
Abstract
Prion diseases are a group of rare neurodegenerative disorders that develop as a result of the conformational conversion of normal prion protein (PrPC) to the disease-associated isoform (PrPSc). The mechanism that actually causes disease remains unclear. However, the mechanism underlying the conformational transformation of prion protein is partially understood—in particular, there is strong evidence that copper ions play a significant functional role in prion proteins and in their conformational conversion. Various models of the interaction of copper ions with prion proteins have been proposed for the Cu (II)-binding, cell-surface glycoprotein known as prion protein (PrP). Changes in the concentration of copper ions in the brain have been associated with prion diseases and there is strong evidence that copper plays a significant functional role in the conformational conversion of PrP. Nevertheless, because copper ions have been shown to have both a positive and negative effect on prion disease onset, the role played by Cu (II) ions in these diseases remains a topic of debate. Because of the unique properties of paramagnetic Cu (II) ions in the magnetic field, their interactions with PrP can be tracked even at single atom resolution using nuclear magnetic resonance (NMR) spectroscopy. Various NMR approaches have been utilized to study the kinetic, thermodynamic, and structural properties of Cu (II)-PrP interactions. Here, we highlight the different models of copper interactions with PrP with particular focus on studies that use NMR spectroscopy to investigate the role played by copper ions in prion diseases.
Similar content being viewed by others


Background
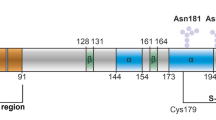
Prion diseases are a family of rare and progressive neurodegenerative disorders that develop as a result of the conformational conversion of the normal form of the transmissible prion protein (PrPC) into the disease-associated form (PrPSc) [1]. These diseases usually take many years to develop; during the incubation period, the disease advances asymptomatically in the brain until initiation of nervous system degeneration and subsequent death [2]. Human (Hu) PrPC is a 209-residue glycoprotein that is attached by a C-terminal glycosylphosphatidylinositol (GPI) to the outer leaflet of the plasma membrane of a brain cell. Prion proteins are highly conserved among mammals [3, 4], where the general structure of globular domain protein, PrPC contains three α-helices and a two-strand antiparallel β-sheets, an NH2-terminal tail consisting of an octapeptide repeat-containing unfolded domain, and GPI attached to the short COOH-terminal tail [5]. Figure 1 shows the structures of various prion proteins.

a Hydrophobic regions of PrP highlighted in yellow. b Comparison of the electrostatic surface between the wild-type PrP and variants. The human PrP protein consists of 253 amino acids. PrPC contains an octapeptide repeat-containing unfolded domain at the N-terminal tail, three α-helices (α1, α2, and α3), antiparallel β-sheets (β1 and β2), and a GPI-anchor signal at the C-terminal tail
Repeated published reports noted that copper may play a significant role in the conversion of PrPC to PrPSc [6,7,8,9] (Fig. 2). Moreover, several reports have shown that cellular prion protein (PrP) may play a crucial role in the redox control of the neuronal environment and in the regulation of copper metabolism in a manner that contributes to disease pathology [7, 10,11,12]. The concentration of copper in humans varies in different organs. A high copper concentration is found in the liver, brain, kidney, and heart [13]. In these organs, copper is essential for the function of several enzymes, including cytochrome C oxidase, catalase, dopamine hydroxylase, uricase, tryptophan dioxygenase, lecithinse, and other monoamine and diamine oxidases as well as superoxide dismutase (SOD) [14,15,16,17,18]. These enzymes are important in oxidation-reduction reactions, transport of oxygen and electrons, and protection of the cell from oxygen radicals [19, 20]. Changes in copper ion concentrations in the brain are associated with several neurological diseases including prion diseases [21,22,23,24]. Gasperini et al. showed that PrPC and copper jointly inhibit N-methyl-d-aspartate receptors (NMDAR) and prevent cell death, thus suggesting a positive role for copper in disease treatment [12]. They also showed that PrPC and copper cooperatively protect neurons from insults and exert neuroprotective effects [12].

Number of published papers using PubMed search engine. The PubMed database was searched for prion diseases (Red) and prion disease (Blue) keywords over the last 25 years. Last updated on September 25, 2018
Advances in medical research and technology, such as nuclear magnetic resonance (NMR) spectroscopy and imaging, have contributed enormously to the detection and management of prion diseases [25,26,27,28,29,30,31,32,33,34,35,36] as was successfully demonstrated in the detection and description of Creutzfeldt-Jakob disease (CJD) [30]. In particular, NMR spectroscopy provided the first three-dimensional (3D) structure of the folded mouse prion protein domain PrP [12, 37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146]. Recently, structures of other PrP-associated diseases were resolved using NMR spectroscopy [147]. In addition, NMR enabled the investigation of the dynamic equilibria between monomeric and oligomeric misfolded states of mammalian PrP [148].
Prion disease
Prion diseases, such as human prion diseases, are a group of progressive neurodegenerative disorders caused by conformational conversion of the α-helix-rich isoform of the prion protein (PrPC), which is the normal form, into the β-sheet rich isoform (PrPSc), which is the disease-associated form [1, 149, 150]. Abnormal folding of the protein (PrPSc) leads to brain damage and causes high fatality rates in both humans and animals [151,152,153,154,155,156,157,158,159,160,161,162,163,164,165,166]. However, the pathogenic mechanism that triggers this abnormal folding leading to prion diseases remains unknown. Prion diseases may take many years to develop with long incubation periods [2, 149]; during this time, the disease grows asymptomatically in the brain until the initiation of nervous-system degeneration and resulting death [2]. The infection causes brain atrophy, spongiform encephalopathy, and cerebellar degeneration. Although prion diseases are rare, they remain an important public health issue requiring attention to their management [167].
Prion diseases can be contracted through sporadic, genetic, and infectious routes [168,169,170,171]. An individual who contracts a prion disease sporadically is exposed to unknown risk factors that vary from one region to another [169]. Some people and animals can inherit prion diseases from their parents, whereas others acquire it from contaminated animal products and feed. The most common types of animal prion diseases are scrapie, bovine spongiform encephalopathy (mad cow disease), and transmissible mink encephalopathy [172, 173]. Examples of human prion diseases are Creutzfeldt-Jakob disease (CJD), Kuru, fatal familial insomnia (FFI), and Gerstmann-Sträussler-Scheinker syndrome (GSS) [147, 174, 175]. Neurological clinical presentations and diagnosis vary among the different human prion diseases. Research has shown that Kuru disease has been eradicated, where it acquired through consumption of the brains of infected humans killed by the disease during the practice of funerary cannibalism [176]. FFI is an autosomal illness characterized by lesions in the thalamus of the brain. GSS is associated with the pathological Q212P mutation, and, like CJD, results in progressive dementia [147, 177]. CJD is associated with mutation in the gene encoding the prion protein [178] and the most common and fatal prion disease (Fig. 3), with about 90% of affected individuals dying within a year of diagnosis. Early symptoms include poor coordination, visual disturbance, and memory problems; later symptoms include blindness, weakness, involuntary movement, and finally coma. Additional file 1: Table S1 summarizes the similarities and differences among the various human prion diseases.

The occurrence ratio of the most common prion diseases
Copper ions in nervous system development and neurodegenerative disorders
Copper ions are found in all living organisms. It is an essential nutrient in humans, animals, and plants [168,169,170,171], where it plays crucial roles in redox chemistry and the actions of enzymes and proteins, especially those related to energy metabolism [169, 179]. Moreover, it is fundamental for normal brain and nervous system development, as it is involved in the synthesis of neurotransmitters and in the production and maintenance of myelin [147, 174, 175].
Copper-containing compounds have also been used in medicine for centuries. Several studies proposed that copper chelators may play a potential therapeutic role in certain inherited diseases of copper homeostasis as well as in neurodegenerative diseases, such as Parkinson’s, Wilson’s, Menkes, Alzheimer’s [147, 177], and prion diseases [172, 173, 180, 181]. Various treatments for prion disease have been trialed. One was based on the use of the copper chelator D-penicillamine (D-PEN), which demonstrated a delay in the onset of prion disease in mice [182, 183]. In another trial, a significant delay in the onset of prion disease was observed when scrapie-infected hamsters were treated with copper. Copper ions inhibit in vitro conversion of prion protein into amyloid fibrils [20]. In cross-correlation analyses, it showed an antiaggregatory effect [184,185,186]. Altered levels of copper and manganese ions have been observed in prion-infected brain tissues [182].
Copper can have one of several oxidation states, including Cu (IV), Cu (III), Cu (II), and Cu(I); the most common states are Cu(I) and Cu (II) [187,188,189]. The coordination chemistry of Cu(I) is different from that of Cu (II), because Cu(I) complexes usually have a lower coordination number (CN) than C (II) complexes have. For example, Cu(I) complexes are usually tetrahedral or square planar with CN = 4, trigonal with CN = 5, or linear with CN = 2, whereas Cu (II) complexes usually have higher coordination numbers (primarily octahedral, with CN = 6). The large number of possible combinations of copper ions allows for a wide range of copper coordination complexes, ranging from monodentate to hexadentate [190,191,192]. Importantly, Cu(I) has d10 configurations and forms colorless and diamagnetic compounds, whereas Cu (II) has d9 configurations and forms colored and paramagnetic compounds [10, 11, 193,194,195,196,197,198,199,200,201,202,203]. There is an important difference in stability of Cu(I) and Cu (II) ions that strictly defines their biological role in the living organisms. The low stability of Cu(I) has led to the relative scarcity of studies on the biological roles of Cu(I) [39], whereas the higher stability of Cu (II) has led to extensive examination of its biological role in neurodegenerative disorders [204].
Studies have highlighted the role of Cu (II) ions in synaptic transmission, axonal targeting, neurite outgrowth as well as in the modulation of signaling cascades induced by neurotrophic factors. Copper not only modulates neurotransmitter receptors at synapses but it can also affect the trafficking of synaptic vesicles and modulate the interaction between proteins involved in secretory vesicle pathways [205]. Copper is clearly important in the normal development of the brain and nervous system [206,207,208,209,210,211]. It follows that copper deficiency can lead to nervous system degeneration. A decrease in copper from its normal levels can lead to several neurodegenerative and other diseases where aggregation of proteins plays a crucial role [187, 212]. On the other hand, an excessive amount of copper (applicable especially to Cu (II) ions) can lead to cytotoxicity, owing to the ability of Cu (II) to initiate redox cycling and to produce reactive oxygen species (ROS) [213, 214].
The biological function of copper ions in many copper-binding proteins and enzymes involves changing copper’s oxidation state through various redox reactions (e.g., Fenton and Haber Weiss reactions).
The enzyme superoxide dismutase 1 (SOD1) is present in almost all eukaryotic cells and a few prokaryotic cells that contain both copper and zinc [40,41,42, 215]. SOD1 catalyzes the dismutation of the superoxide radical to hydrogen peroxide and oxygen [43,44,45,46,47,48,49]. This catalytic cycle is, however, beyond the scope of this review.
Interestingly, amyloid precursor protein (APP), which is found in Alzheimer’s disease patients, can reduce Cu (II) to Cu(I) in a cell-free system [11]; moreover, Cu (II) can be reduced to Cu(I) and remains bound to APP [10, 22, 41, 42, 50, 51]. This suggests that PrP is a target of copper-catalyzed oxidation and that this reaction leads to profound structural changes in the protein. Oxidation therefore must be taken into account as a potential side reaction when considering the role of copper in prion disease [52, 53, 202, 203].
In summary, copper is known to play an important role in neurological development. It can lead to neurodegenerative disorders when present in excess or deficient levels. This suggests that prion diseases may also be affected by the availability or lack of availability of copper in the brain.
