
Abstract
The TECTA gene encodes alpha-tectorin (TECTA), a major noncollagenous component of the tectorial membrane (TM). In humans, mutations in TECTA lead to either dominant (DFNA8/A12) or recessive (DFNB21) forms of nonsyndromic hearing loss. All missense mutations in TECTA that have been reported thus far are associated with the dominant subtype, whereas those leading to recessive deafness are all inactivating mutations. In this paper, we characterize a spontaneous missense mutation (c.1046C > A, p.A349D) arising in the mouse Tecta gene that is, unlike all previously reported missense mutations in TECTA, recessive. The morphological phenotype of the Tecta A349D/A349D mouse resembles but is not identical to that previously described for the \(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \) mouse. As in the \(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \) mouse, the TM is completely detached from the surface of the organ of Corti and spiral limbus, lacks a striated-sheet matrix, and is deficient in both beta-tectorin (Tectb) and otogelin. A significant amount of Tecta is, however, detected in the TM of the Tecta A349D/A349D mouse, and numerous, electron-dense matrix granules are seen interspersed among the disorganized collagen fibrils. Mutated Tecta A349D is therefore incorporated into the TM but presumably unable to interact with either Tectb or otogelin. The Tecta A349D/A349D mouse reveals that missense mutations in Tecta can be recessive and lead to TM detachment and suggests that should similar mutations arise in the human population, they would likely cause deafness.
Similar content being viewed by others


Introduction
Hearing impairment is a common sensory disorder. It has been estimated that genetic factors account for deafness in approximately 1:2,000 newborns and for greater than 50% of severe-to-profound cases of childhood deafness (Morton 1991; Marazita et al. 1993). Hearing impairment also affects the elderly. Nearly 50% of octogenarians have difficulty in communicating without the use of sound amplification, and for many, the cause is genetic (Davis 1989; Petit 1996). Inherited hearing impairment can occur with other coinherited clinical features to form a recognized phenotype (syndromic hearing loss) or appear in isolation (nonsyndromic hearing loss). Nonsyndromic hearing loss accounts for approximately 70% of genetic deafness. It is almost exclusively monogenic and is highly heterogeneous, with some estimates of the number of deafness-causing genes exceeding 100 (Van Camp and Smith 2006).
One of these deafness-causing genes is TECTA, the gene encoding α-tectorin (TECTA). Tecta and Tectb comprise the main noncollagenous proteins of the tectorial membrane, a ribbon-like strip of extracellular matrix that lies over the surface of the organ of Corti. Tecta is a large modular glycoprotein that is post-translationally cleaved to produce three fragments that are covalently associated via disulfide bridges. These three fragments contain the N-terminal entactin G1-like domain, the large zonadhesin-like region containing two partial and three full von Willebrand factor type D repeats, and the C-terminal zona pellucida (ZP) domain (Legan et al. 1997). Tectb (β-tectorin) is a much smaller protein comprising a single ZP domain (Goodyear and Richardson 2002). Otogelin is a third noncollagenous protein of the tectorial membrane. Like TECTA, it is a large, modular glycosylated protein, and it may be involved in the organization of the fibrillar network of the TM (Cohen-Salmon et al. 1997; Simmler et al. 2000).
Mutations in TECTA are responsible for dominant (DFNA8/A12) and recessive (DFNB21) nonsyndromic hearing loss and provide a robust model for genotype–phenotype correlations. Missense mutations cause dominant hearing loss, the phenotype of which depends on the domain and residue affected (Verhoeven et al. 1998; Alloisio et al. 1999; Balciuniene et al. 1999; Moreno-Pelayo et al. 2001; Iwasaki et al. 2002; Pfister et al. 2004; Plantinga et al. 2006; Meyer et al. 2007a). All recessive mutations known thus far are of a truncating nature (i.e., splice site, frameshift, and nonsense mutations or a large deletion of exon 10) and cause moderate to profound prelingual deafness (Mustapha et al. 1999; Naz et al. 2003; Meyer et al. 2007b; Alasti et al. 2008). At present, two transgenic mice have been generated with mutations in Tecta: a mouse with a recessive deletion of the entactin domain of Tecta (\(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \)) and a mouse with the Y1870C mutation in the ZP domain of Tecta (Tecta Y1870C/+). The transgenic mouse homozygous for a deletion in the entactin G1-like domain of Tecta (\(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \)) has a tectorial membrane that lacks all known noncollagenous structures, is primarily composed of randomly organized collagen fibrils, and is completely detached from both the organ of Corti and the spiral limbus (Legan et al. 2000). Mice heterozygous for this deletion have attached tectorial membranes of normal morphology, and heterozygous carriers of recessive, inactivating, human TECTA mutations have normal hearing (Mustapha et al. 1999; Naz et al. 2003; Meyer et al. 2007b, Alasti et al. 2008). The \(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \) mouse reveals a major role for the tectorial membrane in the hearing process: This extracellular matrix ensures that the outer hair cells can effectively respond to basilar membrane motion and deliver feedback with the appropriate gain and timing required for sound amplification (Legan et al. 2000). The Tecta Y1870C mutation is a missense mutation orthologous to that causing dominant, nonsyndromic deafness in an Austrian family (Verhoeven et al. 1998). Mice heterozygous for this mutation show a distinct phenotype: The tectorial membrane is attached to the spiral limbus but the limbal attachment zone is strikingly thinner and less prominent, and the main body of the tectorial membrane has distinctive ‘hump-back’ morphology (Legan et al. 2005). The Tecta Y1870C mutation is semidominant in mice, and mice that are homozygous for this mutation have a phenotype similar to that described for homozygous \(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \) mice; that is, the tectorial membranes are completely detached from the spiral limbus and the surface of the organ of Corti. Functional analysis of the Tecta Y1870C/+ mouse isolates a second role for the tectorial membrane in hearing; this matrix enables the motion of the basilar membrane to optimally drive the inner hair cells at their best frequency. Comparison of the two mouse models reveals that different mutations cause distinctive alterations in the architecture and functional properties of the tectorial membrane.
