
Abstract
Hepatitis B virus surface mutants are of enormous importance because they are capable of escaping detection by serology and can infect both vaccinated and unvaccinated populations, thus putting the whole population at risk. This study aimed to detect and characterise hepatitis B-escaped mutants among blood donors and vaccinees. One thousand serum samples were collected for this study from blood donors and vaccinees. Hepatitis B surface antigen, antibodies and core antibodies were tested using a commercial enzyme-linked immunosorbent assay (ELISA) kit. DNA detection was performed via nested polymerase chain reaction (PCR), and the S gene was sequenced and analysed using bioinformatics. Of the 1,000 samples that were screened, 5.5 % (55/1,000) were found to be HBsAg-negative and anti-HBc- and HBV DNA-positive. All 55 isolates were found to belong to genotype B. Several mutations were found across all the sequences from synonymous and non-synonymous mutations, with the most nucleotide mutations occurring at position 342, where adenine was replaced by guanine, and cytosine at position 46 was replaced by adenine in 96.4 % and 98 % of the isolates, respectively. Mutation at position 16 of the amino acid sequence was found to be common to all the Malaysian isolates, with 85.7 % of the mutations occurring outside the major hydrophilic region. This study revealed a prevalence of 5.5 % for hepatitis B-escaped mutations among blood donors and vaccinated undergraduates, with the most common mutation being found at position 16, where glutamine was substituted with lysine.
Similar content being viewed by others

Introduction
Hepatitis B virus (HBV) is the most common chronic viral infection worldwide, affecting about 2 billion people globally, with 378 million chronic carriers [1]. The HBV genome encodes four genes, called C, X, P and S. The C gene codes for a core protein known as the hepatitis B core antigen (HBcAg), while the X gene encodes the X protein, which is the protein that circulates in the blood of infected individuals when there is active viral replication [2]. HBsAg has a long open-reading frame (ORF), which has three in-frame ‘start’ codons (ATG) that split the gene into three sections: PreS1, PreS2 and S. As a result of the numerous start codons, different-sized polypeptides are produced, known as the large (PreS1 + PreS2 + S), medium (PreS2 + S) or small (S) polypeptides [3].
The hepatitis B surface antigen is a spherical particle that measures 22 mm in diameter. Its determinant is a double-loop structure, which projects from the surface of the virion and forms the key neutralising epitope [4]. This determines the surface antigen known as the ‘a’ determinant gene, and is the main target for the vaccine, including antibodies. However, mutation in the surface protein as a result of amino acid deletions or substitutions, particularly in the region of amino acids 137–147, permits hepatitis B virus replication in vaccinated subjects. This is because antibodies induced by the current vaccine may not recognise changes in the surface antigen as a result of transformation (mutation). HBV surface mutants are of enormous importance because they are capable of infecting both vaccinated and unvaccinated individuals, thus putting the whole population at risk. Clinically important hepatitis B mutants have been reported in all genotypes [5], indicating a wide spread across the various genotypes. Therefore, understanding the prevalence of such mutations would be useful for the design of a diagnostic assay and in the prevention and treatment of HBV. However, extensive molecular characterisation of occult hepatitis B strains in Southeast Asia has not been performed. This study provides new information regarding the phylogenetic analysis of vaccine-escaped hepatitis B strains obtained from blood donors and vaccinated undergraduate volunteers in Malaysia.
Patients and methods
Sample collection
A total of 1,000 serum samples were collected for this study: 500 samples from blood donors at the National Blood Centre Malaysia in Kuala Lumpur and 500 samples from volunteer undergraduate students of the Faculty of Medicine and Health Sciences, Universiti Putra Malaysia (Table 1).
Serological assay
The National Blood Centre Malaysia performed HBsAg testing using an ABBOTT PRISM instrument (Abbott Laboratories, Abbott Park, IL, USA). Each of the samples was re-tested for HBsAg in the laboratory of Medical and Molecular Virology, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia. Anti-HBs, HBsAg and anti-HBc were tested using a commercial enzyme-linked immunosorbent assay (ELISA) kit (DRG International, Inc., New York, USA), according to the manufacturer’s instructions. Twenty randomly selected samples were sent to a commercial laboratory to verify the result of our ELISA. The sensitivity for the serological assays was 10 IU/L optical density (OD ≥ 0.105), 0.5 ng/mL (OD ≥ 0.105) and 2 ng/mL (OD ≤ 0.600).. Sera with repeatedly positive results were considered positive and those with repeatedly negative results were considered negative for each of the serological markers.
