
Abstract
Background
Myeloid cell leukemia-1 (Mcl-1) is an anti-apoptotic protein that regulates apoptosis sensitivity in a variety of cell types. Here we evaluate the roles of Mcl-1 in chemotherapy-associated apoptosis in gastric cancer cells. In addition, our study examined whether Mcl-1 contributed to apoptosis resistance in so-called cancer stem cell (CSC)-like populations in gastric cancer.
Methods
Seven gastric cancer cell lines were used. The expression of Mcl-1 was assessed by either real-time polymerase chain reaction or Western blot analysis. Apoptosis was quantitated by morphological observation and caspase activity measurement. Adenovirus-mediated RNA interference (RNAi) technology was used to knockdown the expression of Mcl-1. The release of cytochrome c was evaluated by subcellular fractionation and immunoblot analysis. To identify and isolate the CSC-like populations, we used the CSC-associated cell surface marker CD44 and flow cytometry.
Results
Six out of the 7 gastric cancer cell lines overexpressed Mcl-1 protein. These Mcl-1-expressing cell lines were relatively resistant to chemotherapeutic agents such as 5-fluorouracil (5-FU) and cisplatin (CDDP). Depletion of Mcl-1 protein by RNAi technology effectively sensitized the cells to anticancer drug-induced mitochondrial cytochrome c release, caspase activation, and apoptosis. In addition, vast amounts of Mcl-1 mRNA were expressed in CD44-positive CSC-like cells. Mcl-1 suppression enhanced the apoptosis in CD44-positive cells to a level equivalent to that in CD44-negative cells, suggesting that Mcl-1 mediates chemotherapy resistance in CSC-like populations.
Conclusion
These results suggest that Mcl-1 mediates the resistance to apoptosis in gastric cancer cells by blocking the mitochondrial pathway of cell death. Mcl-1 depletion appears to be an attractive strategy to overcome chemotherapy resistance in gastric cancer cells.
Similar content being viewed by others

Introduction
Despite the reduction in the incidence of gastric cancer in most areas of the world, gastric cancer remains the second leading cause of cancer deaths worldwide [1]. Surgery is still the most effective treatment for gastric cancer and good survival can be achieved if the tumor is resectable. However, most gastric cancer is either diagnosed at an advanced stage, or relapses after apparently curative surgery. For these patients, systemic chemotherapy is the only treatment option available besides supportive care [2]. Despite recent improvements in response rates owing to new drug combinations, chemotherapy resistance remains a major obstacle to curative treatment. Cancer cell resistance to apoptosis caused by the over-activation of anti-apoptotic machinery is one of the major mechanisms of chemotherapy resistance by which tumor cells escape from anticancer drug-mediated induction of apoptosis.
Myeloid cell leukemia-1 (Mcl-1), a member of the anti-apoptotic bcl-2 family of proteins, is frequently upregulated or overexpressed in malignant cells. Mcl-1 was originally cloned as an early induction gene during differentiation of the myeloid cell line, ML-1 [3]. It localizes to the outer mitochondrial membrane and promotes cell survival by suppressing cytochrome c release from mitochondria via heterodimerization with and neutralization of effector pro-apoptotic Bcl-2 family members, including Bak, Bim, tBid, Bik, PUMA, and NOXA [4–7]. As the most plausible mechanism of the anti-apoptotic action of Mcl-1, it has been suggested that Mcl-1 may function as an anti-apoptotic factor by sequestering Bak on the outer mitochondrial membrane, preventing Bak oligomerization and cytochrome c release from mitochondria [7].
Targeting of the Mcl-1 molecule to enhance sensitivity to apoptosis in malignant cells has been achieved by either the antisense or small inhibitory RNA (siRNA) approach. We previously established an siRNA-mediated Mcl-1 knockdown system and reported that the suppression of Mcl-1 enhanced tumornecrosis factor (TNF)-alpha-related apoptosis inducing ligand (TRAIL)-associated apoptosis in chemotherapy-resistant cholangiocarcinoma cells [8]. Subsequently, we expanded the study to screen the effects of the anti-Mcl-1 siRNA on a variety of malignant cells, including gastric, colorectal, breast, and pancreatic cancer cells. We recently found that the best results for sensitization to chemotherapy by Mcl-1 suppression were obtained with gastric cancer. In support of our results, a recent report suggests that Mcl-1 expression is enhanced in gastric cancer and the expression level predicts the prognosis [9]. In addition, an antisense approach to inhibit Mcl-1 successfully enhanced sensitivity to chemotherapy drugs in gastric cancer cell lines [10].
Tumors consist of a mixture of heterogeneous types of malignant cells. The sensitivity to apoptosis of the different cancer cell types is diverse, and it is widely accepted that a subpopulation of malignant cells, so-called cancer stem cells (CSCs), are relatively resistant to cytotoxic agents as compared to the majority of non-stem cancer cells [11]. These CSCs are thought to be responsible for the high incidence of disease recurrence after complete disappearance of a tumor has been achieved with conventional chemotherapy. Thus, the induction of apoptosis in both non-CSCs and chemotherapy-resistant CSCs appears to be a key to improve cancer treatment results. Currently, some technologies to identify chemotherapy-resistant CSCs have been reported in gastric cancer [12]. Against this background, we evaluated whether Mcl-1 knockdown was effective for sensitizing chemotherapy-induced apoptosis in CSC-like populations in gastric cancer cells. We used a cell surface marker molecule, CD44, which is reported to be a CSC marker in gastric cancer [12–17], to identify chemotherapy-resistant subpopulations. We found that the Mcl-1 expression level was relatively high in CD44-positive gastric cancer cells. Here, we report for the first time that Mcl-1 knockdown effectively enhanced chemotherapy-mediated cytotoxicity in CSC-like populations in gastric cancer cells.