In this study, we have characterized a spontaneous missense mutation (TectaA349D/A349D) in a mouse that is, unlike all hitherto reported missense mutations in TECTA, recessive rather than dominant. The phenotype of this mouse is similar but not identical to that of the \(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \) mouse.
Materials and methods
Mouse matings
Matings set up to generate the different mouse litters, litters A to U, used in this study are summarized in Table 1. To generate litters Q, R, S, and T, 2 × F1 males and 2 × F1 females derived from breeding pair B (Tecta A349D/A349D × 129SvEv+/+) or pair C (Tecta A349D/A349D × C57BL6J+/+) were inbred, respectively, to create an F2 generation. The wild-type strains C57BL6J and 129SvEv were obtained from Harlan UK and Dr. Mark Maconochie, University of Sussex, respectively. The \(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \) and Tecta Y1870/Y1870C mutants used in this study were on a mixed, variable C57BL6J/129SvEv background, and the embryonic stem cell line CCB (Dr. Bill Colledge, University of Cambridge, UK) used to create TG28/1 was derived from a 129SvEv mouse. TG28/1 was a mouse chimera derived from a C57BL6J blastocyst and the 129SvEv-derived embryonic stem (ES) cells and arose during the creation of the \(Tecta^{\Delta {\text{ENT}}} \) mouse line.
Preparation of cochleae
Mice were euthanized according to UK Home Office regulations. Inner ears from the litters described in Table 1 were fixed between postnatal days (P) 11 and P24 for comparison of phenotype by immunofluorescence and light microscopy (see Figs. 2 and 4). Some of these cochleae were additionally examined by electron microscopy. Cochleae from two wild-type, two \(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \), and two Tecta A349D/A349D mice at P80, P77, and P82, respectively, were also examined by electron microscopy to confirm changes seen at the earlier stages persisted into adulthood.
For immunofluorescence microscopy, cochleae were removed and placed in fixative (3.7% formaldehyde in 0.1 M sodium phosphate, pH 7.2) for 2 h. The cochleae were then washed three times in Tris-buffered saline (TBS; 150 mM NaCl, 10 mM Tris HCl, pH 7.2), transferred into 0.5 M ethylenediamine tetraacetic acid (EDTA; pH 8.0), and decalcified for 6–21 days depending on the maturity of the cochleae. After decalcification, the cochleae were washed three times in TBS, equilibrated with 30% sucrose in phosphate-buffered saline (PBS) for 24 h, and embedded in cryo-agar (1% low-gelling temperature agarose, 18% sucrose in PBS). Sections (10 μm thick) were cut at −28°C using a Cryocut 1800 (Reichert-Jung), collected on gelatin-coated slides, dried for 1 h at 37°C, and stored at −20°C until use.
For light and electron microscopy, inner ears were fixed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate, pH 7.2, containing 1% tannic acid for 2 h, rinsed in cacodylate buffer, postfixed in 1% osmium tetroxide for 1–2 h, rinsed in buffer, and decalcified in 0.5 M EDTA as described above. Following decalcification, cochleae were dehydrated through a series of ascending concentrations of ethanol, equilibrated with propylene oxide, and embedded in Epoxy resin. For light microscopy, 1 μm-thick sections were cut with glass knives and stained with 1% Toluidin blue. For electron microscopy, ultrathin sections were cut with a diamond knife, double stained with uranyl acetate and lead citrate, and examined in a Hitachi 7100 transmission microscope.
Antibody staining
Cochlear cryosections were obtained for each mouse of the different litters studied (see Table 1) and stained with polyclonal rabbit antisera specific for Tecta (R9), Tectb (R7), or otogelin (Knipper et al. 2001; Cohen-Salmon et al. 1997). Slides were preblocked with TBS containing 10% horse serum and 1 mM sodium azide for 1 h at room temperature in a humid chamber. Antisera were diluted 1:500 in TBS with 10% horse serum containing 1 mM sodium azide and centrifuged at 14,000 rpm for 5 min prior to use. Diluted sera were applied to sections, and the slides were incubated overnight at room temperature in a humid chamber. Slides were washed three times in TBS, and fluorescein isothiocyanate (FITC)-conjugated swine anti-rabbit antibody (Dako; Ely, Cambs, UK) was added at a dilution of 1:100 in TBS/horse serum. The slides were incubated for 2–3 h at room temperature in a humid chamber. After three washes in TBS, slides were mounted with Vectashield™ (Vector Laboratories, Peterborough, UK). Sections were examined with a Zeiss Axioplan 2 microscope using FITC filters and photographed using a Spot™ Junior camera. All pictures were taken at ×20 magnification.