HBV DNA isolation and detection from serum
HBV DNA was extracted from 200 μl of serum using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions. Briefly, 20 μl of protease was placed into a 1.5-μl tube, and 200 μl of the serum and AL buffer was added to each tube before being vortexed and incubated at 56 °C for 10 min. Then, 200 μl of ethanol was added to enhance DNA precipitation. To eliminate impurities, the trapped DNA was washed in two steps using AW1 and AW2 buffer as the first and second steps, respectively. The DNA was eluted with 50 μl of elution buffer and stored at −20 °C.
Two sets of primers were designed to amplify the S gene in a nested polymerase chain reaction (PCR). The first round targeted a 916-bp segment of the envelope gene and the second round of PCR was performed using the internal primers to amplify 656 bp of the S gene (Table 2). Samples were handled in aseptic conditions and all PCR reactions were carried out in duplicate. In the first round was performed using 20 μl of reaction mixture containing 0.5 μl DNA sample, 10 μl Maxime PCR PreMix Kit (i-pfu) (iNtRON Biotechnology Inc., Korea), 0.5 μl of each forward and reverse primer, and 8.5 μl nuclease-free water. The second round was performed in a 50-μl reaction mixture containing 1 μl DNA sample, 25 μl Maxime PCR PreMix Kit (i-pfu) (iNtRON Biotechnology Inc., Korea), 1.25 μl of each forward and reverse primer, and 21.5 μl nuclease-free water. The PCR conditions comprised an initial denaturation step of 5 min at 94 °C, 30 cycles of 94 °C for 5 min, 94 °C for 30 s, 63.8 °C for 30 s and 72 °C for 60 s, and a final extension at 72 °C for 8 min; both reactions were performed under the same conditions. The products of amplification were recovered from a 1.5 % (w/v) agarose gel using a DNA purification kit (Invitrogen Corporation, San Diego, CA, USA), according to the manufacturer’s instructions.
Sequencing and genotyping
The purified DNA of 55 sera from both blood donors and vaccinated undergraduates was re-amplified using nested PCR as described above, and the products were commercially sequenced (First BASE Laboratories, Selangor, Malaysia). All sequences were submitted to the NCBI GenBank (http://www.ncbi.nlm.nih.gov/genbank) under accession numbers KC953665–KC953699 and KF011208–KF011227 for blood donors and vaccinated undergraduates, respectively (Table 3). The sequences were assembled using BioEdit v7.2.0, after which they were manually edited to remove gaps or ambiguously aligned sites. All sequences were aligned to reference sequences from GenBank using ClustalW via Molecular Evolutionary Genetics Analysis version 5 (MEGA 5) software [6]. Phylogenetic analysis was carried out using the maximum likelihood (ML) method. Reference sequences were selected from the GenBank database. For genotyping, our sequences were aligned with S gene sequences from established HBV genotypes A–I from the NCBI GenBank. The accession numbers are as follows: genotype A (AY161138, AJ309371), B (AB0738554, AB073830), C (AF223960, X75656), D (AY161159, AB090270), E (X75657, AB074845), F (AF223963, AB036908), G (AF405706, M74499), H (AY090457, AY090460) and I (FJ023667, AF241407). Evolutionary history was inferred using the ML method, selecting topology with a superior log likelihood rate. A distinct gamma distribution was used to model evolutionary rate differences among sites (+G, parameter = 0.7185). Genotypes were determined based on the genetic distance and clustering of the DNA sequences [7].
Results
Of the 1,000 samples that were screened, 5.5 % (55/1,000) were found to be HBsAg-negative and anti-HBc-positive, all of which were found to be positive for HBV DNA by nested PCR. Additionally, 84.8 % (848/1,000) of the samples were anti-HBs-positive. The results of the nested PCR showed the presence of a 656-bp band that was specific for the hepatitis B S gene fragment, indicating a positive result (Fig 1). Of the 5.5 % (55/1,000) of the samples that were positive, 87.3 % (48/55) were also hepatitis B surface antibody-positive, suggesting immunity as a result of previous infection [8], while 12.7 % (7/55) were anti-HBs-negative. The latter group is referred to as isolated anti-core, as has been previously described [9]. However, none of these samples were hepatitis B surface antigen-positive.