Materials and methods
Reagents
5-Fluorouracil (5-FU) and cisplatin (CDDP) were provided by Nippon Kayaku (Tokyo, Japan). The following antibodies were used for immunoblot analysis; anti-Mcl-1 (Santa Cruz Biotechnology, Santa Cruz, CA), anti-β-actin (Sigma, St. Louis, MO), and anti-cytochrome c (BD Pharmingen, San Diego, CA, USA).
Cells and cell culture
The human gastric cancer cell lines MKN45, MKN1, MKN74, AZ521, NUGC2, FU97, and IM95 were obtained from the Health Science Research Resources Bank (Osaka, Japan). MKN45, MKN1, and MKN74 were grown in RPMI1640 medium supplemented with 10 % fetal bovine serum (FBS). NUGC cells were grown in RPMI1640 medium supplemented with 15 % FBS. AZ521, FU97, and IM95 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % FBS. All media contained 100,000 units/l penicillin and 100 mg/l streptomycin (GIBCO, Life Technologies, Carlsbad, UK).
Adenovirus-mediated short hairpin (sh) RNA introduction
We used adenovirus vectors containing Ad5/35 chimeric fiber protein [18] and the vector plasmid pAdHM34 and the shuttle vector plasmid pHMCMV-GFP1, which have been described previously [19]. pHMCMV-GFP1 contains the cytomegalovirus (CMV) promoter, the GFP gene derived from pEGFP-N1 (Clontech, Palo Alto, CA, USA), and the bovine growth hormone (BGH) poly(A) signal. An siRNA expression unit containing human U6 promoter and two BspMI cloning sites was excised from the HIV-U6i-GFP plasmid by EcoRI digestion, then subcloned into the EcoRI site in pHMCMV-GFP1, which was located downstream of the BGH poly(A) signal. This vector was designated pHMCMV-GFP-U6i. The double-stranded (ds) oligonucleotides for the shRNA can be directly subcloned into the two BspMI sites of the pHMCMV-GFP-U6i [20]. Shuttle vector plasmids containing ds-oligonucleotides for Mcl-1; 5′-ACG GGA CTG GCT AGT TAA ACT TCA AGA GAG TTT AAC TAG CCA GTC CCG T-3′ [8], or GL3B (firefly luciferase, control) were constructed. The adenovirus vectors, AdHM34-Mcl-1 p906 and AdHM34-GL3B (control), were constructed by an improved in vitro ligation method as described previously [21]. Both adenovirus vectors were propagated in 293 cells and the viral titers were determined using Adeno-X Rapid Titer Kit (Clontech) according to the manufacturer’s instructions.
Immunoblot analysis
Cells were lysed by incubation on ice for 30 min in lysis buffer containing 20 mM Tris–HCl (pH 7.5), 1 % Triton X-100, 150 mM NaCl, 10 % glycerol, 1 mM Na3VO4, 50 mM NaF, 100 mM phenylmethylsulfonyl fluoride, 1 mM phenylmethylsulfonyl fluoride (PMSF), and a protease inhibitor mixture (Complete Protease Inhibitor Cocktail; Roche Diagnostics Japan, Tokyo, Japan). After centrifugation at 14,000g [8] for 15 min at 4 °C, the supernatants were collected. Samples were resolved by 12.5 % sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE), transferred to a nitrocellulose membrane, and blotted with primary antibodies. Horseradish peroxidase-conjugated secondary antibodies (Biosource International, CA) were used at appropriate concentrations. Bound antibodies were visualized using a chemiluminescent substrate (ECL; GE Health Care Sciences, Amersham Place, UK) and exposed to Kodak X-Omat film (Kodak, Rochester, NY).
Quantitative real-time reverse transcription polymerase chain reaction (PCR)
Total RNA was isolated using TRIzol reagent (Invitrogen, Life Technologies, Carlsbad, UK) according to the manufacturer’s instructions. The RNA samples were treated with deoxyribonuclease I (Invitrogen) and a reverse transcription PCR (RT-PCR) reaction was performed using Super Script First Strand Synthesis System for RT-PCR (Invitrogen). After agarose gel electrophoresis, the expected base pair PCR products were identified, and the PCR products were eluted using a QIAquick Gel Extraction kit (Qiagen, Qiagen Japan, Tokyo, Japan). The PCR products were prepared as standards for quantitative PCR at the concentrations of 109, 108, 107, 106, 105, and 104 copies/μl. Real-time PCR was performed using an ABI PRISM 7500 fast and SYBR green system (Applied Biosystems, Foster City, CA). PCR primer for 18s RNA was used as an internal control. The PCR primers for Mcl-1 were: forward, 5′-ATG CTT CGG AAA CTG GAC AT-3′; reverse, 5′-TCC TGA TGC CAC CTT CTT CTA GG-3′.