Molecular genetic methods
Genomic deoxyribonucleic acid (DNA) was extracted from Tecta A349D/A349D and Tecta +/A349D mice following standard conditions (Blin and Stafford 1976; Sambrook et al. 1989). Primers to amplify by the polymerase chain reaction (PCR) all the exons and intron–exon boundaries of the mouse Tecta gene were designed using Oligo 4.0 (Molecular Biology Insights, West Cascade, CO, USA) and are provided in Table 2. PCR reactions were carried out as reported elsewhere (del Castillo et al. 2002). PCR products were purified with the QIAquick PCR purification kit (Qiagen, Hilden, Germany) followed by direct sequencing using the BigDye Terminator v3.1 Cycle Sequencing Ready Reaction Kit in an ABI PRISM 3100 genetic analyzer (Applied Biosystems, Foster City, CA, USA). Genomic DNA was extracted from tail snips obtained from each pup of litters Q, R, S, and T (n = 48; Malumbres et al. 1997). DNA from the 18 mice strains used as controls (CBA, C3HeB/FeJ, BALB/c, 101/H, 129X1/SvJ, A/J, BxD-1/Ty, CE/J, DA/HuSn, DBA/2J, DDY/Jc1, FL/1Re, LP/J, NON/LTJ, RBG/Dn, ST/bJ, SWR/J, and C58/J) were a kind gift from Professor Karen Steel (Sanger Institute, Cambridge, UK). Each sample was screened for the novel mutation c.1046C > A by digesting exon 6 amplimers with the restriction enzyme AvaII and running the digestion products on agarose gels. When the mutation is present in the homozygous state, the 581-bp product is cleaved into two fragments of 331 and 250 bp. In heterozygous samples, the 581-bp band corresponding to the uncleaved wild-type allele is also visible.
Reverse transcriptase polymerase chain reaction
Total ribonucleic acid (RNA) was isolated from ten cochleae from Tecta+/+ or Tecta A349D/A349D mice using Trizol (Invitrogen, Paisley, UK). Randomly primed first-strand complementary DNA (cDNA) was synthesized from 2 μg of total RNA using avian myeloblastosis virus reverse transcriptase, and a control reaction was performed without reverse transcriptase (Promega, Southampton, UK). Reactions were diluted to 100 μl with water, and 2-μl aliquots were used for PCR with primer pairs MmTectaZPF1 and MmTectaZPR1, amplifying an 883-bp product spanning six exons encoding the ZP domain of Tecta. Aliquots were also used for PCR with primers MmTectbZPF1 and Mm TectbZPR1, amplifying an 844-bp product spanning eight exons encoding the ZP domain of Tectb, and with MmOtogF1 and MmOtogR1, amplifying an 853-bp product spanning exons 3 to 10 of Otog. PCR was performed in 25-μl volumes using 0.5 U Taq polymerase (Bioline, London, UK) and 12.5 pmol of each primer. Cycle times and temperatures were as follows: 94°C for 2 min, followed by 35 cycles of 94°C for 15 s, 58°C for 15 s, and 72°C for 1 min, finishing at 72°C for 5 min. Control PCR reactions were performed without adding aliquots of the reverse transcriptase reactions. The primer pairs used were MmTectaZPF1: CAGGAATTCGCTAGCCGTGACCTGCAAGGCAGCCCAG; MmTectaZPR1: GTCGGATCCGCTAGCCTCGAGTCACCTTTTTCTCCTAATGGGTC; MmTectbZPF1: CAGGAATTCGCTAGCCAAATCATGCACTCCGAATAAAGC; MmTectbZPR1: TCGGATCCGCTAGCCTCGAGTCAGCGCTTCCGTTTGTCACAGTTC; MmOtogF1: GCAGTGGAGGATGTCACAACAGT; and MmOtogR1: GGAGGCCTCAACAGTACTTCACA
Results
A mouse with a spontaneous mutation affecting the tectorial membrane
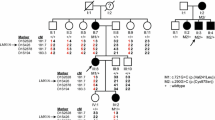
The breeding program used to isolate the spontaneous mutation affecting the tectorial membrane is summarized in Figure 1. A cross between a chimeric male mouse transmitting the \(Tecta^{\Delta {\text{ENT}}} \) mutation through the germ line (\(Tecta^{{ + \mathord{\left/ {\vphantom { + {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \)) with two female mice obtained from an in-house C57BL6J-breeding colony was carried out (\(Tecta^{{ + \mathord{\left/ {\vphantom { + {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \times Tecta^{{ + \mathord{\left/ {\vphantom { + + }} \right. \kern-\nulldelimiterspace} + }} \)). While inbreeding F1 mice derived from this cross, we identified three pairs of mice producing F2 progeny that were heterozygous for the deletion of the entactin G1-like domain of Tecta (\(Tecta^{{ + \mathord{\left/ {\vphantom { + {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \)) but with tectorial membranes that were completely detached from both the spiral limbus and the surface of the organ of Corti, a phenotype similar to that described for \(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \) mice (Legan et al. 2000). The three “wild-type” mice from these pairs that did not carry the \(Tecta^{\Delta {\text{ENT}}} \) mutation (one male and two females) were crossed, and a proportion (11 of 27) of the F2 progeny, all confirmed to lack the \(Tecta^{\Delta {\text{ENT}}} \) mutation, were found to have detached tectorial membranes. Two male and two female F2 mice that were derived from these matings of “wild-type” Tecta +/+ mice and judged to have impaired hearing on the basis of the Preyer reflex were then inbred. All of the resulting progeny (F3) from the two breeding pairs that were examined were found to have detached tectorial membranes. Mice derived from one of these two pairs were subsequently maintained as an inbred mutant colony.

