. Am J Physiol Endocrinol Metab 309:E503–E510. doi:10.1152/ajpendo.00262.2015
Brown-Borg HM, Borg KE, Meliska CJ, Bartke A (1996) Dwarf mice and the ageing process. Nature 384:33. doi:10.1038/384033a0
Canto C et al (2010) Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab 11:213–219. doi:10.1016/j.cmet.2010.02.006
Carafa V et al (2016) Sirtuin functions and modulation: from chemistry to the clinic. Clin Epigenet 8:61. doi:10.1186/s13148-016-0224-3
Caron A, Richard D, Laplante M (2015) The roles of mTOR complexes in lipid metabolism. Annu Rev Nutr 35:321–348. doi:10.1146/annurev-nutr-071714-034355
Chang HC, Guarente L (2014) SIRT1 and other sirtuins in metabolism. Trends Endocrinol Metab 25:138–145. doi:10.1016/j.tem.2013.12.001
Chang GR, Chiu YS, Wu YY, Chen WY, Liao JW, Chao TH, Mao FC (2009a) Rapamycin protects against high fat diet-induced obesity in C57BL/6J mice. J Pharmacol Sci 109:496–503
Chang GR et al (2009b) Long-term administration of rapamycin reduces adiposity, but impairs glucose tolerance in high-fat diet-fed KK/HlJ mice. Basic Clin Pharmacol Toxicol 105:188–198. doi:10.1111/j.1742-7843.2009.00427.x
Chung HY, Kim HJ, Kim KW, Choi JS, Yu BP (2002) Molecular inflammation hypothesis of aging based on the anti-aging mechanism of calorie restriction. Microsc Res Tech 59:264–272. doi:10.1002/jemt.10203
Colman RJ, Beasley TM, Kemnitz JW, Johnson SC, Weindruch R, Anderson RM (2014) Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat Commun 5:3557. doi:10.1038/ncomms4557
Cornu M, Albert V, Hall MN (2013) mTOR in aging, metabolism, and cancer. Curr Opin Genet Dev 23:53–62. doi:10.1016/j.gde.2012.12.005
Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, Seeley RJ (2006) Hypothalamic mTOR signaling regulates food intake. Science 312:927–930. doi:10.1126/science.1124147
Cybulski N, Polak P, Auwerx J, Ruegg MA, Hall MN (2009) mTOR complex 2 in adipose tissue negatively controls whole-body growth. Proc Natl Acad Sci USA 106:9902–9907. doi:10.1073/pnas.0811321106
de Magalhaes JP (2014) Why genes extending lifespan in model organisms have not been consistently associated with human longevity and what it means to translation research. Cell Cycle 13:2671–2673. doi:10.4161/15384101.2014.950151
de Magalhaes JP, Faragher RG (2008) Cell divisions and mammalian aging: integrative biology insights from genes that regulate longevity. BioEssays 30:567–578. doi:10.1002/bies.20760
Escriva F et al (2007) Effect of age and moderate food restriction on insulin sensitivity in Wistar rats: role of adiposity. J Endocrinol 194:131–141. doi:10.1677/joe.1.07043
Fabbiano S, Suarez-Zamorano N, Rigo D, Veyrat-Durebex C, Stevanovic Dokic A, Colin DJ, Trajkovski M (2016) Caloric restriction leads to browning of white adipose tissue through Type 2 immune signaling. Cell Metab 24:434–446. doi:10.1016/j.cmet.2016.07.023
Fang Y et al (2013) Duration of rapamycin treatment has differential effects on metabolism in mice. Cell Metab 17:456–462. doi:10.1016/j.cmet.2013.02.008
Fernandes M et al (2016) Systematic analysis of the gerontome reveals links between aging and age-related diseases. Hum Mol Genet 25:4804–4818. doi:10.1093/hmg/ddw307
Flurkey K, Papaconstantinou J, Miller RA, Harrison DE (2001) Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc Natl Acad Sci USA 98:6736–6741. doi:10.1073/pnas.111158898
Fontana L, Partridge L, Longo VD (2010) Extending healthy life span–from yeast to humans. Science 328:321–326. doi:10.1126/science.1172539
Foukas LC et al (2013) Long-term p110alpha PI3K inactivation exerts a beneficial effect on metabolism. EMBO Mol Med 5:563–571. doi:10.1002/emmm.201201953
Foukas LC et al (2006) Critical role for the p110alpha phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature 441:366–370. doi:10.1038/nature04694
Fraenkel M et al (2008) mTOR inhibition by rapamycin prevents beta-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes 57:945–957. doi:10.2337/db07-0922
Gabriely I et al (2002) Removal of visceral fat prevents insulin resistance and glucose intolerance of aging: an adipokine-mediated process? Diabetes 51:2951–2958