Cell viability assay
Cell viability was evaluated using the CellTiter 96 Aqueous One Solution cell proliferation assay (Promega, Madison, WI). Five thousand cells/well were seeded in 96-well plates and treated with or without either 5-FU or CDDP for 48 h. MTS [3,4-(5-dimethylthiazol-2-yl)-5-(3-carboxy-methoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt] was added and the MTS assay was performed according to the manufacturer’s instructions (Promega, Madison, WI).
Quantitation of apoptosis by 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) nuclear staining
Apoptosis was quantitated by assessing the characteristic nuclear changes of apoptosis (i.e., chromatin condensation and nuclear fragmentation), using the nuclear binding dye DAPI and fluorescence microscopy [22].
Caspase activity assay
Caspase-3 activity was evaluated by using a caspase colorimetric assay kit (Clontech) according to manufacturer’s instructions. In brief, 2 × 106 cells were lysed in 50 μl of lysis buffer. The lysate was mixed with 50 μl of the reaction mix containing 50 μM (final concentration) of acetyl-Asp-Glu-Val-Asp p-nitroanilide (Ac-DEVD-pNA) as a substrate for caspase 3. After 1-h incubation at 37 °C, the increase in absorbance of enzymatically released pNA at 405 nm was measured in a microplate reader.
Mitochondrial cytochrome-c release
Cytosolic extracts for the immunoblot assay of cytochrome c were obtained as described by Leist et al. [23]. Briefly, at the desired time points, the culture medium was exchanged for permeabilization buffer (210 mM d-mannitol, 70 mM sucrose, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 5 mM succinate, 0.2 mM ethylene glycol tetraacetic acid (EGTA), 0.15 % bovine serum albumin, 80 μg/ml digitonin, pH 7.2). The permeabilization buffer was removed by the centrifuge for 10 min at 13,000g. Supernatants representing the cytosolic extract were employed for the immunoblot analysis.
Flow cytometry
To identify and isolate the CD44-positive fraction and CD44-negative fraction, cells were removed from the culture dish with trypsin and ethylenediamine tetraacetic acid (EDTA), washed, and incubated with allophycocyanin (APC)-labeled anti-human CD44 (mouse IgG2b; BD Pharmingen) for 1 h at 37 °C. After washing, the cells were analyzed by using a FACS Vantage SE fluorescence activated cell sorter (Becton–Dickinson, Franklin Lakes, NJ). To identify and isolate the side population (SP) cell fraction and the main population (MP) cell fraction, cells were removed from the culture dish with trypsin and EDTA, washed, and suspended at 1 × 106/ml in 2 % FBS + HANKS Balanced Salt Solution (GIBCO). Then the cells were labeled with Hoechst 33342 dye (Dojinkagaku, Tokyo, Japan). The labeled cells were incubated for 60 min at 37 °C. After being washed once with the HANKS solution, the cells were counterstained with 1 μg/ml propidium iodide, to label dead cells. The cells were analyzed by using a FACS Vantage SE fluorescence activated cell sorter (Becton–Dickinson). After collecting 105 events, SP cells, which showed no Hoechst 33342 dye fluorescence, and MP cells, which included high Hoechst 33342 fluorescence, were defined as described previously [24].
Statistical analysis
Data are presented as means ± SD. To assess the statistical significance of differences, the t-test was performed. P values of <0.01 were considered significant.
Results
Gastric cancer cell lines express different levels of Mcl-1 protein and show varying sensitivity to 5-FU
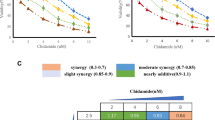
We examined Mcl-1 protein levels and sensitivity to 5-FU-associated cytotoxicity in seven gastric cancer cell lines (Fig. 1a). Mcl-1 protein was identified in all cell lines by immunoblot analysis; however, the Mcl-1 expression level was different in each cell line. Relatively high Mcl-1 levels were identified in six cell lines, while one cell line, AZ521, showed an extremely low level of Mcl-1 (Fig. 1b). Interestingly, the cell lines expressing high levels of Mcl-1 revealed relatively low sensitivity to the chemotherapeutic drug 5-FU, as the half maximal inhibitory concentration (IC50), the values of these cells were >50 μg/ml (Fig. 1b). In contrast, AZ521 cells, which showed low Mcl-1 levels, were considered to be 5-FU-sensitive, as their IC50 value was 12.5 μg/ml (Fig. 1b). Quantitative analysis revealed that the relative expression of Mcl-1/β-actin in 5-FU-sensitive AZ521 cells was significantly lower than that in the cell lines MKN45, MKN1, and MKN74, although it appeared that the sensitivity to 5-FU in MKN45, MKN1, and MKN74 cells was not correlated with the relative expression of Mcl-1/β-actin in these cell lines (Fig. 1c). These results led us to presume that Mcl-1 is a key mediator of chemotherapy resistance in many gastric cancers. Thus, we next attempted to suppress Mcl-1 expression by siRNA technology. We have previously established siRNA- or shRNA-mediated Mcl-1 suppression and found that the nucleotide position at 906–927 from the ATG site on the Mcl-1 mRNA sequence was the most effective target lesion [7]. Thus, an shRNA construct containing the nucleotide position 906–927 in adenovirus vector (Ad-P906) was prepared to obtain high efficiency of shRNA introduction. As shown in Fig. 2a, Ad-P906 effectively suppressed Mcl-1 protein levels at a multiplicity of infection (MOI) of 100–300, as compared to corresponding treatment with the control vector (Ad-control).