The biological roles of copper ions in neurodegenerative disorders
Mis-folded protein aggregates have been associated in several neurodegenerative disorders including Parkinson’s, Alzheimer’s, and prion disease [54]. Proteins’ aggregation rate depends on protein concentration and the ratio of the presence of metal ions like Cu2+, Zn2+, Co2+, Cr3+, and Ni2+ [55,56,57]. Tau protein (TP) and α-Synuclein are examples of biomolecules whose aggregation rates are dependent on their concentration and the metal ion coordination properties [58,59,60]. TP aggregation found in the neuronal cells of Alzheimer’s disease patients [59, 60] while α-Synuclein aggregation is associated with Parkinson’s disease progression [58].
TP found in the neuronal cells of the central nervous system and its aggregation is associated with Alzheimer’s disease [61]. TP is a neuronal microtubule-associated protein and plays a key role in microtubule stabilization in neuronal cells [62]. In general, TP aggregation initiated when the protein gets into the hyperphosphorylated form, which could result in microtubule (MT) assembly decomposition [54, 63]. TP aggregation is promoted in the presence of high Cu2+ concentration in the brain [61, 64]. A number of binding sites of Cu2+ with TP have been reported [65,66,67]. This binding leads to activation of GSK3β kinase [68] or activation of CDK5 [64] supporting the progress of Alzheimer’s disease [69].
α-Synuclein protein abundant in the brain, mainly in the presynaptic terminals and is involved the release of neurotransmitters, regulates glucose and dopamine level [70, 71]. Misfolded α-synuclein aggregation is the major component of Lewy neurites (LNs) and Lewy bodies (LBs), which are pathological hallmarks of Parkinson’s disease and other neurodegenerative synucleinopathies [72,73,74]. The aggregation rates of α-synuclein affected by many factors for instance α-synuclein concentration, pH, post-translational modifications (PTM), and metal ions as Cu2+, Zn2+, Al3+, Fe3+, Ca2+, and Mg2+ [75, 76]. High Cu2+ concentrations have been reported in the cerebrospinal fluid of Parkinson’s disease patients [77], which accelerates the aggregation rate by promoting the nucleation [69, 78]. α-Synuclein–Cu (II) complexes formed through a high-affinity copper-binding site or low-affinity copper-binding sites. The high-affinity of copper-binding sites is located at the N-terminus with residues Met1, Asp2, and Met5. The low-affinity copper-binding sites are located at the N-terminus residue His50 or at the C-terminal part with residues Asp119, Asp121, Asn122, and Glu123 [78,79,80].
In summary, association between Cu2+ and its effect on protein aggregation had been repeatedly reported [64, 67, 78]. Misfolding protein aggregations are common in many neurodegenerative diseases. This suggests that Parkinson and Alzheimer’s diseases may also be affected by the availability or lack of availability of copper in the brain.
Roles of copper ions in prion diseases
PrP is known to bind copper ions, and this binding interaction may affect PrPC function and its conformational transformation to the PrPSc form. However, there is contradictory evidence concerning whether copper ions are beneficial or deleterious to the development of prion diseases [81,82,83,84,85].
Both in vitro and in vivo evidence has been reported for PrP binding to copper ions. Hornshaw et al. showed the first link between copper and prion proteins in 1995 [86] in an investigation of the binding between different transition metals and synthetic peptides. They hypothesized that copper ions bind to the N-terminal octapeptide tandem repeat sequence that corresponds to three or four copies of human PrP (PHGGGWGQ) [87,88,89]. Although Hornshaw et al. conducted in vitro experiments, their results suggested that PrP might be a copper-binding protein in vivo and that PrP preferentially binds copper over other metals [90, 91, 180]. Another study showed that copper ions bind to His96 and His111 of wild-type PrP at pH 5.5, whereas it interacts with His111 at pH 7.5 [202]. Pathological point mutations alter copper coordination under acidic conditions and metal is then anchored to His111 [202]. Additional studies have since confirmed that PrP specifically and preferentially binds copper compared with other transition metals [92, 93]. Some reports have claimed that interaction with Mn causes conversion of PrPc to PrPres, as detected by in vitro studies. Near-infrared spectroscopy coupled with multivariate analysis suggested that (i) PrP binds both Mn and Cu differently, (ii) PrP-Cu, and not PrP-Mn, protects the metal from the water, increasing protein stability, and (iii) PrP-Cu remains stable in solution, whereas PrP-Mn undergoes changes leading to fibril formation [94].
Later studies have shown that the binding of copper to PrP can affect its conformational transition to the infectious form. Takeuchi et al. (1996) showed that PrP requires copper to remain “normal” and non-infective. They suggested that a lack of copper might contribute to prion diseases [95, 96]. A similar study showed that the interaction of Cu (II) ions with PrP promotes a shift from a predominant α-helical structure of PrPC to the β-sheet structure of the infectious isoform, PrPSc, thus suggesting a negative role for copper ions in disease onset [97]. The results do not support Takeuchi’s proposal that the interaction of copper with prion proteins may lead to conformational changes (formation of an α-helical structure on the C-terminal side) that prevent aggregation. Zheng et al. studied the impact of the G127V mutation on the structural and dynamical properties of PrP using NMR and molecular dynamic methods [189]. They concluded that replacement of G127 by V destabilizes the β-sheet and affects the geometric stacking of the α-helices inside the prion molecule.
Studies performed in cell culture models and animal models have provided evidence both for and against the role of copper in promoting the development of prion diseases. For example, several studies have shown that copper functions as an antioxidant agent in copper-containing PrP, which enhances neuronal survival [98]. In contrast, Hijazi et al. found that copper plays a protective role in prion diseases, as they observed a significant delay in prion disease onset in scrapie-infected hamsters treated with copper ions, whereas administration of copper ions to normal hamsters promoted cerebellar PrPC accumulation [12, 39, 99,100,101,102,103,104,105,106,107,108,109,110,111,112, 202]. Moreover, the accumulation of the disease-related conformation (PrPSc) is significantly decreased in scrapie-affected neuroblastoma cells cultured in the presence of copper. On the other hand, normal neuroblastoma cells cultured in the presence of copper exhibited inhibition of the internalization of PrPSc [113]. In agreement with this result, Toni et al. reported that copper modifies PrPC expression and pathways in cultured neurons and that PrP mRNA expression in GN11 neurons is significantly decreased by the addition of copper ions at physiological concentrations [114]. These results suggest that extracellular copper can be used to control the amount of cellular PrP and may be an effective strategy to decrease the expression of PrPC, consequently decreasing the possibility of its conversion to the pathological isoform PrPSc [115].
The contradictory results from the studies described above indicate that the role played by copper in the development of prion diseases is unclear. Further research is needed to resolve these contradictions. Structural biology approaches, in general, and NMR spectroscopy, in particular, have the potential to be very useful in the study of copper ion coordination with PrP to help elucidate the role played by copper ions in prion diseases [116].
NMR spectroscopy
NMR spectroscopy is a powerful analytical tool. It is able to differentiate the unique magnetic environment of a nucleus in a single molecule’s various positions at the atomic level [117, 118]. Moreover, NMR can be used in structural elucidation as well as for kinetics and thermodynamics studies [99, 119, 120]. Most importantly, NMR provides information on the environment of specific atom sites and their neighboring attached atoms using in two dimensions [108, 121]. Thus, NMR spectroscopy is extensively used in a wide range of applications, including organic chemistry [108], biochemistry, polymer chemistry [122], inorganic chemistry [122], structural biology [52], physics [61, 123,124,125,126,127], biology, and drug discovery [52, 128, 129]. Through NMR experiments, researchers can study samples in the solid state [130,131,132], gel phase [133,134,135,136], tissue state [137,138,139], gas phase, and solution state [140,141,142,143]; these approaches have been used to investigate molecular structures, concentration levels, and molecular dynamics [144,145,146]. Moreover, the continuous development of NMR experimental methods and NMR machinery, such as dynamic nuclear polarization (DNP) and high-field NMR spectrometers, has continuously enhanced research on the physical and chemical properties of samples [216,217,218].
The main disadvantage of NMR spectroscopy is its low sensitivity, making milligrams of a sample necessary for useful NMR measurements. The low natural abundance of both 15N and 13C also has to be overcome in the application of NMR spectroscopy to biological samples, such as in the study of proteins. Proteins isotopically labeled with 13C and/or 15N are therefore often used in protein NMR experiments. NMR spectroscopy uses many multidimensional approaches to resolve protein structures, their dynamics and to enhance the resolution of complicated NMR spectrum [219,220,221,222,223].
There are also several limitations to the use of NMR spectroscopy as an analytical tool to study the interaction between copper ions and prion proteins. Generally, paramagnetic ions such as Cu (II) cause a significant broadening in the NMR resonance even at a very low concentration, and this broadening hinders NMR studies at a stoichiometric ratio. Consequently, NMR studies of PrP are typically performed at low copper-ion-to-PrP ratios. Diamagnetic Cu(I) ions that facilitate the use of NMR studies at higher copper-to-PrP ratios are unstable compared with Cu (II) ions and can be easily oxidized to Cu (II) under physiological conditions. However, this problem can be overcome by adding reducing reagents to the NMR tube under inert conditions and then sealing the NMR tube to prevent oxidation.
Two-dimensional NMR spectroscopy
NMR experiments are not only limited to the one-dimensional (1D) space. They can be extended to different types of multidimensional approaches. Two-dimensional (2D) NMR spectroscopy can be used for many applications including molecule identification and structural elucidation, as has been done for PrP and their biologically important complexes with transition metals and other proteins [224]. In general, 2D NMR can be used to overcome the problem of overlapping resonances by dispersing the overlapping chemical shift in a second dimension. The additional resolution offers a practical solution to detecting and identifying specific sites within macromolecule, as in the case of Cu (II) ions [223]. Such identification is not possible with the 1D approach. For example, various homo-nuclear 2D 1H-1H-NMR experiments, including total correlation spectroscopy (TOCSY) [225,226,227,228,229,230,231,232,233,234], correlation spectroscopy (COSY) [219, 234,235,236,237,238,239,240,241], and heteronuclear experiments such as 1H,13C-single quantum coherence (1H-13C-HSQC) and heteronuclear multiple bond correlation (HMBC) have been routinely used in to assign protein signals and to study protein interactions with ligands in drugs and small molecules [242]. Here, we present heteronuclear single-quantum coherence spectroscopy (HSQC) as an example of the most powerful approaches used to assign signals and to probe ligand protein interactions [243]. HSQC is a type of through-bond correlation spectroscopy that utilizes heteronuclear correlations and enhancement of the signal coming from the nucleus of lower sensitivity, such as 13C or 15N by transferring the nuclear spin polarization from the more sensitive nucleus (usually 1H) via J-coupling. The general output of HSQC is 2D spectra of the chemical shift of one nucleus, such as 1H, which is usually detected in the directly measured dimension, and the chemical shift of the other nucleus, such as 13C, which is recorded in the indirect dimension. The 1H,13C-HSQC spectrum coordinates the chemical shift of protons and nitrogen or carbon atoms that are directly covalently bonded, providing only one cross peak for each H-N or H-C coupled pair. Thus, HSQC is useful for the assignment of the protein backbone and side-chain NH signals are assigned by 1H,15N-HSQC. Moreover, utilizing the sensitivity of the 1H atom is an effective approach to reducing the experimental time for nuclei with low natural abundances and/or sensitivities, such as 15N and 13C. The experimental time necessary for HSQC experiments is usually shorter than for 1D, 13C, and 15N NMR experiments. Indeed, HSQC was used to study the interaction of copper with PrP [52, 123, 219, 244,245,246].