Lineage diagram illustrating how the Tecta A349D/A349D mouse was derived. A chimeric male mouse (TG28/1) transmitting the \(Tecta^{\Delta {\text{ENT}}} \) allele through the germ line was mated with two wild-type C57BL6J mice, A and B. Three breeding pairs (\(Tecta^{{ + \mathord{\left/ {\vphantom { + {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \times Tecta^{{ + \mathord{\left/ {\vphantom { + + }} \right. \kern-\nulldelimiterspace} + }} \)) set up from the resultant F1 mice produced some \(Tecta^{{ + \mathord{\left/ {\vphantom { + {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \) F2 mice that had detached tectorial membranes. Inbreeding the Tecta +/+ F1 mice resulted in some Tecta +/+ F2 mice with detached tectorial membranes. F2 mice from this cross that were judged by the Preyer reflex to be hearing impaired were set up as breeding pairs, and all resulting F3 progeny were found to have detached tectorial membranes. Preyer reflex tests were not performed on the chimeric male mouse, its two female C57BL6J partners, or the F1 generation, and their inner ears were not retained for histological analysis.
These observations suggested a recessive spontaneous mutation had arisen either in Tecta or in another component of the tectorial membrane. This spontaneous mutation could have arisen in the chimeric male mouse or in the female partners that were derived from the wild-type C57BL6J colony and thought to be siblings. It is unlikely to have occurred in the F1 generation, as the same mutation would have to have arisen in three independent events. The detached tectorial membrane phenotype of these homozygous spontaneous mutant mice (Tecta A349D/A349D mice, see below) closely resembled that of the \(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \) mice (Fig. 2A–C) at the light microscope level, but both immunofluorescence (Fig. 2D–L) and electron microscopy (Fig. 3B) revealed subtle differences. Using polyclonal sera raised to native forms of chick Tecta and Tectb or recombinant otogelin, a significant amount of Tecta was detectable in the detached tectorial membrane of the spontaneous mutant mice relative to that of the \(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \) mouse (Fig. 2D–F), although neither mouse strain had detectable levels of either Tectb or otogelin in their detached tectorial membranes (Fig. 2G–L). Reverse transcriptase PCR analysis, however, confirmed the expression of Tecta, Tectb, and otogelin transcripts in developing cochleae of the spontaneous mutant mice (Fig. 3A). Transmission electron microscopy of the detached tectorial membranes in the spontaneous mutant mice revealed a complete lack of the striated-sheet matrix that is a characteristic of the tectorial membranes of wild-type mice and numerous clumps of dense granular material interspersed among the collagen fibrils (Fig. 3B, panels a, b, c, d). Similar clumps of dense material were only rarely detected in the detached tectorial membranes of the \(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \) mouse (Fig. 3B, panels c, d).

Phenotype of the Tecta A349D/A349D mouse. Toluidin blue-stained 1-μm-thick sections (A–C), anti-Tecta- (D–F), anti-Tectb- (G–I), and anti-otogelin (J–L) stained cryosections from A, D, G, J wild-type mice, B, E, H, K \(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \) mice (litter U), and C, F, I, L Tecta A349D/A349D mice (litter A1). Arrows indicate the tectorial membrane. Arrow in J shows otogelin staining in the sulcal region of the wild-type tectorial membrane. Scale bars = 100 μm.

Reverse transcriptase PCR analysis and ultrastructure of the tectorial membrane. A Tecta, Tectb, and Otog messenger RNAs (mRNA) are expressed in Tecta A349D/A349D cochleae. Randomly primed first-strand cDNA was prepared from total cochlear RNA from Tecta +/+ (WT) and Tecta A349D/A349D (A349D) mice, and products specific for Tecta, Tectb, and Otog mRNAs were amplified by PCR. C1 Reverse transcriptase reaction control, in which the reverse transcription reaction was performed without reverse transcriptase and then amplified by the PCR, C2 control PCR performed without an aliquot of the reverse transcriptase reaction. B Electron micrographs illustrating the ultrastructural appearance of the tectorial membrane in wild-type (a, b), \(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \) (c, d), and Tecta A349D/A349D (e, f) mice at postnatal days P80, P77, and P82, respectively. Collagen fibrils (arrows) are embedded in a striated-sheet matrix (SSM) in the tectorial membrane of wild-type mice (a, b). In Tecta ΔENT/ΔENT (c, d) and Tecta A349D/A349D (e, f) mice, the striated-sheet matrix is absent, and large electron dense granules (arrowheads) are visible. These granules are more numerous in the Tecta A349D/A349D mouse than in the \(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \) mouse (compare c and e). Scale bars for a, c, e = 1 μm, for b, d, f = 200 nm.
To test the hypothesis that the spontaneous mutant had a novel mutation in either Tecta or Tectb, we crossed the mice that we assumed to be homozygous for the spontaneous mutation with Tectb −/−, \(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \), and Tecta Y1870C/Y1870C mice. We also crossed Tectb −/− mice with \(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \) and Tecta Y1870C/Y1870C mice and \(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \) mice with Tecta Y1870C/Y1870C mice. Mice were analyzed by light and immunofluorescence microscopy (Fig. 4 and Table 1).