Gangloff YG et al (2004) Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol Cell Biol 24:9508–9516. doi:10.1128/MCB.24.21.9508-9516.2004
Garcia D, Shaw RJ (2017) AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell 66:789–800. doi:10.1016/j.molcel.2017.05.032
Godfrey P, Rahal JO, Beamer WG, Copeland NG, Jenkins NA, Mayo KE (1993) GHRH receptor of little mice contains a missense mutation in the extracellular domain that disrupts receptor function. Nat Genet 4:227–232. doi:10.1038/ng0793-227
Gross DN, van den Heuvel AP, Birnbaum MJ (2008) The role of FoxO in the regulation of metabolism. Oncogene 27:2320–2336. doi:10.1038/onc.2008.25
Guarente L (2011) Sirtuins, aging, and metabolism. Cold Spring Harb Symp Quant Biol 76:81–90. doi:10.1101/sqb.2011.76.010629
Guevara-Aguirre J et al (2011) Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci Transl Med 3:70ra13. doi:10.1126/scitranslmed.3001845
Hagiwara A et al (2012) Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab 15:725–738. doi:10.1016/j.cmet.2012.03.015
Harrington LS et al (2004) The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol 166:213–223. doi:10.1083/jcb.200403069
Harrison DE et al (2009) Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460:392–395. doi:10.1038/nature08221
Holzenberger M et al (2003) IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 421:182–187. doi:10.1038/nature01298
Houde VP, Brule S, Festuccia WT, Blanchard PG, Bellmann K, Deshaies Y, Marette A (2010) Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes 59:1338–1348. doi:10.2337/db09-1324
Houtkooper RH, Pirinen E, Auwerx J (2012) Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 13:225–238. doi:10.1038/nrm3293
Hsu PP et al (2011) The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 332:1317–1322. doi:10.1126/science.1199498
Hubbard BP et al (2013) Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 339:1216–1219. doi:10.1126/science.1231097
Hubbard BP, Sinclair DA (2014) Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol Sci 35:146–154. doi:10.1016/j.tips.2013.12.004
Huffman DM, Moellering DR, Grizzle WE, Stockard CR, Johnson MS, Nagy TR (2008) Effect of exercise and calorie restriction on biomarkers of aging in mice. Am J Physiol Regul Integr Comp Physiol 294:R1618–R1627. doi:10.1152/ajpregu.00890.2007
Junnila RK, List EO, Berryman DE, Murrey JW, Kopchick JJ (2013) The GH/IGF-1 axis in ageing and longevity. Nat Rev Endocrinol 9:366–376. doi:10.1038/nrendo.2013.67
Kaeberlein M, Kennedy BK (2009) Ageing: a midlife longevity drug? Nature 460:331–332. doi:10.1038/460331a
Kaeberlein M, McVey M, Guarente L (1999) The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 13:2570–2580
Kaeberlein M, Creevy KE, Promislow DE (2016) The dog aging project: translational geroscience in companion animals. Mamm Genome 27:279–288. doi:10.1007/s00335-016-9638-7
Kapahi P, Chen D, Rogers AN, Katewa SD, Li PW, Thomas EL, Kockel L (2010) With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab 11:453–465. doi:10.1016/j.cmet.2010.05.001
Kenyon CJ (2010) The genetics of ageing. Nature 464:504–512. doi:10.1038/nature08980
Kenyon C (2011) The first long-lived mutants: discovery of the insulin/IGF-1 pathway for ageing. Philos Trans R Soc Lond B Biol Sci 366:9–16. doi:10.1098/rstb.2010.0276
Kim SH, Reaven G (2013) Sex differences in insulin resistance and cardiovascular disease risk. J Clin Endocrinol Metab 98:E1716–E1721. doi:10.1210/jc.2013-1166
Kitamura T, Kahn CR, Accili D (2003) Insulin receptor knockout mice. Annu Rev Physiol 65:313–332. doi:10.1146/annurev.physiol.65.092101.142540
Koketsu Y et al (2008) Hepatic overexpression of a dominant negative form of raptor enhances Akt phosphorylation and restores insulin sensitivity in K/KAy mice. Am J Physiol Endocrinol Metab 294:E719–E725. doi:10.1152/ajpendo.00253.2007
Krebs M et al (2007) The Mammalian target of rapamycin pathway regulates nutrient-sensitive glucose uptake in man. Diabetes 56:1600–1607. doi:10.2337/db06-1016