Panel A shows the first-round polymerase chain reaction (PCR) gel; panel B shows the second-round PCR gel from the sample of students; panel C shows the controls and blank used in the experiment; panel D shows the second-round PCR from blood donors. A 656-bp DNA product was amplified using primers specific for the hepatitis B S gene. All lanes show a PCR product of the expected size (B, C and D), except lane A, which showed no band in the first round PCR. LD: 100-bp molecular weight marker; PC: positive control; NTC: no-template control
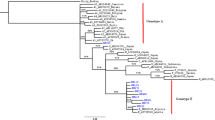
Multiple sequence alignment of Malaysia vaccine-escaped hepatitis B strains revealed a sequence homology of 99 % and a sequence identity that ranged from 83 to 100 %, and pairwise distances ranging from 0.000 to 0.177, with an overall mean distance of 0.029. All of our isolates belonged to genotype B, with some aberrant sequences showing a distant relationship (Figs. 2 and 3). The majority of the Malaysian isolates from blood donors fell into the first cluster (blue), and were similar to the Panama isolate B2 and other isolates from Japan, China and Taiwan. Two of our isolates fell within the second cluster (red), together with isolates from Canada, the Philippines and Indonesia. In the third cluster (green), isolate UPMD197 showed a distant relationship with other isolates, but was similar to isolate KC315324 from Thailand. Malaysian isolates from vaccines are phylogenetically related to other isolates from China, Taiwan and Japan, except for isolates UPMS340, UPMS139, UPMS186, UPMS343 and UPMS388, which were distantly related and belong to a different clade and sub-clades (Fig. 4). The most commonly occurring nucleotide mutations were the replacement of adenine at position 342 by guanine (A342G) and cytosine at position 46 by adenine (C46A); these were present in 96.4 % (53/55) and 98 % (54/55) of the isolates, respectively.

Molecular phylogeny of the S gene of 35 Malaysian isolates from vaccinated individuals and reference sequences from established genotypes (A to H) from the NCBI GenBank. The blue triangles indicate aberrant strains. The evolutionary relationship was inferred using the maximum likelihood (ML) method based on the Kimura model. Malaysian isolates are indicated by HBV UPM D followed by isolate number and all belong to genotype B, with the exception of the aberrant strains

Phylogeny of the S gene of 35 Malaysian isolates from blood donors and reference strains from established genotypes (A to H) from the NCBI GenBank. The blue triangles indicate an aberrant genotype. The evolutionary relationship was inferred using the ML method based on the Kimura model. Malaysian isolates are indicated by HBV UPM D followed by isolate number and all belong to genotype B, with the exception of the aberrant strains

Molecular phylogenetic relationship of Malaysian hepatitis B virus (HBV) isolates from vaccinated individuals with reference isolates from the NCBI GenBank. The evolutionary relationship was inferred using the ML method based on the Jukes–Cantor model. Malaysian isolates are separated into two distinct clusters marked in frames (red and blue)
Aligned deduced amino acid sequence showed corresponding mutations at various points of the amino acid sequences of the surface protein, including substitutions to deletions. The mutation at amino acid 16 is common to all of the Malaysian isolates, resulting in the substitution of glutamine (Q) with lysine (K) in 53 of the isolates, while the remaining two isolates contained an arginine residue or a stop codon. A total of 105 amino acid mutations were found in all 55 Malaysian isolates; 85.7 % (90/105) were outside the major hydrophilic region (MHR) and 14.3 % (15/105) were within the MHR, of which 40 % (6/150) were within the a determinant region (Table 4). The location of amino acid mutations within the MHR (100–160 aa) and the a determinant region (124–147 aa) are shown in Fig. 5.