Mcl-1 levels and chemotherapy sensitivity in gastric cancer cell lines. a Survival of various gastric cancer cell lines in the presence of 5-fluorouracil (5-FU). The cells were grown on 96-well plates. After 48-h incubation with the indicated concentrations of 5-FU, the cells were submitted to the MTS assay, as described in “Materials and methods”. b Mcl-1 expression levels determined by immunoblot analysis. Six out of the seven cell lines tested expressed high levels of Mcl-1 protein, while the Mcl-1 level in AZ521 cells was negligible. The 5-FU half maximal inhibitory concentration (IC50) value for each cell line is indicated below the relevant lane. Note that the AZ521 cell line exhibits the lowest IC50 among these cell lines. c Relative expression of Mcl-1 in four representative cell lines (MKN45, MKN1, MKN74, and AZ521) was quantified by densitometry

Mcl-1 depletion by adenovirus-mediated anti-Mcl-1 short hairpin RNA (sh RNA) introduction. The cells were subjected to adenovirus-mediated shRNA introduction. Either AdHM34-Mcl-1 p906 (Ad-P906) or AdHM34-GL3B (Ad-control) virus was applied to the culture medium at a condition of 100–300 multiplicity of infection (MOI). a Mcl-1 levels determined by immunoblot analysis. An MOI of 300 was identified as a sufficient condition to induce significant suppression of Mcl-1 protein. b MKN45, IM95, and AZ521 cells grown in 10-cm plates were treated with either Ad-P906 or Ad-Control viruses at 300 MOI. Two days after the virus transfection, the cells were re-plated in 96-well plates. c MTS assay revealed no significant difference in cytotoxicity between P906 and control to MKN45, AZ501, and IM95. d The cells were incubated with and without either 5-FU or cisplatin (CDDP) for 48 h. The MTS assay was performed as described in “Materials and methods”. e IC50 values of gastric cancer cell lines for 5-FU and CDDP. N.S. not significant
Short hairpin RNA directed against Mcl-1 enhances chemotherapy sensitivity in gastric cancer cells expressing high levels of Mcl-1
We next evaluated whether Mcl-1 suppression enhanced 5-FU sensitivity (Fig. 2b). We chose three cell lines;two Mcl-1-enriched cell lines, MKN45 and IM95, and one Mcl-1-low cell line, AZ521. We confirmed that the viability with Ad-P906 virus infection was comparable to Ad-control in each of these cell lines when cultured without 5-FU (Fig. 2c). As expected, in the two cell lines with high Mcl-1 expression, Ad-P906 dramatically enhanced sensitivity to 5-FU and CDDP (Fig. 2d). The 5-FU IC50 values for MKN45 was significantly decreased by Ad-P906 transfection, from 78.6 to 26.2 μg/ml. That of IM95 was significantly decreased by Ad-P906 transfection, from 87.7 to 40.2 μg/ml (Fig. 2e). Similarly, the CDDP IC50 values for MKN45 was significantly decreased by Ad-P906 transfection, from 5.38 to 1.99 μg/ml. That of IM95 was significantly decreased by Ad-P906 transfection, from 7.21 to 2.51 μg/ml (Fig. 2e). In contrast, Ad-P906 transfection did not sensitize Mcl-1-low AZ521 cells to 5-FU or CDDP (Fig. 2e). Although AZ521 cells appeared to be relatively sensitive to these drugs as compared to MKN45 and IM95, the reason why Mcl-1 suppression did not enhance chemotherapy sensitivity was unknown. The contribution of other anti-apoptotic molecules, i.e., Bcl-2, Bcl-xL, and Bcl-xs, remains to be elucidated.
Mcl-1 suppression enhances 5-FU-mediated mitochondrial cytochrome c release, caspase activation, and apoptosis
Because Mcl-1 is a well-known anti-apoptotic molecule, we observed some key processes of apoptotic signaling to confirm that Mcl-1 depletion enhanced chemotherapy-mediated apoptosis in MKN45 cells. Figure 3a shows the immunoblot analysis of cytosolic extract of MKN45 cells with and without 5-FU treatment. In samples transfected with control vectors (lanes 1, 3, and 5), cytosolic cytochrome c was not observed without 5-FU treatment; however, significant cytochrome c bands were identified after 5-FU treatment, suggesting cytochrome c release from mitochondria to cytosol, a key phenomenon of apoptosis. Transfection with Ad-P906 dramatically enhanced cytochrome c release by 5-FU (30 and 40 μg/ml), while Ad-P906 itself slightly induced cytochrome c release in the absence of 5-FU. Caspase-3 activity (Fig. 3b) was also induced by 5-FU treatment, and this was significantly enhanced by Ad-P906 transfection. Finally, when apoptosis was morphologically quantitated by nuclear DAPI staining, we found that 5-FU-induced apoptosis was markedly enhanced by Ad-P906 (Fig. 3c). Thus, Mcl-1 suppression enhances 5-FU-mediated apoptosis by modulating mitochondrial cytochrome c release and enhancing caspase 3 activity.