NMR studies of Cu(I) and Cu (II) ions-prion interactions
NMR is the method of choice for studying protein structures and dynamics and for investigating protein-metal ion interactions [247]. The protein binding sites for paramagnetic species such as Cu (II) ions can be examined by monitoring the line broadening of NMR resonance signals; the signals of the protein binding sites are more affected than are other signals. Indeed, NMR spectroscopy was used frequently to study the interaction of copper with PrP [248]. For example, Wells et al. used NMR to investigate how Cu (II) ions interact with the full length of PrP under acidic conditions at pH 5.5. The results showed that the protein binds with two copper ions while all six histidine residues in the unfolded N-terminal act as ligands (Fig. 4) [41, 246, 247, 250,251,252]. The interaction between a diamagnetic ion such as Cu(I) and other molecules such as proteins can be observed by monitoring the ordinary chemical shift change (change of the location of the cross-peak on the spectrum) and the interaction causing a change in the chemical shift value of nuclei within residues of the binding site. Indeed, detecting the interaction between Cu(I) and proteins has become a common approach [41, 247, 252], and the interaction between Cu(I) ions and PrP has been successfully studied using NMR spectroscopy techniques [253,254,255]. Taking into account the fact that Cu(I) is diamagnetic, NMR studies of its complexes with prions could be easier and more accurate because Cu(I) ions do not cause signal broadening [116].

Various binding models of Cu (II) ion coordination with the full-length prion protein at a pH 5.5 and b pH 7.4, demonstrating the coordination by an exchange of histidine imidazoles. From [249] with permission from Portland Press Ltd
Various NMR spectroscopy approaches have been utilized to study the interaction of copper with PrP. Recently, 1H-15N HSQC NMR spectroscopy was employed to study the interaction between the PrP’s copper-bound octarepeat domain [249, 256,257,258,259]. The results suggest a molecular foundation for the role of copper in mediating the cis interaction in prion proteins and suggest that the global domain can regulate the N-terminus, whereas the disruption of the cis-interaction occurs by mutation or by direct competition with globular domain ligands, contributing to protein dysregulation and prion disease [52, 260,261,262,263,264,265,266,267]. 1H NMR has been used to study the interactions of copper with different peptides corresponding to PrP, including 2-, 3-, and 4-octarepeat sequences [265]. The resulting NMR spectra show a clear broadening of the histidine 1H residues in each octarepeat coordinated with the Cu (II) ion, with the four octarepeat peptides cooperatively binding to four Cu (II) ions. Two-dimensional 1H-1H TOCSY NMR spectroscopy has been used to study the interaction between copper and the residue 91–127 fragment of the human prion protein (hPrP) [268,269,270,271,272,273]. In agreement with previous results, NMR spectra from that study show that copper ions selectively bind His-96 and His-111 (Fig. 5) [274, 275]. Interestingly, the results confirm that the protein undergoes a conformational change after binding Cu (II) ions in the presence of sodium dodecyl sulfate (SDS) micelles; the binding strongly stabilizes the α-helical conformation of the peptide backbone [202]. Some researchers hypothesize that copper binding to the prion protein can be protective against the conversion of the protein to its infectious form [260].

a Aromatic region of the 1H 1D NMR spectra of hPrP91–127 at pH 7.2 in the presence of 40 mM sodium dodecyl sulfate (SDS) in the absence (black) and presence (green) of Cu2+. b 2D TOCSY spectra of the aliphatic region with a copper-to-ligand ratio of 1:10. c The aliphatic region of the 1H–1H 2D TOCSY spectra with a metal-to-ligand ratio of 1:1.1 [231, 260,261,262,263,264,265]
NMR spectroscopy has also been used to analyze the interactions between copper and PrP at different pH values. The interaction of Cu (II) ions with full-length PrP has been investigated under mildly acidic conditions. The results show that two Cu (II) ions bind all six histidine residues of the unfolded N-terminal domain and the N-terminal amine coordinate as ligands [260]. Different copper-protein coordination models have been reported under different pH conditions [202, 276,277,278,279]. For instance, at pH 7.4, PrP may interact with a fifth or even sixth coordination site in the flexible region between the octarepeats and the PrP globular C-terminal domain involving His96 and His111 [280]. Similar studies have shown that PrP binds between five and six equivalent units of Cu (II) at pH 7.4, indicating that the interaction of copper with PrP is highly dependent on pH [280]. These reports have proposed that PrP functions may be associated with its ability to bind copper in a pH-dependent fashion [280].
Although the coordination geometry of the different copper-PrP interaction models [281] has been the focus of many studies, surprisingly few investigations have focused on the structural changes induced by the binding of Cu(I) to PrP [265]. Badrick et al. investigated the interaction between Cu(I) ions and the hPrP by using both 1D and 2D 1H NMR experiments. The results show that Cu(I) interacts with PrP in a manner different from that of Cu (II), with the Cu(I) interaction representing a tetrahedral model in which copper coordinates with two imidazoles attached to His96 and His111 and two sulfurs (Met109 and Met112) [282, 283]. The interaction between the copper ions and Met residues has sparked a strong debate in the literature concerning possible direct binding with sulfur atoms [284]. Several reports have ruled out the possibility of copper interacting with Met109 or Met112 [285,286,287]. However, Shearer et al. demonstrated that copper interacts with both Met residues in PrP under mildly basic conditions [265, 284]. These contradictory results may be explained by considering the factors that might lead to different copper-PrP coordination models. Different factors should be considered in explaining that different copper-prion interactions, such as the pH, copper oxidation state, and copper/protein ratio, may lead to different copper-PrP coordination models. For example, the copper oxidation state is a very important factor that determines copper complexation because Cu(I) normally adopts a tetrahedral coordination geometry, whereas Cu (II) prefers an octahedral or square planar coordination geometry [288, 289]. Cu(I) ions can be oxidized simply to Cu (II), and Cu (II) can be reduced to Cu(I), thus enabling copper to be involved in electron transfer reactions and copper-protein interactions and potentially leading to conformational changes associated with changes in the oxidation state [290]. To elucidate the role of copper in prion diseases, further investigations should be conducted to study the relationship between electron transfer reactions and the conformational transformation associated with copper-protein interactions.
Conclusion
Prion diseases are a group of fatal neurodegenerative disorders that occur when prion proteins change their conformation from the normal PrPC form to the disease-specific PrPSc structure. These diseases affect both humans and animals. Animals acquire prion diseases from contaminated feed or other animals, whereas humans can contract prion diseases genetically, sporadically, or via acquisition from infected animals and humans. Although the disease pathology is not completely understood, there is general agreement that the abnormal disease-associated protein conformation (PrPSc) causes prion diseases through the degeneration of the nervous system and leads to death at an advanced stage. It has been repeatedly reported that copper ion may play a major role in structural conversion from a healthy (native) α-helix rich PrP isoform to the predominantly β-sheet conformation (PrPSc). The conversion could be developed by the exposure of the protein to high concentrations of Cu (II) ions.
It is well established that an excessive amount of copper (especially Cu (II) ions) can lead to cytotoxicity, owing to the ability of Cu (II) to initiate redox cycling and produce reactive oxygen species (ROS). However, despite the wide range of studies on copper interaction with prion proteins, the mechanisms by which Cu (II) ions induced protein misfolding and aggregation remains unknown.
The proper application of the NMR spectroscopy techniques could lead to better insight if the studies include both protein function and structure. A gradual titration of prion proteins with different concentration levels of Cu (II) ions could lead to the most optimal concentration as we believe like other bioactive molecules with low or high concentrations could lead to abnormal conditions. To evaluate the copper redox effects, it is also important to study the interaction of PrP with different copper oxidation states, particularly ion (I\II) interactions. The NMR spectroscopy offers atomic-level insights into the interactions of copper ions (I\II) with PrP under physiological conditions (like pH ~ 7.4), enabling researchers to study the role played by copper and other ions in the progress of the prion disease.
Abbreviations
- 1D:
-
One-dimensional
- 1H-13C-HSQC:
-
1H,13C-single quantum coherence
- 2D:
-
Two-dimensional
- APP:
-
Amyloid precursor protein
- CJD:
-
Creutzfeldt-Jakob disease
- COSY:
-
Correlation spectroscopy
- Cu(2+):
-
Copper (II) ions
- DNP:
-
Dynamic nuclear polarization
- D-PEN:
-
D-penicillamine
- FFI:
-
Fatal familial insomnia
- GPI:
-
Glycosylphosphatidylinositol
- GSS:
-
Gerstmann-Sträussler-Scheinker syndrome
- HMBC:
-
Heteronuclear multiple bond correlation
- HSQC:
-
Heteronuclear single-quantum coherence spectroscopy
- NMDAR:
-
N-methyl-d-aspartate receptors
- NMR:
-
Nuclear magnetic resonance
- PrP:
-
Prion protein
- PrPC :
-
Normal prion protein
- PrPSc :
-
Disease-associated isoform pf prion protein
- ROS:
-
Reactive oxygen species
- SDS:
-
Sodium dodecyl sulfate
- SOD:
-
Superoxide dismutase
- SOD1:
-
Enzyme superoxide dismutase 1
- TOSCY:
-
Total correlation spectroscopy
References
Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D et al (1993) Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A 90(23):10962–10966
Shinkai-Ouchi F, Yamakawa Y, Hara H, Tobiume M, Nishijima M, Hanada K et al (2010) Identification and structural analysis of C-terminally truncated collapsin response mediator protein-2 in a murine model of prion diseases. Proteome Sci 8