Multiple sequence alignment of deduced amino acids of hepatitis surface antigen major hydrophilic region (MHR) of 55 Malaysian sequences compared with a reference sequence from the NCBI GenBank. The antigenic region (a determinant) is boxed (red). The MHR is from 100 to 160 aa and the a determinant from 124 to 147 aa
Discussion
In this study, HBV DNA was detected in HBsAg-negative serum using nested PCR, which is in line with previous studies that demonstrated the same result in peripheral blood mononuclear cells, serum and liver samples that were negative for HBsAg, HBV DNA levels were less than 104 copies/mL [10], which is considerably lower than those that are HBsAg-positive. Our study revealed a 5.5 % prevalence of the hepatitis B core antibody among blood donors and vaccinated undergraduate students, which is similar to that reported in Italy [11], Egypt [12], Germany, the UK [13] and the USA [14], but less than that reported in Korea [15] and Greece [16]. This study revealed a 100 % prevalence of vaccine-escaped hepatitis among hepatitis B core antibody-positive blood donors and vaccinated undergraduates. This is similar to the frequency reported in China [17]. Additionally, other studies have reported the identification of vaccine-escaped hepatitis B virus more frequently in individuals with anti-HBc-positive serology than in those with anti-HBc-negative serology, which is in accordance with our findings. On the other hand, a prevalence of 33.3 % was reported in northeast China [17], compared to 0.11 % in Taiwan [18]. Occult or vaccine-escaped hepatitis B infections have significant clinical importance, since they can become reactivated when the immune system is suppressed and can be transmitted through the transfusion of blood or blood products, organ transplant and sexual intercourse. It may also enhance the progression of liver fibrosis and, subsequently, hepatocellular carcinoma.
Our results showed that all isolates belonged to a single genotype, B, which is in agreement with previous studies that confirmed genotypes B and C as the most prevalent in Asia [19]. However, in Malaysia, genotype B was reported to be dominant over C [20], which is in contrast to the findings in asymptomatic carriers in China, where genotype C was reported as being dominant [5].
Most of the sequences were found to exhibit a close relationship with reference isolate 14 (JX869999) from Panama, which belongs to genotype B, serotype adw 2 and was isolated from plasma. Other isolates, including UPMD301 and UPMD170, were closely related to reference isolates from Canada, the Philippines and Indonesia. One of the Malaysian isolates, UPMD179, was found to be distantly related to all other isolates, but to have a close relationship with isolate M13008 (KC315324) from Thailand, which belong to genotype B, serotype adw, isolated from serum [21]. On the other hand, sequences from vaccinated Malaysian individuals were separated into two clusters, the majority of which were found to have a close evolutionary relationship with reference isolates from China, Taiwan and Japan. However, some sequences from vaccinated individuals were distantly related to other sequences belonging to different clades and sub-clades as a result of frequent mutations within the S gene.
The hepatitis B viral DNA polymerase enzyme lacks proofreading ability, making it highly susceptible to nucleotide misincorporation during reverse transcription or replication [22]. Mutations in the S gene are the most concerning from a clinical point of view, because of the difficulty in establishing a diagnosis, leading to the progression of undiagnosed cases to occult chronic infection, liver failure and, subsequently, hepatocellular carcinoma. The development of mutations in the S gene of HBV interferes with the viral antigen recognition by antibodies in serological assays, as well as with the response to recombinant hepatitis B vaccine. Several types of mutations were implicated, many of which affect amino acids within the a determinant region of the S gene; however, our results show that the majority (94.3 %) of mutations occurred outside the major hydrophilic region. As all of our sequences have mutations outside the a determinant region, it can be concluded from our findings that mutations outside the a determinant region may play a significant role in vaccine-escaped hepatitis B mutant infection as a result of the failure to detect HBsAg in the sera. Only 6/105 (5.7 %) of the mutations were located within the a determinant region: T125K, D144G, T125A, S132C and M133L. Therefore, our findings suggest that mutations within the a determinant region, as well as other regions of the S gene, may play a significant role in the diagnosis and detection of vaccine-escaped hepatitis B infection, which is in agreement with the findings from previous studies. However, we found mutations at positions 133 and 144 in isolates UPMD170 and UPMD375, respectively, which concurred with reports from China and Turkey [23] and were described as ‘immune-selected mutations’ [24]. However, the mutation at position 133 aa creates an N-linked glycosylation site, which can effectively rescue the effects of the G145R mutant virion and, to some extent, other immune-escaped mutants [25]. Premature stop codons were found in isolates UPMD47, UPMD95, UPMD348, UPMS45, UPMS139, UPMS186, UPMS388 and UPMS389, which might affect HBsAg production, as described previously [26].