Mcl-1 suppression enhances 5-FU-mediated mitochondrial cytochrome c release, caspase 3 activation, and apoptosis. Either AdHM34-Mcl-1 p906 (Ad-P906) or AdHM34-GL3B (Ad-control) virus was applied to the culture medium of MKN45 cells at the condition of 300 multiplicity of infection (MOI). a Cytosolic extracts were obtained as described in “Materials and methods”. Mitochondrial cytochrome c release was assessed by observing cytosolic cytochrome c by immunoblot analysis. b Caspase 3 activity was measured by a Ac-DEVD-pNA-based fluorometric assay as described in “Materials and methods”. Data are expressed as means ± SD from 3 independent experiments. P < 0.05 for AdHM34-Mcl-1 p906 (P906)-transfected 5-FU-treated group versus AdHM34-GL3B (control)-transfected 5-FU-treated group. c Apoptosis was quantitated by DAPI (4′,6-diamidino-2-phenylindole dihydrochloride) nuclear staining and fluorescence microscopy as described in “ Materials and methods”. Data are expressed as means ± SD from 3 independent experiments. P < 0.05 for Ad-P906-transfected group versus Ad-control-transfected group
The Mcl-1 level was enhanced in chemotherapy-resistant CD44-positive cells, and inhibition of Mcl-1 enhanced chemotherapy sensitivity in both CD44-positive and -negative cells
Because chemotherapy resistance or apoptosis resistance is one of the most important features of CSCs, it is accepted that targeting CSCs is important to achieve better treatment efficiency. The cell surface molecule CD44 has recently been reported as a useful marker to identify CSC-like populations in gastric cancer cells [17]. Thus, we attempted to evaluate the roles of the Mcl-1 molecule in chemotherapy resistance in CD44-positive CSC-like cells. We identified 20–25 % of CD44-positive cells in MKN-45 cells by flow cytometry (Fig. 4a). Interestingly, the expression level of Mcl-1 mRNA was fivefold higher in CD44-positive cells as compared to that in CD44-negative cells (Fig. 4b). As expected, CD44-positive cells were relatively resistant to 5-FU-mediated cytotoxicity as compared to CD44-negative cells (Fig. 4c). Consistently, when MKN-45 cells were cultured with 5-FU, the ratio of CD44+ cells in MKN45 cells cultured with 5-FU was significantly increased as compared to that in MKN45 cells before the culture (Fig. 4d). Lastly, suppression of Mcl-1 by shRNA technology overcame the chemotherapy resistance in CD44-positive cells, as 5-FU-mediated apoptosis was enhanced by Mcl-1 suppression in CD44-positive cells, and the magnitude of apoptosis reached a value equivalent to that in the CD44-negative cells (Fig. 4e) in parallel with the downmodulation of Mcl-1 expression in Ad-P906-transfected CD44+ and CD44− MKN45 cells (Fig. 4f).

Enhanced Mcl-1 expression level contributes to chemotherapy resistance in CD44-positive gastric cancer cells. MKN45 cells were labeled with anti-human CD44 antibodies and either the CD44-positive or the CD44-negative fraction was isolated by flow cytometry. a The cells were separated into CD44-positive and CD44-negative fractions. SSC-W: APC side scatter width APC-labeled, FSC-H forward scatter height. b Mcl-1 mRNA levels were quantitated by real-time polymerase chain reaction (PCR) technology as described in “Materials and methods”. Data are expressed as means ± SD from 3 independent experiments. P < 0.01 for CD44-positive versus CD44-negative group. c Either CD44-positive or CD44-negative cells were incubated on 96-well plates with or without the indicated concentrations of 5-FU for 48 h. Cell viability was assessed by MTS assay as described in “Materials and methods”. CD44-positive cells were markedly resistant to 5-FU-mediated cytotoxicity as compared to CD44-negative cells. d The ratio of CD44+ cells in MKN45 cells cultured with 5-FU was significantly increased as compared to that in MKN45 cells before the culture. e Either CD44-positive or -negative cells were treated with AdHM34-Mcl-1 p906 (P906) or AdHM34-GL3B (control) adenovirus vector. After incubation with 30 mg/ml of 5-FU for 48 h, the growth inhibition rate was evaluated by MTS assay as described in “Materials and methods”. Note that P906 introduction enhanced 5-FU-associated apoptosis in CD44-positive cells to a level equivalent to that in CD44-negative cells. f The Mcl-1 expression level in CD44-positive cells induced by Ad-P906 was almost same as that in CD44-negative cells induced by Ad-P906
To further strengthen our hypothesis, we isolated SP cells, which are thought to be enriched CSCs [25], and MP cells from MKN45 cells, using FACS (Fig. 5a), and assessed the expression of Mcl-1 mRNA. As expected, we found that approximately 2–3 % of total cells were SP cells (Fig. 5a), and also as expected, the expression of Mcl-1 mRNA in SP cells was significantly higher than that in MP cells (Fig. 5b). Furthermore, Mcl-1-rich SP cells were significantly resistant to 5-FU-induced cytotoxicity as compared to MP cells (Fig. 5c), but Ad-P906-mediated knockdown of Mcl-1 in SP cells restored the sensitivity of 5-FU-induced cytotoxicity as compared to Ad-control virus-infected SP cells (Fig. 5c). Collectively, these results suggest that Mcl-1 plays pivotal roles in regulating chemotherapy resistance in CD44-positive gastric cancer cells.