Kurt TD, Jiang L, Fernández-Borges N, Bett C, Liu J, Yang T et al (2015) Human prion protein sequence elements impede cross-species chronic wasting disease transmission. J Clin Invest 125(4):1485–1496
Oliveira-Martins JB, Yusa S-i, Calella AM, Bridel C, Baumann F, Dametto P et al (2010) Unexpected tolerance of α-cleavage of the prion protein to sequence variations. PLoS One 5(2):e9107
Schätzl HM, Da Costa M, Taylor L, Cohen FE, Prusiner SB (1995) Prion protein gene variation among primates. J Mol Biol 245(4):362–374
Collinge J (2001) Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci 24:519–550
Morot-Gaudry-Talarmain Y, Rezaei H, Guermonprez L, Treguer E, Grosclaude J (2003) Selective prion protein binding to synaptic components is modulated by oxidative and nitrosative changes induced by copper (II) and peroxynitrite in cholinergic synaptosomes, unveiling a role for calcineurin B and thioredoxin. J Neurochem 87(6):1456–1470
Suhre MH, Hess S, Golser AV, Scheibel T (2009) Influence of divalent copper, manganese and zinc ions on fibril nucleation and elongation of the amyloid-like yeast prion determinant Sup35p-NM. J Inorg Biochem 103(12):1711–1720
Huang S, Chen L, Bladen C, Stys PK, Zamponi GW (2018) Differential modulation of NMDA and AMPA receptors by cellular prion protein and copper ions. Mol Brain 11(1):62
Bellingham SA, Guo B, Hill AF (2015) The secret life of extracellular vesicles in metal homeostasis and neurodegeneration. Biol Cell 107(11):389–418
D'Ambrosi N, Rossi L (2015) Copper at synapse: release, binding and modulation of neurotransmission. Neurochem Int 90:36–45
Gasperini L, Meneghetti E, Pastore B, Benetti F, Legname G (2015) Prion protein and copper cooperatively protect neurons by modulating NMDA receptor through S-nitrosylation. Antioxid Redox Signal 22(9):772–784
Manto M (2014) Abnormal copper homeostasis: mechanisms and roles in neurodegeneration. Toxics. 2(2):327–345
Hodgson EK, Fridovich I (1975) Interaction of bovine erythrocyte superoxide dismutase with hydrogen peroxide. Inactivation of the enzyme. Biochemistry 14(24):5294–5299
Sinet P-M, Garber P (1981) Inactivation of the human CuZn superoxide dismutase during exposure to O2− and H2O2. Arch Biochem Biophys 212(2):411–416
Blech DM, Borders CL (1983) Hydroperoxide anion, HO2−, is an affinity reagent for the inactivation of yeast Cu,Zn superoxide dismutase: modification of one histidine per subunit. Arch Biochem Biophys 224(2):579–586
Borders CL, Fridovich I (1985) A comparison of the effects of cyanide, hydrogen peroxide, and phenylglyoxal on eucaryotic and procaryotic Cu,Zn superoxide dismutases. Arch Biochem Biophys 241(2):472–476
Yim MB, Chock PB, Stadtman ER (1993) Enzyme function of copper, zinc superoxide dismutase as a free radical generator. J Biol Chem 268(6):4099–4105
Harris ED (1983) Chapter 3 - Copper in human and animal health**. In: Rose J (ed) Acknowledgement: Funding for this review was provided by USPHS Grant AM-26604 from the National Institutes of Health Bethesda, Maryland. Trace Elements in Health, Butterworth-Heinemann, pp 44–73
Tisato F, Marzano C, Porchia M, Pellei M, Santini C (2010) Copper in diseases and treatments, and copper-based anticancer strategies. Med Res Rev 30(4):708–749
Basun H, Forssell LG, Wetterberg L, Winblad B (1991) Metals and trace elements in plasma and cerebrospinal fluid in normal aging and Alzheimer's disease. J Neural Transm Park Dis Dement Sect 3(4):231–258
Lutsenko S, Bhattacharjee A, Hubbard AL (2010) Copper handling machinery of the brain. Metallomics. 2(9):596–608
Hershey CO, Hershey LA, Varnes A, Vibhakar SD, Lavin P, Strain WH (1983) Cerebrospinal fluid trace element content in dementia: clinical, radiologic, and pathologic correlations. Neurology. 33(10):1350–1353
Nischwitz V, Berthele A, Michalke B (2008) Speciation analysis of selected metals and determination of their total contents in paired serum and cerebrospinal fluid samples: an approach to investigate the permeability of the human blood-cerebrospinal fluid-barrier. Anal Chim Acta 627(2):258–269
Aguzzi A, Haass C (2003) Games played by rogue proteins in prion disorders and Alzheimer's disease. Science. 302(5646):814–818
Kneipp J, Miller LM, Joncic M, Kittel M, Lasch P, Beekes M et al (2003) In situ identification of protein structural changes in prion-infected tissue. Biochim Biophys Acta 1639(3):152–158
Oppenheim C, Zuber M, Galanaud D, Detilleux M, Bolgert F, Mas JL et al (2004) Spectroscopy and serial diffusion MR findings in hGH-Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry 75(7):1066–1069
Cordery RJ, MacManus D, Godbolt A, Rossor MN, Waldman AD (2006) Short TE quantitative proton magnetic resonance spectroscopy in variant Creutzfeldt-Jakob disease. Eur Radiol 16(8):1692–1698
Vidal C, Meric P, Provost F, Herzog C, Lasmezas C, Gillet B et al (2006) Preclinical metabolic changes in mouse prion diseases detected by 1H-nuclear magnetic resonance spectroscopy. Neuroreport. 17(1):89–93
Macfarlane RG, Wroe SJ, Collinge J, Yousry TA, Jager HR (2007) Neuroimaging findings in human prion disease. J Neurol Neurosurg Psychiatry 78(7):664–670
Galanaud D, Haik S, Linguraru MG, Ranjeva JP, Faucheux B, Kaphan E et al (2010) Combined diffusion imaging and MR spectroscopy in the diagnosis of human prion diseases. AJNR Am J Neuroradiol 31(7):1311–1318
Krasnoslobodtsev AV, Portillo AM, Deckert-Gaudig T, Deckert V, Lyubchenko YL (2010) Nanoimaging for prion related diseases. Prion. 4(4):265–274
Lyubchenko YL, Kim BH, Krasnoslobodtsev AV, Yu J (2010) Nanoimaging for protein misfolding diseases. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2(5):526–543
Letourneau-Guillon L, Wada R, Kucharczyk W (2012) Imaging of prion diseases. J Magn Reson Imaging 35(5):998–1012
McDade EM, Boeve BF, Fields JA, Kumar N, Rademakers R, Baker MC et al (2013) MRS in early and presymptomatic carriers of a novel octapeptide repeat insertion in the prion protein gene. J Neuroimaging 23(3):409–413
Ortega-Cubero S, Luquin MR, Dominguez I, Arbizu J, Pagola I, Carmona-Abellan MM et al (2013) Structural and functional neuroimaging in human prion diseases. Neurologia. 28(5):299–308
Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wuthrich K (1996) NMR structure of the mouse prion protein domain PrP(121-231). Nature. 382(6587):180–182
Zahn R, Liu A, Luhrs T, Riek R, von Schroetter C, Lopez Garcia F et al (2000) NMR solution structure of the human prion protein. Proc Natl Acad Sci U S A 97(1):145–150
Sigurdsson EM, Brown DR, Alim MA, Scholtzova H, Carp R, Meeker HC et al (2003) Copper chelation delays the onset of prion disease. J Biol Chem 278(47):46199–46202
Meloni G, Faller P, Vašák M (2007) Redox silencing of copper in metal-linked neurodegenerative disorders reaction of Zn7metallothionein-3 with Cu2+ ions. J Biol Chem 282(22):16068–16078
Alghrably M, Czaban I, Jaremko Ł, Jaremko M (2019) Interaction of amylin species with transition metals and membranes. J Inorg Biochem 191:69–76
Dong X, Svantesson T, Sholts SB, Wallin C, Jarvet J, Gräslund A et al (2019) Copper ions induce dityrosine-linked dimers in human but not in murine islet amyloid polypeptide (IAPP/amylin). Biochem Biophys Res Commun 510(4):520–524
Nishida Y (2011) The chemical process of oxidative stress by copper (II) and iron (III) ions in several neurodegenerative disorders. Monatshefte für Chemie-Chemical Monthly 142(4):375–384
Dudzik CG, Walter ED, Millhauser GL (2011) Coordination features and affinity of the Cu2+ site in the α-synuclein protein of Parkinson’s disease. Biochemistry. 50(11):1771–1777
Spencer WA, Jeyabalan J, Kichambre S, Gupta RC (2011) Oxidatively generated DNA damage after Cu (II) catalysis of dopamine and related catecholamine neurotransmitters and neurotoxins: role of reactive oxygen species. Free Radic Biol Med 50(1):139–147
Chan T, Chow AM, Tang DW, Li Q, Wang X, Brown IR et al (2010) Interaction of baicalein and copper with α-synuclein: electrochemical approach to Parkinson’s disease. J Electroanal Chem 648(2):151–155
Lin C-J, Huang H-C, Jiang Z-F (2010) Cu (II) interaction with amyloid-β peptide: a review of neuroactive mechanisms in AD brains. Brain Res Bull 82(5–6):235–242
Multhaup G (1997) Amyloid precursor protein, copper and Alzheimer's disease. Biomed Pharmacother 51(3):105–111
Qin K, Yang D-S, Yang Y, Chishti MA, Meng L-J, Kretzschmar HA et al (2000) Copper (II)-induced conformational changes and protease resistance in recombinant and cellular PrP effect of protein age and deamidation. J Biol Chem 275(25):19121–19131
Lech T, Sadlik J (2007) Copper concentration in body tissues and fluids in normal subjects of southern Poland. Biol Trace Elem Res 118(1):10–15
Gybina AA, Tkac I, Prohaska JR (2009) Copper deficiency alters the neurochemical profile of developing rat brain. Nutr Neurosci 12(3):114–122
Emwas AHM, Al-Talla ZA, Guo X, Al-Ghamdi S, Al-Masri HT (2013) Utilizing NMR and EPR spectroscopy to probe the role of copper in prion diseases. Magn Reson Chem 51(5):255–268
Uriu-Adams JY, Scherr RE, Lanoue L, Keen CL (2010) Influence of copper on early development: prenatal and postnatal considerations. Biofactors. 36(2):136–152
Sorrentino G, Bonavita V (2007) Neurodegeneration and Alzheimer's disease: the lesson from tauopathies. Neurol Sci 28(2):63–71
Kodaka M (2004) Interpretation of concentration-dependence in aggregation kinetics. Biophys Chem 109(2):325–332
Hedberg YS, Dobryden I, Chaudhary H, Wei Z, Claesson PM, Lendel C (2019) Synergistic effects of metal-induced aggregation of human serum albumin. Colloids Surf B: Biointerfaces 173:751–758
Yuan Y, Niu F, Liu Y, Lu N (2014) Zinc and its effects on oxidative stress in Alzheimer's disease. Neurol Sci 35(6):923–928
Tsukita K, Sakamaki-Tsukita H, Tanaka K, Suenaga T, Takahashi R (2019) Value of in vivo alpha-synuclein deposits in Parkinson's disease: a systematic review and meta-analysis. Mov Disord 34(10):1452–1463
Bernabeu-Zornoza A, Coronel R, Palmer C, Monteagudo M, Zambrano A, Liste I (2019) Physiological and pathological effects of amyloid-beta species in neural stem cell biology. Neural Regen Res 14(12):2035–2042
Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, Salloway S et al (2016) Alzheimer's disease. Lancet. 388(10043):505–517
Lippens G, Sillen A, Landrieu I, Amniai L, Sibille N, Barbier P et al (2007) Tau aggregation in Alzheimer's disease: what role for phosphorylation? Prion. 1(1):21–25
Alonso AD, Cohen LS, Corbo C, Morozova V, ElIdrissi A, Phillips G et al (2018) Hyperphosphorylation of tau associates with changes in its function beyond microtubule stability. Front Cell Neurosci 12(338)
Zhu Y, Wang J (2015) Wogonin increases β-amyloid clearance and inhibits tau phosphorylation via inhibition of mammalian target of rapamycin: potential drug to treat Alzheimer’s disease. Neurol Sci 36(7):1181–1188