Kumar A, Harris TE, Keller SR, Choi KM, Magnuson MA, Lawrence JC Jr (2008) Muscle-specific deletion of rictor impairs insulin-stimulated glucose transport and enhances Basal glycogen synthase activity. Mol Cell Biol 28:61–70. doi:10.1128/MCB.01405-07
Kumar A et al (2010) Fat cell-specific ablation of rictor in mice impairs insulin-regulated fat cell and whole-body glucose and lipid metabolism. Diabetes 59:1397–1406. doi:10.2337/db09-1061
Lagouge M et al (2006) Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 127:1109–1122. doi:10.1016/j.cell.2006.11.013
Lakshminarasimhan M, Rauh D, Schutkowski M, Steegborn C (2013) Sirt1 activation by resveratrol is substrate sequence-selective. Aging (Albany NY) 5:151–154. doi:10.18632/aging.100542
Lamming DW et al (2012) Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 335:1638–1643. doi:10.1126/science.1215135
Lamming DW, Ye L, Sabatini DM, Baur JA (2013) Rapalogs and mTOR inhibitors as anti-aging therapeutics. J Clin Invest 123:980–989. doi:10.1172/JCI64099
Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149:274–293. doi:10.1016/j.cell.2012.03.017
Liao CY, Rikke BA, Johnson TE, Diaz V, Nelson JF (2010) Genetic variation in the murine lifespan response to dietary restriction: from life extension to life shortening. Aging Cell 9:92–95. doi:10.1111/j.1474-9726.2009.00533.x
Lindberg-Larsen R, Moller N, Schmitz O, Nielsen S, Andersen M, Orskov H, Jorgensen JO (2007) The impact of pegvisomant treatment on substrate metabolism and insulin sensitivity in patients with acromegaly. J Clin Endocrinol Metab 92:1724–1728. doi:10.1210/jc.2006-2276
List EO et al (2011) Endocrine parameters and phenotypes of the growth hormone receptor gene disrupted (GHR-/-) mouse. Endocr Rev 32:356–386. doi:10.1210/er.2010-0009
Lopez-Guadamillas E, Munoz-Martin M, Martinez S, Pastor J, Fernandez-Marcos PJ, Serrano M (2016) PI3Kalpha inhibition reduces obesity in mice. Aging (Albany NY) 8:2747–2753. doi:10.18632/aging.101075
Lopez-Lluch G, Navas P (2016) Calorie restriction as an intervention in ageing. J Physiol 594:2043–2060. doi:10.1113/JP270543
Ma XM, Blenis J (2009) Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol 10:307–318. doi:10.1038/nrm2672
Madiraju AK et al (2014) Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 510:542–546. doi:10.1038/nature13270
Makki K et al (2014) Beneficial metabolic effects of rapamycin are associated with enhanced regulatory cells in diet-induced obese mice. PLoS ONE 9:e92684. doi:10.1371/journal.pone.0092684
Martin-Montalvo A et al (2013) Metformin improves healthspan and lifespan in mice. Nat Commun 4:2192. doi:10.1038/ncomms3192
Martins R, Lithgow GJ, Link W (2016) Long live FOXO: unraveling the role of FOXO proteins in aging and longevity. Aging Cell 15:196–207. doi:10.1111/acel.12427
Masoro EJ (2009) Caloric restriction-induced life extension of rats and mice: a critique of proposed mechanisms. Biochim Biophys Acta 1790:1040–1048. doi:10.1016/j.bbagen.2009.02.011
Masternak MM et al (2012) Metabolic effects of intra-abdominal fat in GHRKO mice. Aging Cell 11:73–81. doi:10.1111/j.1474-9726.2011.00763.x
Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ (2013) Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 494:256–260. doi:10.1038/nature11808
Miller RA et al (2014) Rapamycin-mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell 13:468–477. doi:10.1111/acel.12194
Mitchell SJ et al (2016) Effects of sex, strain, and energy intake on hallmarks of aging in mice. Cell Metab 23:1093–1112. doi:10.1016/j.cmet.2016.05.027
Mitchell SJ et al (2014) The SIRT1 activator SRT1720 extends lifespan and improves health of mice fed a standard diet. Cell Rep 6:836–843. doi:10.1016/j.celrep.2014.01.031
Morris BJ, Willcox DC, Donlon TA, Willcox BJ (2015) FOXO3: a major gene for human longevity–a mini-review. Gerontology 61:515–525. doi:10.1159/000375235
Murakami M et al (2004) mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol Cell Biol 24:6710–6718. doi:10.1128/MCB.24.15.6710-6718.2004
Nelson JF, Strong R, Bokov A, Diaz V, Ward W (2012) Probing the relationship between insulin sensitivity and longevity using genetically modified mice. J Gerontol A 67:1332–1338. doi:10.1093/gerona/gls199