Enhanced Mcl-1 expression level contributes to chemotherapy resistance in SP cells in gastric cancer cells. a MKN45 cells were labeled with Hoechst red and Hoechst blue, and either the SP or the MP population was isolated by flow cytometry for further experiment. SP side population, MP main population. b Mcl-1 mRNA levels were quantitated by a real-time PCR method as described in “Materials and methods”. Data are expressed as means ± SD from 3 independent experiments. P < 0.05 for the SP versus the MP group. c Either SP or MP cells were incubated on 96-well plates with or without the indicated concentrations of 5-FU for 48 h. Cell viability was assessed by the MTS assay as described in “Materials and methods”. Either SP or MP cells were treated with Ad-P906 or Ad-control adenovirus vector. After incubation with 30 mg/ml of 5-FU for 48 h, the growth inhibition rate was evaluated by the MTS assay. Note that P906 introduction enhanced 5-FU-associated apoptosis in SP cells to a level equivalent to that in MP cells
Discussion
The principal findings of this study relate to the molecular mechanisms by which gastric cancer cells exert apoptosis resistance to chemotherapeutic agents. The results demonstrated that: (1) Mcl-1-expressing gastric cancer cells were relatively resistant to chemotherapeutic agents such as 5-FU and CDDP; (2) suppression of Mcl-1 protein by shRNA technology successfully enhanced chemotherapy sensitivity; (3) Mcl-1 suppression also enhanced 5-FU-mediated apoptosis-related events, including mitochondrial cytochrome c release and caspase-3 activation; and (4) chemotherapy resistance in the so-called CSC-like CD44-positive population was mediated by enhanced Mcl-1 expression. Our results are consistent with a previous report using antisense-based technology which has shown that Mcl-1 is a therapeutic target in gastric cancer cells [9]. In addition, our study is the first to demonstrate that Mcl-1 expression is relatively enhanced in CD44-positive gastric cancer cells. The present study further demonstrates that Mcl-1 suppression enhances 5-FU-mediated apoptosis in CD44-positive gastric cancer cells, suggesting that Mcl-1 is a key target molecule to overcome chemotherapy resistance in so-called CSCs. Because targeting CSCs is currently recognized as a promising strategy to improve the response rate and/or curative power of chemotherapy, anti-Mcl-1 therapy would appear to be an attractive technology which may contribute to improvement of outcomes in gastric cancer chemotherapy.
Enhanced expression of the anti-apoptotic molecule Mcl-1 has been reported in a variety of malignant cells [26]. For example, cholangiocarcinoma cells upregulate Mcl-1 expression via the interleukin-6-mediated Stat3 pathway [27]. Bcr/Abl-mediated Stat5-dependent Mcl-1 upregulation has been suggested in leukemic cells [28]. Melanoma cells upregulate the Mcl-1 level upon endoplasmic reticulum stress [29]. In gastric cancer, the Mcl-1 level was evaluated by an immunohistochemical technique, and the expression level of Mcl-1 was suggested as a prognostic marker [9, 30]. In addition, the mechanisms by which gastric cancer cells upregulate Mcl-1 expression level were also evaluated recently. These mechanisms include the PI3-kinase-Akt signaling-mediated induction of Mcl-1 [31], and post-transcriptional modification by certain micro-RNAs [32, 33]. The above reports suggest that Mcl-1 plays pivotal roles in the regulation of chemotherapy resistance in gastric cancer. Our results demonstrate that the Mcl-1 expression level varies between gastric cancer cell lines. Most of the cell lines used in the present study expressed high levels of Mcl-1 protein, while AZ521 cells expressed a relatively lower level of Mcl-1 and showed higher sensitivity to 5-FU (Fig. 1b). Thus, our results are consistent with the previous findings, and further confirm the importance of Mcl-1 in regulating apoptosis sensitivity in gastric cancer. The effects of shRNA-mediated Mcl-1 suppression suggest that Mcl-1 would appear to be an attractive molecular target to enhance chemotherapy sensitivity in gastric cancer cells which express high levels of Mcl-1 (Fig. 2).
Our study has demonstrated the roles of Mcl-1 in chemotherapy-associated apoptosis signaling in detail. Mcl-1 is a mitochondria-associated molecule and the major role of this molecule is to suppress other pro-apoptotic Bcl-2 family molecules such as Bak, Bim, tBid, Bik, PUMA, and NOXA [4–7]. Therefore, Mcl-1 inhibits the key process in the mitochondrial pathway of apoptosis, i.e., mitochondrial cytochrome c release. Our results clearly demonstrate that Mcl-1 downregulation induced by shRNA increased 5-FU-induced cytochrome c release in the cytosol (Fig. 3b). In addition, the downstream events of caspase 3 activation and apoptotic nuclear alteration were also inhibited by the shRNA treatment (Fig. 3c). Thus, our study further supports the idea that Mcl-1 is a pivotal anti-apoptotic molecule that prevents mitochondria-mediated apoptosis signaling.