Crouch PJ, Hung LW, Adlard PA, Cortes M, Lal V, Filiz G et al (2009) Increasing cu bioavailability inhibits Abeta oligomers and tau phosphorylation. Proc Natl Acad Sci U S A 106(2):381–386
Zhou LX, Du JT, Zeng ZY, Wu WH, Zhao YF, Kanazawa K et al (2007) Copper (II) modulates in vitro aggregation of a tau peptide. Peptides. 28(11):2229–2234
Ma Q, Li Y, Du J, Liu H, Kanazawa K, Nemoto T et al (2006) Copper binding properties of a tau peptide associated with Alzheimer's disease studied by CD, NMR, and MALDI-TOF MS. Peptides. 27(4):841–849
Soragni A, Zambelli B, Mukrasch MD, Biernat J, Jeganathan S, Griesinger C et al (2008) Structural characterization of binding of Cu (II) to tau protein. Biochemistry. 47(41):10841–10851
Voss K, Harris C, Ralle M, Duffy M, Murchison C, Quinn JF (2014) Modulation of tau phosphorylation by environmental copper. Transl Neurodegener 3(1):24
Poulson BG, Szczepski K, Lachowicz JI, Jaremko L, Emwas A-H, Jaremko M (2020) Aggregation of biologically important peptides and proteins: inhibition or acceleration depending on protein and metal ion concentrations. RSC Adv 10(1):215–227
Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M et al (1993) Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A 90(23):11282–11286
Emamzadeh FN (2016) Alpha-synuclein structure, functions, and interactions. J Res Med Sci 21:29
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature. 388(6645):839–840
Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) Alpha-synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proc Natl Acad Sci U S A 95(11):6469–6473
Bougea A, Stefanis L, Paraskevas GP, Emmanouilidou E, Vekrelis K, Kapaki E (2019) Plasma alpha-synuclein levels in patients with Parkinson's disease: a systematic review and meta-analysis. Neurol Sci 40(5):929–938
Ghosh D, Mehra S, Sahay S, Singh PK, Maji SK (2017) Alpha-synuclein aggregation and its modulation. Int J Biol Macromol 100:37–54
Breydo L, Wu JW, Uversky VN (2012) Alpha-synuclein misfolding and Parkinson's disease. Biochim Biophys Acta 1822(2):261–285
Pall HS, Williams AC, Blake DR, Lunec J, Gutteridge JM, Hall M et al (1987) Raised cerebrospinal-fluid copper concentration in Parkinson's disease. Lancet. 2(8553):238–241
Rasia RM, Bertoncini CW, Marsh D, Hoyer W, Cherny D, Zweckstetter M et al (2005) Structural characterization of copper (II) binding to alpha-synuclein: insights into the bioinorganic chemistry of Parkinson's disease. Proc Natl Acad Sci U S A 102(12):4294–4299
Valensin D, Dell'Acqua S, Kozlowski H, Casella L (2016) Coordination and redox properties of copper interaction with alpha-synuclein. J Inorg Biochem 163:292–300
Bloch DN, Kolkowska P, Tessari I, Baratto MC, Sinicropi A, Bubacco L et al (2019) Fibrils of alpha-Synuclein abolish the affinity of Cu(2+)-binding site to His50 and induce hopping of Cu(2+) ions in the termini. Inorg Chem 58(16):10920–10927
Koppenol WH (2001) The Haber-Weiss cycle–70 years later. Redox Rep 6(4):229–234
Barb W, Baxendale J, George P, Hargrave K (1951) Reactions of ferrous and ferric ions with hydrogen peroxide. Part I.—the ferrous ion reaction. Trans Faraday Soc 47:462–500
Baruch-Suchodolsky R, Fischer B (2009) Aβ40, either soluble or aggregated, is a remarkably potent antioxidant in cell-free oxidative systems. Biochemistry. 48(20):4354–4370
Halliwell B, Gutteridge JM (1990) Role of free radicals and catalytic metal ions in human disease: an overview. Methods Enzymol. 186: Elsevier; p. 1–85
Sanchez-Lopez C, Rossetti G, Quintanar L, Carloni P (2018) Structural determinants of the prion protein N-terminus and its adducts with copper ions. Int J Mol Sci 20(1)
Bertini I, Mangani S, Viezzoli M, Sykes A (1998) Advanced inorganic chemistry. Academic, San Diego, pp 127–250
Quintanar L, Rivillas-Acevedo L, Grande-Aztatzi R, Gómez-Castro CZ, Arcos-López T, Vela A (2013) Copper coordination to the prion protein: insights from theoretical studies. Coord Chem Rev 257(2):429–444
Ling Y, Khade RL, Zhang Y (2011) Structural, EPR superhyperfine, and NMR hyperfine properties of the u−octarepeat binding site in the prion protein. J Phys Chem B 115(11):2663–2670
dos Santos NV, Silva AF, Oliveira VX Jr, Homem-de-Mello P, Cerchiaro G (2012) Copper (II) complexation to 1-octarepeat peptide from a prion protein: insights from theoretical and experimental UV-visible studies. J Inorg Biochem 114:1–7
Viles JH (2012) Metal ions and amyloid fiber formation in neurodegenerative diseases. Copper, zinc and iron in Alzheimer's, Parkinson's and prion diseases. Coord Chem Rev 256(19–20):2271–2284
Sánchez-López C, Rossetti G, Quintanar L, Carloni P (2019) Structural determinants of the prion protein N-terminus and its adducts with copper ions. Int J Mol Sci 20(1):18
McCord JM, Fridovich I (1969) Superoxide dismutase. J Biol Chem 244(22):6049–6055
Fee JA, Gaber BP (1972) Anion binding to bovine erythrocyte superoxide dismutase. J Biol Chem 247(1):60–65
White AR, Multhaup G, Maher F, Bellingham S, Camakaris J, Zheng H et al (1999) The Alzheimer's disease amyloid precursor protein modulates copper-induced toxicity and oxidative stress in primary neuronal cultures. J Neurosci 19(21):9170–9179
Bush AI, Pettingell W, De Paradis M, Tanzi RE, Wasco W (1994) The amyloid beta-protein precursor and its mammalian homologues. Evidence for a zinc-modulated heparin-binding superfamily. J Biol Chem 269(43):26618–26621
Simons A, Ruppert T, Schmidt C, Schlicksupp A, Pipkorn R, Reed J et al (2002) Evidence for a copper-binding superfamily of the amyloid precursor protein. Biochemistry. 41(30):9310–9320
Requena JR, Groth D, Legname G, Stadtman ER, Prusiner SB, Levine RL (2001) Copper-catalyzed oxidation of the recombinant SHa (29–231) prion protein. Proc Natl Acad Sci 98(13):7170–7175
Hornshaw M, McDermott J, Candy J, Lakey J (1995) Copper binding to the N-terminal tandem repeat region of mammalian and avian prion protein: structural studies using synthetic peptides. Biochem Biophys Res Commun 214(3):993–999
Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R et al (1997) The cellular prion protein binds copper in vivo. Nature. 390(6661):684
Hornshaw M, McDermott J, Candy J (1995) Copper binding to the N-terminal tandem repeat regions of mammalian and avian prion protein. Biochem Biophys Res Commun 207(2):621–629
Serra A, Manno D, Filippo E, Buccolieri A, Urso E, Rizzello A et al (2011) SERS based optical sensor to detect prion protein in neurodegenerate living cells. Sensors Actuators B Chem 156(1):479–485
Kállay C, Turi I, Timári S, Nagy Z, Sanna D, Pappalardo G et al (2011) The effect of point mutations on copper (II) complexes with peptide fragments encompassing the 106–114 region of human prion protein. Monatshefte für Chemie-Chemical Monthly 142(4):411–419
Hong L, Simon JD (2011) Insights into the thermodynamics of copper association with amyloid-β, α-synuclein and prion proteins. Metallomics. 3(3):262–266
Chaves JA, Sanchez-López C, Gomes MP, Sisnande T, Macedo B, de Oliveira VE et al (2014) Biophysical and morphological studies on the dual interaction of non-octarepeat prion protein peptides with copper and nucleic acids. JBIC J Biol Inorg Chem 19(6):839–851
Evans EGB (2015) Copper and zinc drive inter-domain structure in the cellular prion protein: UC Santa Cruz
Cereghetti GM, Negro A, Vinck E, Massimino ML, Sorgato MC, Van Doorslaer S (2004) Copper (II) binding to the human Doppel protein may mark its functional diversity from the prion protein. J Biol Chem 279(35):36497–36503
Giese A, Buchholz M, Herms J, Kretzschmar HA (2005) Mouse brain synaptosomes accumulate copper-67 efficiently by two distinct processes independent of cellular prion protein. J Mol Neurosci 27(3):347–354
Hijazi N, Shaked Y, Rosenmann H, Ben-Hur T, Gabizon R (2003) Copper binding to PrPC may inhibit prion disease propagation. Brain Res 993(1–2):192–200
Kourie J, Kenna B, Tew D, Jobling MF, Curtain C, Masters C et al (2003) Copper modulation of ion channels of PrP [106–126] mutant prion peptide fragments. J Membr Biol 193(1):35–45
Li A, Dong J, Harris DA (2004) Cell surface expression of the prion protein in yeast does not alter copper utilization phenotypes. J Biol Chem 279(28):29469–29477
Millhauser GL (2004) Copper binding in the prion protein. Acc Chem Res 37(2):79–85
Rachidi W, Mangé A, Senator A, Guiraud P, Riondel J, Benboubetra M et al (2003) Prion infection impairs copper binding of cultured cells. J Biol Chem 278(17):14595–14598
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A et al (1997) Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science 276(5321):2045–2047
Miura T, Hori-i A, Takeuchi H (1996) Metal-dependent [alpha]-helix formation promoted by the glycine-rich octapeptide region of prion protein. FEBS Lett 396(2–3):248–252
Stöckel J, Safar J, Wallace AC, Cohen FE, Prusiner SB (1998) Prion protein selectively binds copper (II) ions. Biochemistry. 37(20):7185–7193
Zheng Z, Zhang M, Wang Y, Ma R, Guo C, Feng L et al (2018) Structural basis for the complete resistance of the human prion protein mutant G127V to prion disease. Sci Rep 8(1):13211
Alahmari F, Dey S, Emwas A-H, Davaasuren B, Rothenberger A (2019) Layered copper thioaluminate K2Cu3AlS4: synthesis, crystal structure, characterization and solid-state 27Al and 39K NMR studies. J Alloys Compd 776:1041–1047
Alahmari F, Davaasuren B, Emwas A-H, Costa PMFJ, Rothenberger A (2019) Tris (ethylenediamine) nickel (II) thio-hydroxogermanate monohydrate: synthesis, crystal structure, 1H NMR, EPR, optical and magnetic properties. Inorg Chim Acta 488:145–151
Brown DR (2001) Copper and prion disease. Brain Res Bull 55(2):165–173
Davaasuren B, Emwas A-H, Rothenberger A (2017) MAu2GeS4-Chalcogel (M = Co, Ni): heterogeneous intra- and intermolecular hydroamination catalysts. Inorg Chem 56(16):9609–9616
Emwas A-H, Roy R, McKay RT, Tenori L, Saccenti E, Gowda GAN et al (2019) NMR spectroscopy for metabolomics research. Metabolites. 9(7):123
Toni M, Massimino ML, Griffoni C, Salvato B, Tomasi V, Spisni E (2005) Extracellular copper ions regulate cellular prion protein (PrPC) expression and metabolism in neuronal cells. FEBS Lett 579(3):741–744
Mattar SM, Emwas AH, Calhoun LA (2004) Spectroscopic studies of the intermediates in the conversion of 1, 4, 11, 12-tetrahydro-9, 10-anthraquinone to 9, 10-anthraquinone by reaction with oxygen under basic conditions. J Phys Chem A 108(52):11545–11553
Ellegood J, McKay RT, Hanstock CC, Beaulieu C (2007) Anisotropic diffusion of metabolites in peripheral nerve using diffusion weighted magnetic resonance spectroscopy at ultra-high field. J Magn Reson 184(1):20–28