The most prevalent mutation found in this Malaysian isolate was the substitution of glutamine at position 16, which is in contrast to the previous report of G145R being the most common mutation [27]. None of our isolates contained a mutation at position 145 of the amino acid sequence and, yet, HBsAg could not be detected using commercial ELISA kits. Thus, it can be speculated that mutants with defects in the S gene of the HBV genome at position 16 lead to the loss of HBsAg expression, which is the main target for serological diagnosis. This might be the result of post-transcriptional effects of mutations on HBsAg expression, as described previously [28], resulting in HBV-escaped mutant infection; however, further functional studies in cell culture are required. This suggests that mutations outside the MHR can also lead to the development of escaped mutant strains of hepatitis B and subsequent serological diagnostic failure, which concur with previous studies [29]. However, the findings from this study are region-specific.
Limitations
We were not able to obtain a known G145R escaped mutant to use as a control, and, thus, we relied on sequencing of the gene. The ability of the S region to secrete surface antigen was not determined in vitro, but carriers of this escaped mutant are being followed up.
References
Thun MJ, DeLancey JO, Center MM, Jemal A, Ward EM (2010) The global burden of cancer: priorities for prevention. Carcinogenesis 31(1):100–110
Ie SI, Thedja MD, Roni M, Muljono DH (2010) Prediction of conformational changes by single mutation in the hepatitis B virus surface antigen (HBsAg) identified in HBsAg-negative blood donors. Virol J 7(1):326
Beck J, Nassal M (2007) Hepatitis B virus replication. World J Gastroenterol 13(1):48–64
Stirk HJ, Thornton JM, Howard CR (1992) A topological model for hepatitis B surface antigen. Intervirology 33(3):148–158
Ma Q, Wang Y (2012) Comprehensive analysis of the prevalence of hepatitis B virus escape mutations in the major hydrophilic region of surface antigen. J Med Virol 84(2):198–206
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28(10):2731–2739
Norder H, Couroucé AM, Coursaget P, Echevarria JEM, Lee SD, Mushahwar IK, Robertson BH, Locarnini S, Magnius LO (2003) Genetic diversity of hepatitis B virus strains derived worldwide: genotypes, subgenotypes, and HBsAg subtypes. Intervirology 47(6):289–309
Lee H, Kiang P, Watanabe P, Halon P, Shi L, Church DR (2013) Hepatitis B virus infection and immunizations among Asian American college students: infection, exposure, and immunity rates. J Am Coll Health 61(2):67–74
Hudu SA, Malik YA, Niazlin MT, Harmal NS, Alshrari AS, Sekawi Z (2013) Isolated hepatitis B core antibody positive among vaccinated cohort in Malaysia. Ann Saudi Med 33(6):591–594
Carman WF, Van Deursen FJ, Mimms LT, Hardie D, Coppola R, Decker R, Sanders R (1997) The prevalence of surface antigen variants of hepatitis B virus in Papua New Guinea, South Africa, and Sardinia. Hepatology 26(6):1658–1666
Manzini P, Girotto M, Borsotti R, Giachino O, Guaschino R, Lanteri M, Testa D, Ghiazza P, Vacchini M, Danielle F, Pizzi A, Valpreda C, Castagno F, Curti F, Magistroni P, Abate ML, Smedile A, Rizzetto M (2007) Italian blood donors with anti-HBc and occult hepatitis B virus infection. Haematologica 92(12):1664–1670
Badrawy H, Bakry R (2013) Anti-HBc and HBV-DNA detection in blood donors negative for hepatitis B virus surface antigen. Am J Mol Biol 3:62–66
Soldan K, Davison K, Dow B (2005) Estimates of the frequency of HBV, HCV, and HIV infectious donations entering the blood supply in the United Kingdom, 1996 to 2003. Euro Surveill 10(2):17–19
Kleinman SH, Kuhns MC, Todd DS, Glynn SA, McNamara A, DiMarco A, Busch MP; Retrovirus Epidemiology Donor Study (2003) Frequency of HBV DNA detection in US blood donors testing positive for the presence of anti-HBc: implications for transfusion transmission and donor screening. Transfusion 43(6):696–704
Seo DH, Whang DH, Song EY, Kim HS, Park Q (2011) Prevalence of antibodies to hepatitis B core antigen and occult hepatitis B virus infections in Korean blood donors. Transfusion 51(8):1840–1846