Our study has demonstrated, for the first time, that the Mcl-1 level is enhanced in the so-called CSC population. Recently, various technologies and cell surface markers to identify gastric CSCs have been reported [12–17]. Takaishi et al. [17] identified CD44 as a CSC marker in gastric cancer cells. They demonstrated that CD44-positive gastric cancer cells had superiority over CD44-negative cells in forming either in vitro spheres or in vivo tumors, suggesting CD44-positive cells have so-called CSC-like properties. Ishimoto et al. [15] demonstrated that CD44-positive slow-cycling stem cells were located in the squamo-columnar junction (SCJ) in normal mouse stomach. They suggested that gastric tumors had derived from the CD44-positive stem cells, and, using a mouse carcinogenesis model, they found that CD44 suppression induced gastric tumor growth [14, 15]. More recently, identification of CD44-positive human gastric CSCs from patients’ tumor tissues and peripheral blood has been reported [12]. Accordingly, we used CD44 as a CSC marker, and succeeded in identifying that the CD44-positive subpopulation was relatively resistant to 5-FU treatment as compared to CD44-negative cells. Interestingly, the expression level of Mcl-1 level was dramatically higher in CD44-positive cells than in CD44-negative cells (≥5-fold) even in Mcl-1 overexpressing MKN-45 cells (Fig. 4a). Therefore, we hypothesize that Mcl-1 plays pivotal roles in chemotherapy resistance in CD44-positive CSC-like cells. Indeed, the suppression of Mcl-1 by shRNA technology successfully canceled the chemotherapy resistance in CD44-positive cells. Taken together, these results suggest that Mcl-1 plays pivotal roles in mediating apoptosis resistance in CD44-positive CSC-like cells. Recently, another stem cell marker, leucine-rich repeat containing G-protein coupling receptor 5 (Lgr5), has been reported to mark gastric gland stem cells [34]. Unfortunately, no useful antibody to identify the human Lgr5 marker is currently available. Thus, evaluation of Mcl-1 status in Lgr5-positive gastric cancer cells will be performed in a future study.
In conclusion, our study suggests that Mcl-1 plays pivotal roles in the process by which gastric cancer cells exhibit resistance to chemotherapy-mediated apoptosis. The suppression of Mcl-1 appears to be an attractive therapeutic strategy to overcome Mcl-1-mediated chemotherapy resistance. Chemotherapy-resistant CD44-positive CSC-like cells uniquely overexpress Mcl-1; therefore, it appears that the inhibition of Mcl-1 will be a promising technology to target CSC-like populations.
References
Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, et al. Cancer statistics, 2006. CA Cancer J Clin. 2006;56(2):106–30.
Middleton G, Cunningham D. Current options in the management of gastrointestinal cancer. Ann Oncol. 1995. 6(Suppl 1):17–25 (discussion 25–6, review).
Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci USA. 1993;90(8):3516–20.
Clohessy JG, Zhuang J, de Boer J, Gil-Gomez G, Brady HJ. Mcl-1 interacts with truncated Bid and inhibits its induction of cytochrome c release and its role in receptor-mediated apoptosis. J Biol Chem. 2006;281(9):5750–9.
Han J, Goldstein LA, Hou W, Rabinowich H. Functional linkage between NOXA and Bim in mitochondrial apoptotic events. J Biol Chem. 2007;282(22):16223–31.
Shimazu T, Degenhardt K, Nur E, Kamal A, Zhang J, Yoshida T, Zhang Y, et al. NBK/BIK antagonizes MCL-1 and BCL-XL and activates BAK-mediated apoptosis in response to protein synthesis inhibition. Genes Dev. 2007;21(8):929–41.
Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19(11):1294–305.
Taniai M, Grambihler A, Higuchi H, Werneburg N, Bronk SF, Farrugia DJ, et al. Mcl-1 mediates tumor necrosis factor-related apoptosis-inducing ligand resistance in human cholangiocarcinoma cells. Cancer Res. 2004;64(10):3517–24.
Maeta Y, Tsujitani S, Matsumoto S, Yamaguchi K, Tatebe S, Kondo A, et al. Expression of Mcl-1 and p53 proteins predicts the survival of patients with T3 gastric carcinoma. Gastric Cancer. 2004;7(2):78–84.
Wacheck V, Cejka D, Sieghart W, Losert D, Strommer S, Crevenna R, et al. Mcl-1 is a relevant molecular target for antisense oligonucleotide strategies in gastric cancer cells. Cancer Biol Ther. 2006;5(10):1348–54.
Siddique HR, Saleem M. Role of BMI1, a stem cell factor in cancer recurrence and chemoresistance: preclinical and clinical evidences. Stem Cells. 2012. doi:10.1002/stem.1035.
Dhingra S, Feng W, Brown RE, Zhou Z, Khoury T, Zhang R, Tan D. Clinicopathologic significance of putative stem cell markers, CD44 and nestin in gastric adenocarcinoma. Int J Clin Exp Pathol. 2011;4(8):733–41.
Chen T, Yang K, Yu J, Meng W, Yuan D, Bi F, et al. Identification and expansion of cancer stem cells in tumor tissues and peripheral blood derived from gastric adenocarcinoma patients. Cell Res. 2011;22(1):248–58.
Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(−) and thereby promotes tumor growth. Cancer Cell. 2011;19(3):387–400.