McKay RT, Saltibus LF, Li MX, Sykes BD (2000) Energetics of the induced structural change in a Ca2+ regulatory protein: Ca2+ and troponin I peptide binding to the E41A mutant of the N-domain of skeletal troponin C. Biochemistry. 39(41):12731–12738
Amniai L, Barbier P, Sillen A, Wieruszeski J-M, Peyrot V, Lippens G et al (2009) Alzheimer disease specific phosphoepitopes of tau interfere with assembly of tubulin but not binding to microtubules. FASEB J 23(4):1146–1152
Giustiniani J, Guillemeau K, Dounane O, Sardin E, Huvent I, Schmitt A et al (2015) The FK506-binding protein FKBP52 in vitro induces aggregation of truncated tau forms with prion-like behavior. FASEB J 29(8):3171–3181
Abdul Jameel AG, Van Oudenhoven V, Emwas A-H, Sarathy SM (2018) Predicting octane number using nuclear magnetic resonance spectroscopy and artificial neural networks. Energy Fuel 32(5):6309–6329
Alahmari F, Davaasuren B, Emwas A-H, Rothenberger A (2018) Thioaluminogermanate M (AlS2)(GeS2) 4 (M= Na, Ag, Cu): synthesis, crystal structures, characterization, ion-exchange and solid-state 27Al and 23Na NMR spectroscopy. Inorg Chem 57(7):3713–3719
Batool F, Parveen S, Emwas A-H, Sioud S, Gao X, Munawar MA et al (2015) Synthesis of fluoroalkoxy substituted arylboronic esters by iridium-catalyzed aromatic C-H borylation. Org Lett 17(17):4256–4259
Alahmari F, Dey S, Emwas A-H, Davaasuren B, Rothenberger A (2019) Ultra-low thermal conductivity in Na/Sb chalcobismuthates: synthesis, crystal structures, optical properties and 23Na NMR spectroscopy. New J Chem 43(27):10814–10820
Medina SC, Farinha ASF, Emwas A-H, Tabatabai A, Leiknes T (2020) A fundamental study of adsorption kinetics of surfactants onto metal oxides using quartz crystal microbalance with dissipation (QCM-D). Colloids Surf A Physicochem Eng Asp 586:124237
Alkordi MH, Haikal RR, Hassan YS, Emwas A-H, Belmabkhout Y (2015) Poly-functional porous-organic polymers to access functionality–CO 2 sorption energetic relationships. J Mater Chem A 3(45):22584–22590
Atiqullah M, Al-Harthi MA, Anantawaraskul S, Emwas A-HM (2015) Ethylene homo-and copolymerization chain-transfers: a perspective from supported (nBuCp) 2ZrCl2 catalyst active Centre distribution. J Chem Sci 127(4):717–728
Chisca S, Duong P, Emwas A-H, Sougrat R, Nunes SP (2015) Crosslinked copolyazoles with a zwitterionic structure for organic solvent resistant membranes. Polym Chem 6(4):543–554
Bahuleyan BK, De Kumar S, Sarath PU, Furquan SA, Masihullah JK, Emwas AH et al (2012) Effect of aluminium nitride on the properties of polyethylene obtained by in situ polymerization using Ni (II) diimine complex. Macromol Res 20(7):772–775
Liu Z, Dong X, Zhu Y, Emwas A-H, Zhang D, Tian Q et al (2015) Investigating the influence of mesoporosity in zeolite beta on its catalytic performance for the conversion of methanol to hydrocarbons. ACS Catal 5(10):5837–5845
Alezi D, Belmabkhout Y, Suyetin M, Bhatt PM, Weseliński ŁJ, Solovyeva V et al (2015) MOF crystal chemistry paving the way to gas storage needs: aluminum-based soc-MOF for CH4, O2, and CO2 storage. J Am Chem Soc 137(41):13308–13318
Al-Bloushi M, Davaasuren B, Emwas AH, Rothenberger A (2015) Synthesis and characterization of the quaternary thioaluminogermanates A (AlS2)(GeS2)(A= Na, K). Z Anorg Allg Chem 641(7):1352–1356
Caro JA, Wand AJ (2018) Practical aspects of high-pressure NMR spectroscopy and its applications in protein biophysics and structural biology. Methods. 148:67–80
Matlahov I, van der Wel PCA (2018) Hidden motions and motion-induced invisibility: dynamics-based spectral editing in solid-state NMR. Methods 148:123–135
Quinn CM, Polenova T (2017) Structural biology of supramolecular assemblies by magic-angle spinning NMR spectroscopy. Q Rev Biophys 50
Yadav DK, Lukavsky PJ (2016) NMR solution structure determination of large RNA-protein complexes. Prog Nucl Magn Reson Spectrosc 97:57–81
Bechmann M, Mueller N (2017) Nonlinear effects in NMR. Annual Reports on NMR Spectroscopy. 92: Elsevier; p. 199–226
Bian J, Jiang M, Cui J, Liu X, Chen B, Ji Y et al (2017) Universal quantum control in zero-field nuclear magnetic resonance. Phys Rev A 95(5):052342
Fundo JF, Galvis-Sanchez A, Madureira AR, Carvalho A, Feio G, Silva CL et al (2016) NMR water transverse relaxation time approach to understand storage stability of fresh-cut ‘Rocha’pear. LWT. 74:280–285
Ilc G, Giachin G, Jaremko M, Jaremko L, Benetti F, Plavec J et al (2010) NMR structure of the human prion protein with the pathological Q212P mutation reveals unique structural features. PLoS One 5(7):e11715
Larda ST, Simonetti K, Al-Abdul-Wahid MS, Sharpe S, Prosser RS (2013) Dynamic equilibria between monomeric and oligomeric misfolded states of the mammalian prion protein measured by 19F NMR. J Am Chem Soc 135(28):10533–10541
Mead S (2019) Prion diseases and possible treatments. J Neurol Sci 405:70
Fremuntova Z, Mosko T, Soukup J, Kucerova J, Kostelanska M, Hanusova ZB et al (2020) Changes in cellular prion protein expression, processing and localisation during differentiation of the neuronal cell line CAD 5. Biol Cell 112(1):1–21
Geissen M, Krasemann S, Matschke J, Glatzel M (2007) Understanding the natural variability of prion diseases. Vaccine. 25(30):5631–5636
Fernandez-Borges N, Erana H, Venegas V, Elezgarai SR, Harrathi C, Castilla J (2015) Animal models for prion-like diseases. Virus Res 207:5–24
Geschwind MD (2015) Prion diseases. Continuum (Minneapolis, Minn) 21(6 Neuroinfectious Disease):1612–1638
Huang WJ, Chen WW, Zhang X (2015) Prions mediated neurodegenerative disorders. Eur Rev Med Pharmacol Sci 19(21):4028–4034
Lukic A, Uphill J, Brown CA, Beck J, Poulter M, Campbell T et al (2015) Rare structural genetic variation in human prion diseases. Neurobiol Aging 36(5):2004.e1–2004.e8
Nhat Tran Thanh L, Narkiewicz J, Aulic S, Salzano G, Hoa Thanh T, Scaini D et al (2015) Synthetic prions and other human neurodegenerative proteinopathies. Virus Res 207:25–37
Marandi Y, Farahi N, Sadeghi A, Sadeghi-Hashjin G (2012) Prion diseases - current theories and potential therapies: a brief review. Folia Neuropathol 50(1):46–49
Norrby E (2011) Prions and protein-folding diseases. J Intern Med 270(1):1–14
Sikorska B, Liberski PP (2012) Human prion diseases: from Kuru to variant Creutzfeldt-Jakob disease. Subcell Biochem 65:457–496
Solomon IH, Biasini E, Harris DA (2012) Ion channels induced by the prion protein mediators of neurotoxicity. Prion. 6(1):40–45
Syed M, Nourizadeh-Lillabadi R, Press CM, Alestrom P (2011) Prion protein function and the disturbance of early embryonic development in zebrafish. Prion. 5(2):88–92
Akhvlediani T, Gochitashvili N, Tsertsvadze T (2007) Prion diseases--mysterious persistent infections. Georgian Med News 146:38–42
Fornai F, Ferrucci M, Gesi M, di Poggio AB, Giorgi FS, Biagioni F et al (2006) A hypothesis on prion disorders: are infectious, inherited, and sporadic causes so distinct? Brain Res Bull 69(2):95–100
Ironside JW. Human prion diseases: biology and transmission by blood. In: Mayr WR, editor. Isbt Science Series, Vol 1, No 1: State of the Art Presentations. ISBT Science Series. 12006. p. 15–20
Ironside JW, Ritchie DL, Head MW (2005) Phenotypic variability in human prion diseases. Neuropathol Appl Neurobiol 31(6):565–579
Rachidi W, Riondel J, McMahon HM, Favier A (2005) Prion protein and copper: a mysterious relationship. Pathol Biol 53(4):244–250
Sakudo A, Onodera T (2011) Tissue- and cell type-specific modification of prion protein (PrP)-like protein Doppel, which affects PrP endoproteolysis. Biochem Biophys Res Commun 404(1):523–527
Figini M, Alexander DC, Redaelli V, Fasano F, Grisoli M, Baselli G et al (2015) Mathematical models for the diffusion magnetic resonance signal abnormality in patients with prion diseases. NeuroImage Clinical 7:142–154
Krakauer DC, Zanotto PMD, Pagel M (1998) Prion's progress: patterns and rates of molecular evolution in relation to spongiform disease. J Mol Evol 47(2):133–145
Lodi R, Parchi P, Tonon C, Manners D, Capellari S, Strammiello R et al (2009) Magnetic resonance diagnostic markers in clinically sporadic prion disease: a combined brain magnetic resonance imaging and spectroscopy study. Brain. 132:2669–2679
Tranchant C, Geranton L, Guiraud-Chaumeil C, Mohr M, Warter JM (1999) Basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Neurology. 52(6):1244–1249
Collins SJ, Lawson VA, Masters CL (2004) Transmissible spongiform encephalopathies. Lancet. 363(9402):51–61
Edskes HK, Wickner RB (2004) Transmissible spongiform encephalopathies - prion proof in progress. Nature. 430(7003):977–979
Basset-Leobon C, Uro-Coste E, Peoc'h K, Haik S, Sazdovitch V, Rigal M et al (2006) Familial Creutzfeldt-Jakob disease with an R208H-129V haplotype and Kuru plaques. Arch Neurol 63(3):449–452
Giles K, Olson SH, Prusiner SB (2017) Developing therapeutics for PrP prion diseases. Cold Spring Harbor perspectives in medicine 7(4):a023747
Mead S, Whitfield J, Poulter M, Shah P, Uphill J, Campbell T et al (2009) A novel protective prion protein variant that colocalizes with Kuru exposure. N Engl J Med 361(21):2056–2065
Stewart LA, Rydzewska LH, Keogh GF, Knight RS (2008) Systematic review of therapeutic interventions in human prion disease. Neurology. 70(15):1272–1281
Mancuso M, Siciliano G, Capellari S, Orsucci D, Moretti P, Di Fede G et al (2009) Creutzfeldt-Jakob disease with E200K PRNP mutation: a case report and revision of the literature. Neurol Sci 30(5):417–420
Bilandžić N, Đokić M, Sedak M, Varenina I, Kolanović BS, Oraić D et al (2012) Determination of copper in food of animal origin and fish in Croatia. Food Control 27(2):284–288
Bandmann O, Weiss KH, Kaler SG (2015) Wilson's disease and other neurological copper disorders. Lancet Neurol 14(1):103–113
Zheng W, Monnot AD (2012) Regulation of brain iron and copper homeostasis by brain barrier systems: implication in neurodegenerative diseases. Pharmacol Ther 133(2):177–188
Hart E, Steenbock H, Waddell J, Elvehjem C, Van Donk E, Riising BM (2001) Iron in nutrition: VII. Copper as a supplement to iron for hemoglobin building in the rat (Reprinted from Journal of Biological Chemistry, vol 77, pg 797-812, 1928). J Trace Elem Exp Med 14(2):195–206
Kozlowski H, Luczkowski M, Remelli M, Valensin D (2012) Copper, zinc and iron in neurodegenerative diseases (Alzheimer's, Parkinson's and prion diseases). Coord Chem Rev 256(19–20):2129–2141
Prohaska JR, Bailey WR (1994) Regional specificity in alterations of rat brain copper and catecholamines following perinatal copper deficiency. J Neurochem 63(4):1551–1557
Singh N, Das D, Singh A, Mohan ML (2010) Prion protein and metal interaction: physiological and pathological implications. Curr Issues Mol Biol 12(2):99