Phenotype of compound and double heterozygotes. Toluidin blue-stained 1-μm sections (A, D, G, J, and M), anti-Tecta-stained cryosections (B, E, H, K, and N), and anti-Tectb-stained cryosections (C, F, I, L, and O) from the following double heterozygotes Tecta +/A349D, Tectb +/− mice (litter D, A–C); \(Tecta^{{ + \mathord{\left/ {\vphantom { + {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \), Tectb +/− mice (litter F, D–F); Tecta Y1870C/+, Tectb +/− mice (litter I, G–I); and from the \(Tecta^{{{{\text{A349D}}} \mathord{\left/ {\vphantom {{{\text{A349D}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \) mice (litter J, J–L) and Tecta Y1870C/A349D mice (litter G, M–O) compound heterozygotes. Arrows indicate the tectorial membrane. Scale bars = 100 μm.
Tectb−/− mice have the first four coding exons of Tectb deleted and are functional nulls (Russell et al. 2007). The mutation is recessive, and Tectb−/− mice have no Tectb present in the tectorial membrane, but the production and targeting of Tecta to the tectorial membrane is normal. The tectorial membrane of Tectb−/− mice has a number of structural defects, including a lack of organized striated-sheet matrix and the loss of Hensen’s stripe, but it remains attached to the spiral limbus and lies over the surface of the organ of Corti. The F1 generation from the mating of homozygous spontaneous mutant mice with Tectb−/− mice (litter D) was expected to be heterozygous at each locus. All progeny had tectorial membranes that were attached to the spiral limbus, stretched over the inner sulcus, lay over the organ of Corti, and stained positively for both Tecta and Tectb (Fig. 4A–C). These results indicate complementation, mutations in two different genes, and were consistent with the hypothesis that the spontaneous mutation was recessive and had arisen in the Tecta gene.
As a positive control, \(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \) and Tectb −/− mice, known to have recessive mutations in genes encoding two different tectorial membrane proteins, were mated. As expected, all offspring (litter F) were heterozygous at each locus (\(Tecta^{{ + \mathord{\left/ {\vphantom { + {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \), Tectb +/ −) and had attached tectorial membranes with wild-type morphology (Fig. 4D). The tectorial membranes were positive for Tecta and Tectb by immunofluorescence (Fig. 4E–F). Furthermore, mice doubly heterozygous for the semidominant Tecta Y1870C mutation and the Tectb deletion (litter I; Tecta Y1870C/+, Tectb +/−) had Tecta/Tectb-positive tectorial membranes with the hump-backed morphology characteristic of Tecta Y1870C/+ mice (Fig. 4G–I).
Matings between mice deduced to be homozygous for the spontaneous recessive mutation and mice homozygous for either the recessive \(Tecta^{\Delta {\text{ENT}}} \) or the semidominant Tecta Y1870C mutation further indicated that spontaneous mutation was likely to be in Tecta. All progeny from these two matings (litters G and J) had detached tectorial membranes that lacked the expression of Tectb (Fig. 4J–L,M–O), as in \(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \), Tecta A349D/A349D, and Tecta Y1870C/Y1870C mice. As a positive control for the phenotype arising from the combination of a semidominant and a recessive mutation in Tecta, we mated Tecta Y1870/Y1870C mice with \(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \) mice and found that all progeny (litter P) also had detached tectorial membranes that lacked Tectb (not shown).
Identification of the spontaneous recessive missense mutation, p.Ala349Asp, in the zonadhesin domain of α-tectorin
The results described above strongly suggested the spontaneous mutation was likely to be in Tecta. We therefore sequenced all exons and intron/exon boundaries of the mouse Tecta gene from a mouse assumed to be homozygous for the spontaneous mutation and from the ES cell line used to generate the chimeric male transmitting the \(Tecta^{\Delta {\text{ENT}}} \) mutation. This analysis revealed, after comparison with the genomic Tecta sequence of a C57BL6J mouse used as a reference (Contig accession number NT _ 039472.6 and GenBank entry AK136719.1), that both DNA samples shared several homozygous changes including intronic and silent exonic variations and four amino acid substitutions (see full list of changes summarized in Table 3). All these exonic changes were also present in the Tecta cDNA sequence (NM _ 009347) of a wild-type CD1 mouse and should correspond to single nucleotide polymorphisms. The mouse assumed to be homozygous for the spontaneous mutation presented, however, an additional, previously undescribed homozygous C-to-A transversion at nucleotide 1046 in exon 6 (c.1046C > A, Fig. 5A). This mutation is predicted to cause a missense mutation that replaces an alanine by an aspartic acid at position 349 of Tecta (p.Ala349Asp, A349D). This residue is fully conserved between the protein sequences of human, mouse, rat, and chicken Tecta (Fig. 5C). The A349D mutation is located in the first full von Willebrand type D repeat (D1) of Tecta (Fig. 5B). Thus far, no missense mutations have been reported in this repeat in the human TECTA. The mutation creates a novel restriction site for the enzyme AvaII at exon 6. We took advantage of this finding to develop a screening test specific for the mutation (see “MATERIALS AND METHODS”). This test allowed us to verify: (1) that this mutation was not present in 18 different nonassociated mouse strains (see “MATERIALS AND METHODS”) and (2) that mice predicted to be heterozygous for the spontaneous mutation were heterozygous for the c.1046C > A mutation. In addition, this test was used to analyze the offspring (litters Q, R, S, and T, see Table 1) from the mating between mice heterozygous for the spontaneous mutation. Litters Q and R, as well as S and T, were expected to be identical in genotype as two male and two female siblings that were considered to be heterozygous for the spontaneous mutation were bred together. Of the 48 pups from the four F2 generations (litters Q, R, S, and T), 14 (29%) displayed detached tectorial membranes, and 34 (71%) displayed wild-type-looking tectorial membranes. These data indicate that the tectorial membrane phenotypes segregate in the expected 1:3 Mendelian ratio. The test showed: (1) the presence of the c.1046C > A mutation in a homozygous state in all mice with a detached tectorial membrane and (2) among the 34 pups with a normal tectorial membrane phenotype, 18 (38% out of 48) were Tecta +/A349D and 16 (33% out of 48) were Tecta +/+. The observed distribution (14 × A349D/A349D, 18 × +/A349D, 16 × +/+) did not show significant differences with that expected for a sample of 48 mice (12 × −/−, 24 × +/−, 12 × +/+) and a recessive Mendelian segregation (P value = 0.4533, chi-squared test). Taken together, these findings indicate that the A349D mutation is recessive and that it is responsible for the altered tectorial membrane phenotype seen in the Tecta A349D/A349D mouse.