Nojima A et al (2013) Haploinsufficiency of akt1 prolongs the lifespan of mice. PLoS ONE 8:e69178. doi:10.1371/journal.pone.0069178
Onken B, Driscoll M (2010) Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans Healthspan via AMPK, LKB1, and SKN-1. PLoS ONE 5:e8758. doi:10.1371/journal.pone.0008758
Ortega-Molina A et al (2012) Pten positively regulates brown adipose function, energy expenditure, and longevity. Cell Metab 15:382–394. doi:10.1016/j.cmet.2012.02.001
Ortega-Molina A et al (2015) Pharmacological inhibition of PI3K reduces adiposity and metabolic syndrome in obese mice and rhesus monkeys. Cell Metab 21:558–570. doi:10.1016/j.cmet.2015.02.017
Panici JA, Harper JM, Miller RA, Bartke A, Spong A, Masternak MM (2010) Early life growth hormone treatment shortens longevity and decreases cellular stress resistance in long-lived mutant mice. FASEB J 24:5073–5079. doi:10.1096/fj.10-163253
Park SJ et al (2012) Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 148:421–433. doi:10.1016/j.cell.2012.01.017
Parkinson C, Drake WM, Roberts ME, Meeran K, Besser GM, Trainer PJ (2002) A comparison of the effects of pegvisomant and octreotide on glucose, insulin, gastrin, cholecystokinin, and pancreatic polypeptide responses to oral glucose and a standard mixed meal. J Clin Endocrinol Metab 87:1797–1804. doi:10.1210/jcem.87.4.8432
Plank M, Wuttke D, van Dam S, Clarke SA, de Magalhaes JP (2012) A meta-analysis of caloric restriction gene expression profiles to infer common signatures and regulatory mechanisms. Mol BioSyst 8:1339–1349. doi:10.1039/c2mb05255e
Polak P, Hall MN (2009) mTOR and the control of whole body metabolism. Curr Opin Cell Biol 21:209–218. doi:10.1016/j.ceb.2009.01.024
Polak P, Cybulski N, Feige JN, Auwerx J, Ruegg MA, Hall MN (2008) Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab 8:399–410. doi:10.1016/j.cmet.2008.09.003
Pryor R, Cabreiro F (2015) Repurposing metformin: an old drug with new tricks in its binding pockets. Biochem J 471:307–322. doi:10.1042/BJ20150497
Risson V et al (2009) Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. J Cell Biol 187:859–874. doi:10.1083/jcb.200903131
Rovira J et al (2008) Effect of mTOR inhibitor on body weight: from an experimental rat model to human transplant patients. Transpl Int 21:992–998. doi:10.1111/j.1432-2277.2008.00710.x
Rudman D et al (1990) Effects of human growth hormone in men over 60 years old. N Engl J Med 323:1–6. doi:10.1056/NEJM199007053230101
Samaras TT, Storms LH (1992) Impact of height and weight on life span. Bull World Health Organ 70:259–267
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307:1098–1101. doi:10.1126/science.1106148
Selman C et al (2008) Evidence for lifespan extension and delayed age-related biomarkers in insulin receptor substrate 1 null mice. FASEB J 22:807–818. doi:10.1096/fj.07-9261com
Selman C, Partridge L, Withers DJ (2011) Replication of extended lifespan phenotype in mice with deletion of insulin receptor substrate 1. PLoS ONE 6:e16144. doi:10.1371/journal.pone.0016144
Selman C et al (2009) Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 326:140–144. doi:10.1126/science.1177221
Semba RD, Nicklett EJ, Ferrucci L (2010) Does accumulation of advanced glycation end products contribute to the aging phenotype? J Gerontol A 65:963–975. doi:10.1093/gerona/glq074
Shah OJ, Wang Z, Hunter T (2004) Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol 14:1650–1656. doi:10.1016/j.cub.2004.08.026
Sinclair DA, Guarente L (2014) Small-molecule allosteric activators of sirtuins. Annu Rev Pharmacol Toxicol 54:363–380. doi:10.1146/annurev-pharmtox-010611-134657
Slack C (2017) Ras signalling in aging and metabolic regulation nutrition and healthy aging. Preprint:1-11. doi:10.3233/NHA-160021
Slack C, Alic N, Foley A, Cabecinha M, Hoddinott MP, Partridge L (2015) The Ras-Erk-ETS-signaling pathway is a drug target for longevity. Cell 162:72–83. doi:10.1016/j.cell.2015.06.023