Yen C-F, Harischandra DS, Kanthasamy A, Sivasankar S (2016) Copper-induced structural conversion templates prion protein oligomerization and neurotoxicity. Sci Adv 2(7):e1600014
Scheiber IF, Mercer JF, Dringen R (2014) Metabolism and functions of copper in brain. Prog Neurobiol 116:33–57
Brazier MW, Volitakis I, Kvasnicka M, White AR, Underwood JR, Green JE et al (2010) Manganese chelation therapy extends survival in a mouse model of M1000 prion disease. J Neurochem 114(2):440–451
De Gregorio G, Biasotto F, Hecel A, Luczkowski M, Kozlowski H, Valensin D (2019) Structural analysis of copper (I) interaction with amyloid β peptide. J Inorg Biochem
Strausak D, Mercer JF, Dieter HH, Stremmel W, Multhaup G (2001) Copper in disorders with neurological symptoms: Alzheimer’s, Menkes, and Wilson diseases. Brain Res Bull 55(2):175–185
Rivera-Mancía S, Pérez-Neri I, Ríos C, Tristán-López L, Rivera-Espinosa L, Montes S (2010) The transition metals copper and iron in neurodegenerative diseases. Chem Biol Interact 186(2):184–199
Bolognin S, Drago D, Messori L, Zatta P (2009) Chelation therapy for neurodegenerative diseases. Med Res Rev 29(4):547–570
Slivarichová D, Mitrová E, Ursínyová M, Uhnáková I, Koscová S, Wsólová L (2011) Geographic accumulation of Creutzfeldt-Jakob disease in Slovakia-environmental metal imbalance as a possible cofactor. Cent Eur J Public Health 19(3):158
Mitteregger G, Korte S, Shakarami M, Herms J, Kretzschmar HA (2009) Role of copper and manganese in prion disease progression. Brain Res 1292:155–164
Hodak M, Chisnell R, Lu W, Bernholc J (2009) Functional implications of multistage copper binding to the prion protein. Proc Natl Acad Sci 106(28):11576–11581
Desai V, Kaler SG (2008) Role of copper in human neurological disorders. Am J Clin Nutr 88(3):855S–858S
Viles JH, Klewpatinond M, Nadal RC (2008) Copper and the structural biology of the prion protein. Portland Press Limited
Varela-Nallar L, González A, Inestrosa NC (2006) Role of copper in prion diseases: deleterious or beneficial? Curr Pharm Des 12(20):2587–2595
Quaglio E, Chiesa R, Harris DA (2001) Copper converts the cellular prion protein into a protease-resistant species that is distinct from the scrapie isoform. J Biol Chem 276(14):11432–11438
Prince RC, Gunson DE (1998) Prions are copper-binding proteins. Trends Biochem Sci 23(6):197–198
Yamamoto N, Kuwata K (2009) Difference in redox behaviors between copper-binding octarepeat and nonoctarepeat sites in prion protein. JBIC J Biol Inorg Chem 14(8):1209–1218
Giachin G, Mai PT, Tran TH, Salzano G, Benetti F, Migliorati V et al (2015) The non-octarepeat copper binding site of the prion protein is a key regulator of prion conversion. Sci Rep 5:15253
O'Sullivan J, Comerford E, Rachidi W, Scott M, Hooper NM, McMahon HE (2015) The effects of the cellular and infectious prion protein on the neuronal adaptor protein X11α. Biochim Biophys Acta Gen Subj 1850(11):2213–2221
Bocharova OV, Breydo L, Salnikov VV, Baskakov IV (2005) Copper (II) inhibits in vitro conversion of prion protein into amyloid fibrils. Biochemistry. 44(18):6776–6787
Giese A, Levin J, Bertsch U, Kretzschmar H (2004) Effect of metal ions on de novo aggregation of full-length prion protein. Biochem Biophys Res Commun 320(4):1240–1246
Kjaergaard CH, Jones SM, Gounel SB, Mano N, Solomon EI (2015) Two-electron reduction versus one-electron oxidation of the type 3 pair in the multicopper oxidases. J Am Chem Soc 137(27):8783–8794
Serrano-Plana J, Garcia-Bosch I, Company A, Costas M (2015) Structural and reactivity models for copper oxygenases: cooperative effects and novel reactivities. Acc Chem Res 48(8):2397–2406
Arnesano F, Banci L, Bertini I, Mangani S, Thompsett AR (2003) A redox switch in CopC: an intriguing copper trafficking protein that binds copper (I) and copper (II) at different sites. Proc Natl Acad Sci 100(7):3814–3819
Artés JM, López-Martínez M, Díez-Pérez I, Sanz F, Gorostiza P (2014) Conductance switching in single wired redox proteins. Small. 10(13):2537–2541
Hepel M, Stobiecka M, Peachey J, Miller J (2012) Intervention of glutathione in pre-mutagenic catechol-mediated DNA damage in the presence of copper (II) ions. Mutat Res Fundam Mol Mech Mutagen 735(1–2):1–11
Gutiérrez AGP, Zeitouny J, Gomila A, Douziech B, Cosquer N, Conan F et al (2014) Insights into water coordination associated with the Cu II/Cu I electron transfer at a biomimetic Cu centre. Dalton Trans 43(17):6436–6445
K-i S, Maeshima H, Yoshida H, Satsuma A, Hattori T (2000) Spectroscopic characterisation of Cu–Al 2 O 3 catalysts for selective catalytic reduction of NO with propene. Phys Chem Chem Phys 2(10):2435–2439
Collery P, Maymard I, Theophanides T, Khassanova L, Collery T, Collery P, et al. Metal ions in biology and medicine, Vol 10. Metal Ions in Biology and Medicine, Vol 10. Metal Ions in Biology and Medicine. 102008
Roat-Malone RM, Roat-Malone RM. Bioinorganic chemistry: a short course2002. i-xvii, 1–348 p
Brewer GJ (2009) Risks of copper and iron toxicity during aging in humans. Chem Res Toxicol 23(2):319–326
Abuhijleh AL, Ali HA, Emwas A-H (2009) Synthesis, spectral and structural characterization of dinuclear rhodium (II) complexes of the anticonvulsant drug valproate with theophylline and caffeine. J Organomet Chem 694(22):3590–3596
Nageeb A, Al-Tawashi A, Mohammad Emwas A-H, Abdel-Halim Al-Talla Z, Al-Rifai N (2013) Comparison of artemisia annua bioactivities between traditional medicine and chemical extracts. Curr Bioact Compd 9(4):324–332
Al-Talla Z, Akrawi SH, Emwas A (2011) Solid state NMR and bioequivalence comparison of the pharmacokinetic parameters of two formulations of clindamycin. Int J Clin Pharmacol Ther 49(7):469–476
Chu S, Maltsev S, Emwas A-H, Lorigan GA (2010) Solid-state NMR paramagnetic relaxation enhancement immersion depth studies in phospholipid bilayers. J Magn Reson 207(1):89–94
Jackson MD, Chae SR, Mulcahy SR, Meral C, Taylor R, Li P et al (2013) Unlocking the secrets of Al-tobermorite in Roman seawater concrete. Am Mineral 98(10):1669–1687
Jackson MD, Moon J, Gotti E, Taylor R, Chae SR, Kunz M et al (2013) Material and elastic properties of Al-tobermorite in ancient Roman seawater concrete. J Am Ceram Soc 96(8):2598–2606
Mroue KH, Emwas A-HM, Power WP (2010) Solid-state 27Al nuclear magnetic resonance investigation of three aluminum-centered dyes. Can J Chem 88(2):111–123
Bouhrara M, Ranga C, Fihri A, Shaikh RR, Sarawade P, Emwas A-H et al (2013) Nitridated fibrous silica (KCC-1) as a sustainable solid base nanocatalyst. ACS Sustain Chem Eng 1(9):1192–1199
Sahloul N, Emwas A, Power W, Penlidis A (2005) Ethyl acrylate-hydroxyethyl acrylate and hydroxyethyl acrylate-methacrylic acid: reactivity ratio estimation from cross-linked polymer using high resolution magic angle spinning spectroscopy. J Macromol Sci Pure Appl Chem A42(10):1369–1385
Atiqullah M, Winston M, Bercaw J, Hussain I, Fazal A, Al-Harthi M et al (2012) Effects of a vanadium post-metallocene catalyst-induced polymer backbone inhomogeneity on UV oxidative degradation of the resulting polyethylene film. Polym Degrad Stab 97(7):1164–1177
Karbach FF, Macko T, Duchateau R (2016) Preparation of ethylene/1-hexene copolymers from ethylene using a fully silica-supported tandem catalyst system. Macromolecules. 49(4):1229–1241
Kirchheim A, Dal Molin D, Fischer P, Emwas A-H, Provis JL, Monteiro PJM (2011) Real-time high-resolution X-ray imaging and nuclear magnetic resonance study of the hydration of pure and Na-doped C3A in the presence of sulfates. Inorg Chem 50(4):1203–1212
Emwas A-H, Luchinat C, Turano P, Tenori L, Roy R, Salek RM et al (2015) Standardizing the experimental conditions for using urine in NMR-based metabolomic studies with a particular focus on diagnostic studies: a review. Metabolomics. 11(4):872–894
Asghar S, Shahzadi T, Alazmi M, Gao X, Emwas A-H, Saleem RS et al (2018) Iridium-catalyzed regioselective borylation of substituted biaryls. Synthesis. 50(11):2211–2220
Jameel AGA, Naser N, Issayev G, Touitou J, Ghosh MK, Emwas A-H et al (2018) A minimalist functional group (MFG) approach for surrogate fuel formulation. Combust Flame 192:250–271
Fine M, Cinar M, Voolstra CR, Safa A, Rinkevich B, Laffoley D et al (2019) Coral reefs of the Red Sea—challenges and potential solutions. Reg Stud Mar Sci 25:100498
Omar H, Moosa B, Alamoudi K, Anjum DH, Emwas A-H, El Tall O et al (2018) Impact of pore–walls ligand assembly on the biodegradation of mesoporous Organosilica nanoparticles for controlled drug delivery. ACS Omega 3(5):5195–5201
Yang P, Alsufyani M, Emwas AH, Chen C, Khashab NM (2018) Lewis acid guests in a {P8W48} archetypal polyoxotungstate host: enhanced proton conductivity via metal-oxo cluster within cluster assemblies. Angew Chem Int Ed 57(40):13046–13051
Li S, Winters H, Jeong S, Emwas A-H, Vigneswaran S, Amy GL (2016) Marine bacterial transparent exopolymer particles (TEP) and TEP precursors: characterization and RO fouling potential. Desalination. 379:68–74
Li S, Winters H, Villacorte L, Ekowati Y, Emwas A-H, Kennedy M et al (2015) Compositional similarities and differences between transparent exopolymer particles (TEPs) from two marine bacteria and two marine algae: significance to surface biofouling. Mar Chem 174:131–140
Linenberger KJ, Emwas A-H, Peat I, Lorigan GA, Bretz SL, editors. Using NMR to determine the structure of a peptide: an inquiry approach for an upper level undergraduate laboratory. ABSTRACTS OF PAPERS OF THE AMERICAN CHEMICAL SOCIETY; 2009: AMER CHEMICAL SOC 1155 16TH ST, NW, WASHINGTON, DC 20036 USA
Das SK, Xu S, Emwas A-H, Lu YY, Srivastava S, Archer LA (2012) High energy lithium–oxygen batteries–transport barriers and thermodynamics. Energy Environ Sci 5(10):8927–8931
Decken A, Mattar S, Emwas A (2005) 1, 4, 11, 12-Tetrahydro-9, 10-anthraquinone. Acta Crystallogr Sect E: Struct Rep Online 61(3):o641–o6o2
Elbaz AM, Gani A, Hourani N, Emwas A-H, Sarathy SM, Roberts W (2015) TG/DTG, FT-ICR mass spectrometry, and NMR spectroscopy study of heavy fuel oil. Energy Fuel 29(12):7825–7835
Emwas A-HM, Merzaban JS, Serrai H (2015) Theory and applications of NMR-based metabolomics in human disease diagnosis. Applications of NMR Spectroscopy: Elsevier, pp 93–130
Emwas A-HM, Antakly T, Saoudi A-H, Al-Ghamdi S, Serrai H (2015) Magnetic resonance spectroscopy and imaging in breast cancer prognosis and diagnosis. applications of NMR spectroscopy: Volume 3: Elsevier; p. 4–35
Emwas A-H, Saunders M, Ludwig C, Günther U (2008) Determinants for optimal enhancement in ex situ DNP experiments. Appl Magn Reson 34(3–4):483–494
Chiliveri SC, Deshmukh MV (2016) Recent excitements in protein NMR: large proteins and biologically relevant dynamics. J Biosci 41(4):787–803