Zervou EK, Dalekos GN, Boumba DS, Tsianos EV (2001) Value of anti-HBc screening of blood donors for prevention of HBV infection: results of a 3-year prospective study in Northwestern Greece. Transfusion 41(5):652–658
Fang Y, Shang QL, Liu JY, Li D, Xu WZ, Teng X, Zhao HW, Fu LJ, Zhang FM, Gu H-X (2009) Prevalence of occult hepatitis B virus infection among hepatopathy patients and healthy people in China. J Infect 58(5):383–388
Li L, Chen PJ, Chen MH, Chak KF, Lin KS, Tsai SJL (2008) A pilot study for screening blood donors in Taiwan by nucleic acid amplification technology: detecting occult hepatitis B virus infections and closing the serologic window period for hepatitis C virus. Transfusion 48(6):1198–1206
Purdy MA (2007) Hepatitis B virus S gene escape mutants. Asian J Transfus Sci 1(2):62–70
Meldal BHM, Bon AH, Prati D, Ayob Y, Allain JP (2011) Diversity of hepatitis B virus infecting Malaysian candidate blood donors is driven by viral and host factors. J Viral Hepat 18(2):91–101
Sa-Nguanmoo P, Tangkijvanich P, Tharmaphornpilas P, Rasdjarmrearnsook AO, Plianpanich S, Thawornsuk N, Theamboonlers A, Poovorawan Y (2012) Molecular analysis of hepatitis B virus associated with vaccine failure in infants and mothers: a case–control study in Thailand. J Med Virol 84(8):1177–1185
Carman WF, Zanetti AR, Karayiannis P, Waters J, Manzillo G, Tanzi E, Zuckerman AJ, Thomas HC (1990) Vaccine-induced escape mutant of hepatitis B virus. Lancet 336(8711):325–329
Sayan M, Şentürk Ö, Akhan SÇ, Hülagü S, Cekmen MB (2010) Monitoring of hepatitis B virus surface antigen escape mutations and concomitantly nucleos(t)ide analog resistance mutations in Turkish patients with chronic hepatitis B. Int J Infect Dis 14:e136–e141
Sheldon J, Soriano V (2008) Hepatitis B virus escape mutants induced by antiviral therapy. J Antimicrob Chemother 61(4):766–768
Kwei K, Tang X, Lok AS, Sureau C, Garcia T, Li J, Wands J, Tong S (2013) Impaired virion secretion by hepatitis B virus immune escape mutants and its rescue by wild-type envelope proteins or a second-site mutation. J Virol 87(4):2352–2357
Wakil SM, Kazim SN, Khan LA, Raisuddin S, Parvez MK, Guptan RC, Thakur V, Hasnain SE, Sarin SK (2002) Prevalence and profile of mutations associated with lamivudine therapy in Indian patients with chronic hepatitis B in the surface and polymerase genes of hepatitis B virus. J Med Virol 68(3):311–318
Martin CM, Welge JA, Rouster SD, Shata MT, Sherman KE, Blackard JT (2012) Mutations associated with occult hepatitis B virus infection result in decreased surface antigen expression in vitro. J Viral Hepat 19(10):716–723
Candotti D, Lin CK, Belkhiri D, Sakuldamrongpanich T, Biswas S, Lin S, Teo D, Ayob Y, Allain J-P (2012) Occult hepatitis B infection in blood donors from South East Asia: molecular characterisation and potential mechanisms of occurrence. Gut 61(12):1744–1753
Weinberger KM, Bauer T, Böhm S, Jilg W (2000) High genetic variability of the group-specific a-determinant of hepatitis B virus surface antigen (HBsAg) and the corresponding fragment of the viral polymerase in chronic virus carriers lacking detectable HBsAg in serum. J Gen Virol 81(Pt 5):1165–1174
Acknowledgements
We are grateful to the management and staff of the National Blood Centre Malaysia for making our sample collection. We are also grateful to all of the undergraduate students who volunteered to take part in this study.
Compliance with ethical standards
Ethical approval was obtained from the Medical Research Ethic Committee, Ministry of Health Malaysia registration number NMRR-12-469-11762 and Universiti Putra Malaysia, Faculty of Medicine and Health Sciences Human Ethic Committee ref.: UPM/FPSK/100-9/2-MJKEtikaPen(JMPP_Mei(12)01. Respondent and implied consent was obtained from the participants as appropriate for this study.
Conflict of interest
None to declare.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Hudu, S.A., Harmal, N.S., Saeed, M.I. et al. Naturally occurring hepatitis B virus surface antigen mutant variants in Malaysian blood donors and vaccinees. Eur J Clin Microbiol Infect Dis 34, 1349–1359 (2015). https://doi.org/10.1007/s10096-015-2358-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-015-2358-1