Identification of the p.A349D mutation in Tecta. A Electropherogram depicting the mouse Tecta exon 6 fragment that contains the homozygous recessive mutation c.1046C > A (p.A349D) responsible for the Tecta A349D/A349D phenotype. B Schematic representation of the Tecta protein domains. D0–D4 units represent the von Willebrand factor (vWFD) type D repeats (Zonadhesin-like). The asterisk indicates the position of the p.A349D mutation at the vWFD (D1) repeat. The initial and last amino acid (AA) residues for each domain are indicated. C Multiple protein alignment of alpha-tectorin and homologous sequences in the von Willebrand type D1 repeat partial region. The A349 residue, which is mutated in the Tecta A349D/A349D mouse, is indicated in bold. The alignment comprises the following sequences: Mus musculus (NP_033373), Rattus norvegicus (XP_136124), Homo sapiens (NP_005413), and Gallus gallus (NP_990204).
Discussion
In this study, we have identified a spontaneous recessive missense mutation (p.A349D) occurring in the zonadhesin-like domain of Tecta and shown that it is most likely to be responsible for the observed phenotype. The Tecta A349D mutation is accompanied by several nucleotide variations, including four nonsynonymous changes that are also found in the ES cell line used to generate the chimera, indicating it arose in the chimeric male transmitting the \(Tecta^{\Delta {\text{ENT}}} \) mutation and not in the female partners derived from the wild-type C57BL6J colony. The Tecta A349D mutation was uncovered by virtue of its interaction with the recessive \(Tecta^{\Delta {\text{ENT}}} \) mutation.
The recessive Tecta A349D missense mutation leads to a morphological phenotype with characteristics that are similar but not identical to those caused by the deletion of the entactin-G1 like domain of Tecta in the \(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \) mouse (Legan et al. 2000). While the Tecta A349D/A349D and \(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \) mice both have tectorial membranes that are completely detached from the organ of Corti and spiral limbus and while the TMs of these mice lack both Tectb and otogelin, there are distinct differences. In the Tecta A349D/A349D mouse, numerous electron-dense aggregates are present in the detached TM, and Tecta is readily detectable by immunofluorecence microscopy. In the \(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \) mouse, by contrast, electron-dense aggregates are only occasionally present in the TM, and Tecta is only just detectable. As Tecta is the only major noncollagenous protein present in the TM of the TectaA349D/A349D mouse and as the structure of the collagen fibrils appears normal, the electron dense aggregates are presumably accumulations of mutated TectaA349D protein. Together, these observations suggest that significant amounts of mutated TectaA349D are incorporated into the tectorial membranes but unable to interact with either Tectb or otogelin. Whether this failure to interact is directly or indirectly due to the mutation is unknown. The entactin domain deletion and the missense A349D mutation are in different domains of Tecta, both of which may interact with Tectb, but both mutations may simply prevent the protein from folding correctly and indirectly prevent association with Tectb or otogelin, while allowing differing amounts of protein to be retained within the tectorial membrane as stable aggregates. This interpretation is also possible for the Tecta A349D/Y1870C mouse, in which a combination of the A349D recessive mutation with a semidominant mutation in the ZP domain of Tecta results in detached tectorial membranes with no staining for Tectb. The lack of Tectb or otogelin in the TM of the Tecta A349D/A349D mouse indicates that these proteins are either not secreted or unable to form stable, insoluble matrix structures in the absence of Tecta and provides further evidence that Tecta is the major organizer of the TM’s noncollagenous matrix (Legan et al. 2000). The loss of otogelin from the TM of Tecta A349D/A349D mice is an unlikely account for the absence of Tectb, as a null mutation for otogelin has no effect on the structure of the striated-sheet matrix or the distribution of the major components of the TM (Simmler et al. 2000).