Ishimoto T, Oshima H, Oshima M, Kai K, Torii R, Masuko T, et al. CD44+ slow-cycling tumor cell expansion is triggered by cooperative actions of Wnt and prostaglandin E2 in gastric tumorigenesis. Cancer Sci. 2010;101(3):673–8.
Qiao XT, Gumucio DL. Current molecular markers for gastric progenitor cells and gastric cancer stem cells. J Gastroenterol. 2011;46(7):855–65.
Takaishi S, Okumura T, Tu S, Wang SS, Shibata W, Vigneshwaran R, et al. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells. 2009;27(5):1006–20.
Mizuguchi H, Hayakawa T. Adenovirus vectors containing chimeric type 5 and type 35 fiber proteins exhibit altered and expanded tropism and increase the size limit of foreign genes. Gene. 2002;285(1–2):69–77.
Okada N, Saito T, Masunaga Y, Tsukada Y, Nakagawa S, Mizuguchi H, et al. Efficient antigen gene transduction using Arg-Gly-Asp fiber-mutant adenovirus vectors can potentiate antitumor vaccine efficacy and maturation of murine dendritic cells. Cancer Res. 2001;61(21):7913–9.
Sumimoto H, Yamagata S, Shimizu A, Miyoshi H, Mizuguchi H, Hayakawa T, et al. Gene therapy for human small-cell lung carcinoma by inactivation of Skp-2 with virally mediated RNA interference. Gene Ther. 2005;12(1):95–100.
Mizuguchi H, Kay MA. Efficient construction of a recombinant adenovirus vector by an improved in vitro ligation method. Hum Gene Ther. 1998;9(17):2577–83.
Higuchi H, Bronk SF, Takikawa Y, Werneburg N, Takimoto R, El-Deiry W, et al. The bile acid glycochenodeoxycholate induces trail-receptor 2/DR5 expression and apoptosis. J Biol Chem. 2001;276(42):38610–8.
Leist M, Volbracht C, Fava E, Nicotera P. 1-Methyl-4-phenylpyridinium induces autocrine excitotoxicity, protease activation, and neuronal apoptosis. Mol Pharmacol. 1998;54(5):789–801.
Matsuzaki Y, Kinjo K, Muligan RC, Okano H. Unexpectedly efficient homing capacity of purified murine hematopoietic stem cells. Immunity. 2004;20:87–93.
Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med. 2001;7(9):1028–34.
Akgul C. Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell Mol Life Sci. 2009;66(8):1326–36.
Isomoto H, Kobayashi S, Werneburg NW, Bronk SF, Guicciardi ME, Frank DA, et al. Interleukin 6 upregulates myeloid cell leukemia-1 expression through a STAT3 pathway in cholangiocarcinoma cells. Hepatology. 2005;42(6):1329–38.
Aichberger KJ, Mayerhofer M, Krauth MT, Skvara H, Florian S, Sonneck K, et al. Identification of mcl-1 as a BCR/ABL-dependent target in chronic myeloid leukemia (CML): evidence for cooperative antileukemic effects of imatinib and mcl-1 antisense oligonucleotides. Blood. 2005;105(8):3303–11.
Jiang CC, Lucas K, Avery-Kiejda KA, Wade M, deBock CE, Thorne RF, et al. Up-regulation of Mcl-1 is critical for survival of human melanoma cells upon endoplasmic reticulum stress. Cancer Res. 2008;68(16):6708–17.
Likui W, Qun L, Wanqing Z, Haifeng S, Fangqiu L, Xiaojun L. Prognostic role of myeloid cell leukemia-1 protein (Mcl-1) expression in human gastric cancer. J Surg Oncol. 2009;100(5):396–400.
Yu HG, Ai YW, Yu LL, Zhou XD, Liu J, Li JH, et al. Phosphoinositide 3-kinase/Akt pathway plays an important role in chemoresistance of gastric cancer cells against etoposide and doxorubicin induced cell death. Int J Cancer. 2008;122(2):433–43.
Saito Y, Suzuki H, Tsugawa H, Nakagawa I, Matsuzaki J, Kanai Y, et al. Chromatin remodeling at Alu repeats by epigenetic treatment activates silenced microRNA-512-5p with downregulation of Mcl-1 in human gastric cancer cells. Oncogene. 2009;28(30):2738–44.
Wang HJ, Ruan HJ, He XJ, Ma YY, Jiang XT, Xia YJ, et al. MicroRNA-101 is down-regulated in gastric cancer and involved in cell migration and invasion. Eur J Cancer. 2010;46(12):2295–303.
Barker N, Huch M, Kujala P, van de Wetering M, Snippert HJ, van Es JH, et al. Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell. 2010;6(1):25–36.
Acknowledgments
We acknowledge Nippon Kayaku Co. for providing 5-FU and CDDP. We thank Ms. Naoko Kuriyama for her technical assistance. This work was supported by a Grant-in-Aid for Scientific Research from the Japanese Ministry of Education, Culture, Sports, Science and Technology, and by Scientific Funding from Taiho Pharmaceutical Co. Ltd.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Akagi, H., Higuchi, H., Sumimoto, H. et al. Suppression of myeloid cell leukemia-1 (Mcl-1) enhances chemotherapy-associated apoptosis in gastric cancer cells. Gastric Cancer 16, 100–110 (2013). https://doi.org/10.1007/s10120-012-0153-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-012-0153-6