Wang Y, Xu J, Wang L, Zhang B, Du W (2010) Interaction of the human prion protein PrP106–126 with metal complexes: potential therapeutic agents against prion disease. Chem Eur J 16(45):13339–13342
Frederick KK, Michaelis VK, Corzilius B, Ong T-C, Jacavone AC, Griffin RG et al (2015) Sensitivity-enhanced NMR reveals alterations in protein structure by cellular milieus. Cell. 163(3):620–628
Wang X, He L, Zhao C, Du W, Lin J (2013) Gold complexes inhibit the aggregation of prion neuropeptides. JBIC J Biol Inorg Chem. 18(7):767–778
Hecel A, Valensin D, Kozłowski H (2019) How copper ions and membrane environment influence the structure of the human and chicken tandem repeats domain? J Inorg Biochem 191:143–153
Sandusky P, Raftery D (2005) Use of selective TOCSY NMR experiments for quantifying minor components in complex mixtures: application to the metabonomics of amino acids in honey. Anal Chem 77(8):2455–2463
Banci L, Bertini I, Cantini F, Felli IC, Gonnelli L, Hadjiliadis N et al (2006) The Atx1-Ccc2 complex is a metal-mediated protein-protein interaction. Nat Chem Biol 2(7):367
Féraud B, Govaerts B, Verleysen M, De Tullio P (2015) Statistical treatment of 2D NMR COSY spectra in metabolomics: data preparation, clustering-based evaluation of the metabolomic informative content and comparison with 1 H-NMR. Metabolomics. 11(6):1756–1768
Guennec AL, Giraudeau P, Caldarelli S (2014) Evaluation of fast 2D NMR for metabolomics. Anal Chem 86(12):5946–5954
Tompa P, Fuxreiter M (2008) Fuzzy complexes: polymorphism and structural disorder in protein–protein interactions. Trends Biochem Sci 33(1):2–8
Becker W, Bhattiprolu KC, Gubensäk N, Zangger K (2018) Investigating protein–ligand interactions by solution nuclear magnetic resonance spectroscopy. ChemPhysChem. 19(8):895–906
Skinner AL, Laurence JS (2008) High-field solution NMR spectroscopy as a tool for assessing protein interactions with small molecule ligands. J Pharm Sci 97(11):4670–4695
Larsen E, Olivieri C, Walker C, Manu VS, Gao J, Bernlohr D et al (2018) Probing protein-protein interactions using asymmetric labeling and carbonyl-carbon selective heteronuclear NMR spectroscopy. Molecules 23(8):1937
Cobine PA, McKay RT, Zangger K, Dameron CT, Armitage IM (2004) Solution structure of Cu6 metallothionein from the fungus Neurospora crassa. Eur J Biochem 271(21):4213–4221
Maret W (2011) Metals on the move: zinc ions in cellular regulation and in the coordination dynamics of zinc proteins. Biometals 24(3):411–418
Chong LX, Ash M-R, Maher MJ, Hinds MG, Xiao Z, Wedd AG (2009) Unprecedented binding cooperativity between CuI and CuII in the copper resistance protein CopK from Cupriavidus metallidurans CH34: implications from structural studies by NMR spectroscopy and X-ray crystallography. J Am Chem Soc 131(10):3549–3564
Ou M-H, Chen Y-M, Chang Y-H, Lu W-K, Liu G-C, Wang Y-M (2007) Synthesis, complexation and water exchange properties of Gd (iii)–TTDA-mono and bis (amide) derivatives and their binding affinity to human serum albumin. Dalton Trans 26:2749–2759
Evans EG, Pushie MJ, Markham KA, Lee H-W, Millhauser GL (2016) Interaction between prion protein's copper-bound octarepeat domain and a charged C-terminal pocket suggests a mechanism for N-terminal regulation. Structure. 24(7):1057–1067
Narayanan SP, Nair DG, Schaal D, De Aguiar MB, Wenzel S, Kremer W et al (2016) Structural transitions in full-length human prion protein detected by xenon as probe and spin labeling of the N-terminal domain. Sci Rep 6:28419
De Ricco R, Potocki S, Kozlowski H, Valensin D (2014) NMR investigations of metal interactions with unstructured soluble protein domains. Coord Chem Rev 269:1–12
O'sullivan DB, Jones CE, Abdelraheim SR, Brazier MW, Toms H, Brown DR et al (2009) Dynamics of a truncated prion protein, PrP (113–231), from 15N NMR relaxation: order parameters calculated and slow conformational fluctuations localized to a distinct region. Protein Sci 18(2):410–423
Guantieri V, Venzo A, Di Marco V, Acampora M, Biondi B (2007) Potentiometric and NMR studies on Cd2+ coordination with the histidine-containing Ac184–188NH2 prion protein fragment. Inorg Chim Acta 360(14):4051–4057
Wells MA, Jackson GS, Jones S, Hosszu LL, Craven CJ, Clarke AR et al (2006) A reassessment of copper (II) binding in the full-length prion protein. Biochem J 399(3):435–444
Gaggelli E, Bernardi F, Molteni E, Pogni R, Valensin D, Valensin G et al (2005) Interaction of the human prion PrP (106− 126) sequence with copper (II), manganese (II), and zinc (II): NMR and EPR studies. J Am Chem Soc 127(3):996–1006
Wissler J, Laub M, Jennissen H, editors. 3D-Rapid prototyping molecular image models of cellular prion proteins [PrPC] based on NMR data: metalloregulated interactions with copper-structured RNA biaptamers and relation to RNA chaperones. FASEB JOURNAL; 2003: FEDERATION AMER SOC EXP BIOL 9650 ROCKVILLE PIKE, BETHESDA, MD 20814–3998 USA
Banci L, Bertini I, Ciofi-Baffoni S, Gonnelli L, Su X-C (2003) Structural basis for the function of the N-terminal domain of the ATPase CopA from Bacillus subtilis. J Biol Chem 278(50):50506–50513
Banci L, Bertini I, Ciofi-Baffoni S, Su X-C, Borrelly GP, Robinson NJ (2004) Solution structures of a cyanobacterial metallochaperone INSIGHT INTO AN ATYPICAL COPPER-BINDING MOTIF. J Biol Chem 279(26):27502–27510
Myari A, Hadjiliadis N, Fatemi N, Sarkar B (2004) Copper (I) interaction with model peptides of WD6 and TM6 domains of Wilson ATPase: regulatory and mechanistic implications. J Inorg Biochem 98(9):1483–1494
Abajian C, Yatsunyk LA, Ramirez BE, Rosenzweig AC (2004) Yeast Cox17 solution structure and copper (I) binding. J Biol Chem 279(51):53584–53592
Wernimont AK, Huffman DL, Finney LA, Demeler B, O'Halloran TV, Rosenzweig AC (2003) Crystal structure and dimerization equilibria of PcoC, a methionine-rich copper resistance protein from Escherichia coli. JBIC J Biol Inorg Chem 8(1–2):185–194
Cobine PA, George GN, Jones CE, Wickramasinghe WA, Solioz M, Dameron CT (2002) Copper transfer from the Cu (I) chaperone, CopZ, to the repressor, Zn (II) CopY: metal coordination environments and protein interactions. Biochemistry 41(18):5822–5829
Bonomo RP, Pappalardo G, Rizzarelli E, Tabbì G, Vagliasindi LI (2008) Studies of nitric oxide interaction with mono-and dinuclear copper (II) complexes of prion protein bis-octarepeat fragments. Dalton Trans 29:3805–3816
Martic S, Rains MK, Kraatz H-B (2013) Probing copper/tau protein interactions electrochemically. Anal Biochem 442(2):130–137
Viles JH, Cohen FE, Prusiner SB, Goodin DB, Wright PE, Dyson HJ (1999) Copper binding to the prion protein: structural implications of four identical cooperative binding sites. Proc Natl Acad Sci 96(5):2042–2047
Gaggelli E, Kozlowski H, Valensin D, Valensin G (2006) Copper homeostasis and neurodegenerative disorders (Alzheimer's, prion, and Parkinson's diseases and amyotrophic lateral sclerosis). Chem Rev 106(6):1995–2044
Migliorini C, Sinicropi A, Kozlowski H, Luczkowski M, Valensin D (2014) Copper-induced structural propensities of the amyloidogenic region of human prion protein. JBIC J Biol Inorg Chem 19(4–5):635–645
Berti F, Gaggelli E, Guerrini R, Janicka A, Kozlowski H, Legowska A et al (2007) Structural and dynamic characterization of copper (II) binding of the human prion protein outside the octarepeat region. Chem Eur J 13(7):1991–2001
Hecel A, Migliorini C, Valensin D, Luczkowski M, Kozlowski H (2015) Impact of SDS surfactant on the interactions of Cu2+ ions with the amyloidogenic region of human prion protein. Dalton Trans 44(29):13125–13132
Cox DL, Pan J, Singh RR (2006) A mechanism for copper inhibition of infectious prion conversion. Biophys J 91(2):L11–LL3
Viles JH (2012) Metal ions and amyloid fiber formation in neurodegenerative diseases. Copper, zinc and iron in Alzheimer's, Parkinson's and prion disease. Coord Chem Rev 256:2271–2284
Sarell CJ, Syme CD, Rigby SEJ, Viles JH (2009) Copper (II) binding to amyloid-β fibrils of Alzheimer’s disease reveals a picomolar affinity: stoichiometry and coordination geometry are independent of Aβ oligomeric form. Biochemistry 48(20):4388–4402
Burns CS, Aronoff-Spencer E, Legname G, Prusiner SB, Antholine WE, Gerfen GJ et al (2003) Copper coordination in the full-length, recombinant prion protein. Biochemistry 42(22):6794–6803
Whittal RM, Ball HL, Cohen FE, Burlingame AL, Prusiner SB, Baldwin MA (2000) Copper binding to octarepeat peptides of the prion protein monitored by mass spectrometry. Protein Sci 9(2):332–343
Kramer ML, Kratzin HD, Schmidt B, Römer A, Windl O, Liemann S et al (2001) Prion protein binds copper within the physiological concentration range. J Biol Chem 276(20):16711–16719
Qin K, Yang Y, Mastrangelo P, Westaway D (2002) Mapping Cu (II) binding sites in prion proteins by diethyl pyrocarbonate modification and matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometric footprinting. J Biol Chem 277(3):1981–1990
Klewpatinond M, Davies P, Bowen S, Brown DR, Viles JH (2008) Deconvoluting the Cu2+ binding modes of full-length prion protein. J Biol Chem 283(4):1870–1881
Viles JH, Klewpatinond M, Nadal RC (2008) Copper and the structural biology of the prion protein. Biochem Soc Trans 36(Pt 6):1288–1292
Younan ND, Klewpatinond M, Davies P, Ruban AV, Brown DR, Viles JH (2011) Copper (II) induced secondary structure changes and reduced folding stability of the prion protein. J Mol Biol 410:369–382
Acknowledgments
We thank King Abdullah University of Science and Technology (KAUST) and King Abdullah International Medical Research Center (KAIMRC) for technical support.
Funding
MA, MJ, LJ, and AE are financially supported by KAUST. RA, SA, and AS are financially supported by KAIMRC.
Author information
Authors and Affiliations
Contributions
Literature review: RA MJ AE MA LJ. Figures 1 and 3 construction: MA. Drafting the article: SA AE RA MA. Critical revision of the article: SA MJ AE. Final approval of the version to be published: AE AS MJ. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
Authors declare no relevant competing interests.
Availability of data and materials
All of the supporting data reported are included in this review.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interest
The authors declare that they have no competing interest.
Availability of supporting data
Not applicable
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 22 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alsiary, R.A., Alghrably, M., Saoudi, A. et al. Using NMR spectroscopy to investigate the role played by copper in prion diseases. Neurol Sci 41, 2389–2406 (2020). https://doi.org/10.1007/s10072-020-04321-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-020-04321-9