The dominant DFNA8/A12 missense mutations, which affect different residues and domains of human TECTA, result in distinctive audiological phenotypes (Verhoeven et al. 1998; Alloisio et al. 1999; Balciuniene et al. 1999; Moreno-Pelayo et al. 2001; Iwasaki et al. 2002; Pfister et al. 2004; Plantinga et al. 2006; Meyer et al. 2007a). Thus, missense mutations in the zonadhesin-like region cause high-frequency hearing loss, whereas those in the ZP domain lead to hearing loss in the mid-frequencies. In addition, mutations affecting cysteine residues cause progressive hearing loss, whereas those affecting other residues cause stable hearing loss. The recessive mutations that have been reported thus far in TECTA are all predicted to result in functional null alleles (Mustapha et al. 1999; Naz et al. 2003; Meyer et al. 2007b; Alasti et al. 2008). In contrast to the phenotypic variability seen with dominant TECTA mutations, families segregating recessive mutations share a similar phenotype, a severe hearing loss that is more pronounced in the 1,000–2,000-Hz frequency range and characterized by a flat to a shallow “U”-shaped audiogram, regardless of the location of the mutation. As all known recessive mutations are inactivating mutations, it has been suggested that the similar clinical phenotypes reflect the consequence of null alleles at the TECTA locus (Meyer et al. 2007b). The physiological phenotype of the Tecta A349D/A349D mouse has not yet been investigated. In light of the observed detachment of the tectorial membrane from the organ of Corti and spiral limbus, it is likely to be very similar to that previously described for the \(Tecta^{{{\Delta {\text{ENT}}} \mathord{\left/ {\vphantom {{\Delta {\text{ENT}}} {\Delta {\text{ENT}}}}} \right. \kern-\nulldelimiterspace} {\Delta {\text{ENT}}}}} \) mouse in which there is a severe (∼80 dB) elevation of the compound action potential audiogram threshold in the mid- (5–20 kHz) frequency region (Legan et al. 2000). The Tecta A349D/A349D mouse therefore reveals that recessive missense mutations in TECTA could cause detachment of the TM and, as a likely consequence, early-onset forms of deafness similar to those caused by the inactivating mutations. Whether such mutations occur in the human population remains to be shown.
References
Alasti F, Sanati MH, Behrouzifard AH, Sadeghi A, de Brouwer AP, Kremer H, Smith RJ, Van Camp G. A novel TECTA mutation confirms the recognizable phenotype among autosomal recessive hearing impairment families. Int. J. Pediatr. Otorhinolaryngol. 72:249–255, 2008.
Alloisio N, Morlé L, Bozon M, Godet J, Verhoeven K, Van Camp G, Plauchu H, Muller P, Collet L, Lina-Granade G. Mutation in the zonadhesin-like domain of α-tectorin associated with autosomal dominant non-syndromic hearing loss. Eur. J. Hum. Genet. 7:255–258, 1999.
Balciuniene J, Dahl N, Jalonen P, Verhoeven K, Van Camp G, Borg E, Pettersson U, Jazin E. Alpha-tectorin involvement in hearing disabilities: one gene-two phenotypes. Hum. Genet. 105:211–216, 1999.
Blin N, Stafford DW. A general method for isolation of high molecular weight DNA from eukaryotes. Nucleic Acids Res. 3:2303–2308, 1976.
Cohen-Salmon M, El-Amraoui A, Leibovici M, Petit C. Otogelin: a glycoprotein specific to the acellular membranes of the inner ear. Proc. Natl. Acad. Sci. USA 94:14450–14455, 1997.
Davis AC. The prevalence of hearing impairment and reported hearing disability among adults in Great Britain. Int. J. Epidemiol. 18:911–917, 1989.
del Castillo I, Villamar M, Moreno-Pelayo MA, del Castillo FJ, Alvarez A, Telleria D, Menendez I, Moreno F. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N. Engl. J. Med. 346:243–249, 2002.
Goodyear RJ, Richardson GP. Extracellular matrices associated with the apical surfaces of sensory epithelia in the inner ear: molecular and structural diversity. J. Neurobiol. 53:212–227, 2002.
Iwasaki S, Harada D, Usami S, Nagura M, Takeshita T, Hoshino T. Association of clinical features with mutation of TECTA in a family with autosomal dominant hearing loss. Arch. Otolaryngol. Head Neck Surg. 128:913–917, 2002.
Knipper M, Richardson G, Mack A, Muller M, Goodyear R, Limberger A, Rohbock K, Kopschall I, Zenner HP, Zimmermann U. Thyroid hormone-deficient period prior to the onset of hearing is associated with reduced levels of beta-tectorin protein in the tectorial membrane: implication for hearing loss. J. Biol. Chem. 276:39046–39052, 2001.
Legan PK, Rau A, Keen JN, Richardson GP. The mouse tectorins. Modular matrix proteins of the inner ear homologous to components of the sperm–egg adhesion system. J. Biol. Chem. 272:8791–8801, 1997.
Legan PK, Lukashkina VA, Goodyear RJ, Kossl M, Russell IJ, Richardson GP. A targeted deletion in alpha-tectorin reveals that the tectorial membrane is required for the gain and timing of cochlear feedback. Neuron. 28:273–285, 2000.
Legan PK, Lukashkina VA, Goodyear RJ, Lukashkin AN, Verhoeven K, Van Camp G, Russell IJ, Richardson GP. A deafness mutation isolates a second role for the tectorial membrane in hearing. Nat. Neurosci. 8:1035–1042, 2005.