Suh Y et al (2008) Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc Natl Acad Sci USA 105:3438–3442. doi:10.1073/pnas.0705467105
Taguchi A, Wartschow LM, White MF (2007) Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science 317:369–372. doi:10.1126/science.1142179
Tanti JF, Jager J (2009) Cellular mechanisms of insulin resistance: role of stress-regulated serine kinases and insulin receptor substrates (IRS) serine phosphorylation. Curr Opin Pharmacol 9:753–762. doi:10.1016/j.coph.2009.07.004
Templeman NM et al (2017) Reduced circulating insulin enhances insulin sensitivity in old mice and extends lifespan. Cell Rep 20:451–463. doi:10.1016/j.celrep.2017.06.048
Thankamony A et al (2014) Short-term administration of pegvisomant improves hepatic insulin sensitivity and reduces soleus muscle intramyocellular lipid content in young adults with type 1 diabetes. J Clin Endocrinol Metab 99:639–647. doi:10.1210/jc.2013-3264
Timmers S et al (2011) Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab 14:612–622. doi:10.1016/j.cmet.2011.10.002
Tremblay F, Marette A (2001) Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J Biol Chem 276:38052–38060. doi:10.1074/jbc.M106703200
Um JH et al (2010) AMP-activated protein kinase-deficient mice are resistant to the metabolic effects of resveratrol. Diabetes 59:554–563. doi:10.2337/db09-0482
Um SH et al (2004) Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431:200–205. doi:10.1038/nature02866
van der Lely AJ et al (2012) Long-term safety of pegvisomant in patients with acromegaly: comprehensive review of 1288 subjects in ACROSTUDY. J Clin Endocrinol Metab 97:1589–1597. doi:10.1210/jc.2011-2508
van Heemst D et al (2005) Reduced insulin/IGF-1 signalling and human longevity. Aging Cell 4:79–85. doi:10.1111/j.1474-9728.2005.00148.x
Vermeij WP et al (2016) Restricted diet delays accelerated ageing and genomic stress in DNA-repair-deficient mice. Nature 537:427–431. doi:10.1038/nature19329
Wander SA, Hennessy BT, Slingerland JM (2011) Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. J Clin Invest 121:1231–1241. doi:10.1172/JCI44145
Weiss EP et al (2006) Improvements in glucose tolerance and insulin action induced by increasing energy expenditure or decreasing energy intake: a randomized controlled trial. Am J Clin Nutr 84:1033–1042
Westbrook R, Bonkowski MS, Strader AD, Bartke A (2009) Alterations in oxygen consumption, respiratory quotient, and heat production in long-lived GHRKO and Ames dwarf mice, and short-lived bGH transgenic mice. J Gerontol A 64:443–451. doi:10.1093/gerona/gln075
White UA, Tchoukalova YD (2014) Sex dimorphism and depot differences in adipose tissue function. Biochim Biophys Acta 1842:377–392. doi:10.1016/j.bbadis.2013.05.006
Whiteman EL, Cho H, Birnbaum MJ (2002) Role of Akt/protein kinase B in metabolism. Trends Endocrinol Metab 13:444–451
Wu JJ et al (2013) Increased mammalian lifespan and a segmental and tissue-specific slowing of aging after genetic reduction of mTOR expression. Cell Rep 4:913–920. doi:10.1016/j.celrep.2013.07.030
Xu J, Gontier G, Chaker Z, Lacube P, Dupont J, Holzenberger M (2014) Longevity effect of IGF-1R(±) mutation depends on genetic background-specific receptor activation. Aging Cell 13:19–28. doi:10.1111/acel.12145
Zahr E et al (2008) Rapamycin impairs beta-cell proliferation in vivo. Transplant Proc 40:436–437. doi:10.1016/j.transproceed.2008.02.011
Zhou Y et al (1997) A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse). Proc Natl Acad Sci USA 94:13215–13220
Zoncu R, Efeyan A, Sabatini DM (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12:21–35. doi:10.1038/nrm3025
Acknowledgements
The authors wish to thank the anonymous referees for critical comments and suggestions on the manuscript. Research in L. Foukas’ lab is supported by the Wellcome Trust.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bettedi, L., Foukas, L.C. Growth factor, energy and nutrient sensing signalling pathways in metabolic ageing. Biogerontology 18, 913–929 (2017). https://doi.org/10.1007/s10522-017-9724-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10522-017-9724-6