Malumbres M, Mangues R, Ferrer N, Lu S, Pellicer A. Isolation of high molecular weight DNA for reliable genotyping of transgenic mice. Biotechniques 22:1114–1119, 1997.
Marazita ML, Ploughman LM, Rawlings B, Remington E, Arnos KS, Nance WE. Genetic epidemiological studies of early-onset deafness in the U.S. school-age population. Am. J. Med. Genet. 46:486–491, 1993.
Meyer NC, Nishimura CJ, McMordie S, Smith RJH. Audioprofiling identifies TECTA and GJB2-related deafness segregating in a single extended pedigree. Clin. Genet. 72:130–137, 2007a.
Meyer NC, Alasti F, Nishimura CJ, Imanirad P, Kahrizi K, Riazalhosseini Y, Malekpour M, Kochakian N, Jamali P, Van Camp G, et al. Identification of three novel TECTA mutations in Iranian families with autosomal recessive nonsyndromic hearing impairment at the DFNB21 locus. Am. J. Med. Genet. A. 143:1623–1629, 2007b.
Moreno-Pelayo MA, del Castillo I, Villamar M, Romero L, Hernández-Calvín FJ, Herraiz C, Barberá R, Navas C, Moreno F. A cysteine substitution in the zona pellucida domain of α-tectorin results in autosomal dominant postlingual progressive mid-frequencies hearing loss in a Spanish family. J. Med. Genet. 38:e13, 2001.
Morton NE. Genetic epidemiology of hearing impairment. Ann. NY Acad. Sci. 630:16–31, 1991.
Mustapha M, Weil D, Chardenoux S, Elias S, El-Zir E, Beckmann JS, Loiselet J, Petit C. An alpha -tectorin gene defect causes a newly identified autosomal recessive form of sensorineural pre-lingual non-syndromic deafness, DFNB21. Hum. Mol. Genet. 8:409–412, 1999.
Naz S, Alasti F, Mowjoodi A, Riazuddin S, Sanati MH, Friedman TB, Griffith AJ, Wilcox ER, Riazuddin S. Distinctive audiometric profile associated with DFNB21 alleles of TECTA. J. Med. Genet. 40:360–363, 2003.
Petit C. Genes responsible for human hereditary deafness: symphony of a thousand. Nat. Genet. 14:385–391, 1996.
Pfister M, Thiele H, Van Camp G, Fransen E, Apaydin F, Aydin O, Leistenschneider P, Devoto M, Zenner HP, Blin N, Nürnberg P, Ozkarakas H, Kupka S. A genotype-phenotype correlation with gender-effect for hearing impairment caused by TECTA mutations. Cell Physiol. Biochem. 14(4–6):369–376, 2004.
Plantinga RF, de Brouwer AP, Huygen PL, Kunst HP, Kremer H, Cremers CW. A novel TECTA mutation in a Dutch DFNA8/12 family confirms genotype-phenotype correlation. J. Assoc. Res. Otolaryngol. 7:173–181, 2006.
Russell IJ, Legan PK, Lukashkina VA, Lukashkin AN, Goodyear RJ, Richardson GP. Sharpened cochlear tuning in a mouse with a genetically modified tectorial membrane. Nat. Neurosci. 10:215–223, 2007.
Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual, vol. 2. Cold Spring Harbor, NY, Cold Spring Harbor Laboratory, pp. 9.16–9.19, 1989.
Simmler MC, Cohen-Salmon M, El-Amraoui A, Guillaud L, Benichou JC, Petit C, Panthier JJ. Targeted disruption of otog results in deafness and severe imbalance. Nat. Genet. 24:139–143, 2000.
Van Camp G, Smith RJH. Hereditary hearing loss homepage. 2006. Accessed November 2007, available at http://webhost.ua.ac.be/hhh/.
Verhoeven K, Van Laer L, Kirschhofer K, Legan PK, Hughes DC, Schatteman I, Verstreken M, Van Hauwe P, Coucke P, Chen A, et al. Mutations in the human α-tectorin gene cause autosomal dominant non-syndromic hearing impairment. Nat. Genet. 19:60–62, 1998.
Acknowledgments
We express our gratitude to Professor Karen Steel for the kind donation of DNA samples from the different mouse strains used as a control for the mutation screening. Antiserum to otogelin was kindly provided by Professor Christine Petit, Institute Pasteur, Paris, France. We also thank G. Mackmurdo for his collaboration in the immunohistochemistry assay. S.M.H. is recipient of a fellowship from L’Oréal-UNESCO “for women in Science”. A.M.R. is recipient of a contract from the Centre for Biomedical Research on Rare Diseases (CIBERER). This work was supported by grants from the Spanish Ministerio de Ciencia y Tecnología (SAF2005-06355), Spanish Fondo de Investigaciones Sanitarias (FIS PI-051942; G03/203, CP03/00014), the European Commission (FP6 Integrated Project EUROHEAR, LSHG-CT-2004–512063), and the Wellcome Trust (grant ref no. 071394/Z/03/Z).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Moreno-Pelayo, M.Á., Goodyear, R.J., Mencía, A. et al. Characterization of a Spontaneous, Recessive, Missense Mutation Arising in the Tecta Gene. JARO 9, 202–214 (2008). https://doi.org/10.1007/s10162-008-0116-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10162-008-0116-0