
Abstract
A density functional theory study of the influence of the various functional groups of the molybdopterin ligand on electronic and geometric properties of active-site models for the molybdenum and tungsten cofactors has been undertaken. We used analogous molybdenum and tungsten complexes with increasingly accurate representation of the molybdopterin ligands and compared bond lengths, angles, charge distribution, composition of the binding orbitals, as well as the redox potentials in relation to each other. On the basis of our findings, we suggest using ligand systems including the pyrane and the pyrazine rings, besides the dithiolene function, to obtain sufficiently reliable computational, but also synthetic, models for the molybdenum and tungsten cofactors, whereas the second ring of the pterin might be neglected for efficiency reasons.
Similar content being viewed by others
Introduction
Enzymes depending on molybdenum and tungsten cofactors are ubiquitous and indispensable [1, 2]. They usually catalyze oxygen-transfer reactions from the substrate to water, or vice versa, in the form of two-electron redox reactions: R + H2O → RO + 2H+ + 2e –. These reactions are part of the carbon, nitrogen, and sulfur metabolism.
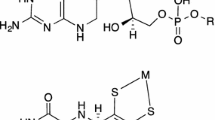
One common feature of these proteins is the unusual molybdopterin ligand (also referred to as pyranopterindithiolate [1], pterindithiolene [3], or pterin-ene-dithiolate [4]), one or two of which are bound to the metal at the active site (Fig. 1), depending on the enzyme family [5, 6]. This ligand is coordinated to the metal by a dithiolene function. The molybdopterin ligand is usually modeled in bioinorganic studies of the molybdenum and tungsten cofactors by using any dithiolene ligand with two substituents on the ene function. Dithiolenes are non-innocent ligands that actively take part in redox reactions at the metal, sometimes even changing the oxidation state of the metal, which has been shown in great detail for a number of ligands (not only dithiolenes) by the Wieghardt group [7–17]. For example, the redox potential of [MoO(S2C2(CN)2)2]−/2− is 1 V higher than that of [MoO(S2C2Me2)2]−/2− [18, 19]. Such a participation in redox processes is accompanied by structural changes of the dithiolene ligand. For instance, the C=C bond length is increased if the metal is oxidized, because electron density is pushed towards the metal, while the C–S bond is shortened. Consequently, the way the molybdopterin ligand is modeled can be expected to have a strong impact on the result of both experimental and theoretical studies, for the reactivity as well as for the structure.

The general structure of the active sites of the molybdenum and tungsten enzymes. Additional ligands not shown here may be oxo, sulfido, hydroxide, and water
In theoretical studies, the ligand is typically modeled with rather simple systems: ethenedithiol (edt; S2C2H2 2−) [20], maleonitrile [mnt; S2C2(CN)2 2−] [21–23], 1,2-dimethyldithiolene (S2C2Me2 2−) [21, 24–26] or benzenedithiol (bdt; S2C6H4 2−) [22, 24–27]. Functional groups of molybdopterin, besides the dithiolene function that binds to the metal, were ignored, probably to reduce the computational load. Notable exceptions to this are the study of McNamara et al. [28] in which a simplified form of molybdopterin without the pyrane ring was used, and the study of Joshi and Enemark [29] about the folding angle and electronic effects, in which the full molybdopterin ligand (without the phosphate group) was used.
In this paper, we present a systematic theoretical study [density functional theory (DFT) calculations] of various and increasingly accurate models of the molybdopterin ligand. The earliest group of synthetic models for the molybdenum- and tungsten-dependent oxidoreductases that is still used today is based on a metal (Mo or W) coordinated by two dithiolene ligands and one oxo ligand in oxidation states +4 and +5 or with two oxo ligands in oxidation state +6, respectively [30–52]. For this type of model, quite a substantial amount of experimental data is available. Therefore, we focus on this group of compounds, although more accurate models of the proteins are now known [53–58]. Thus, we studied models of the form [MO(dithiolene)2]n− (M is Mo, W; n = 0, 1, 2). In oxidation state +6 of the metal, a second oxo ligand is usually present in the synthetic models [42–46, 49–52]. To simplify the calculations and the comparisons, we abstained from including this second oxo ligand and kept the same coordination number and ligand set throughout all three oxidation states. The aim of our study is to ascertain which parts of molybdopterin need to be included in the calculations to obtain very accurate structures, energies, reactivities, and reduction potentials of the models.
Methods
All calculations were performed with the Gaussian 03 (revision C.02) software package [59]. All structures were fully optimized without any restrictions and the minima were verified by analytical frequency calculations.
Geometry optimizations, population analysis of molecular orbitals, and partial-charge distribution (with the natural population analysis and natural bond analysis methods [60–64]) were carried out at the DFT level using the B3LYP method (Becke’s three-parameter-hybrid functional [65], combined with the correlation functional of Lee et al. [66]; unrestricted formalism for the open-shell systems). The geometry parameters (Table S1) and the coordinates of the fully optimized structures can be found in Table S4.
The LANL2DZ [67] basis set, including the Los Alamos relativistic effective core potentials of Hay and Wadt [68], was used for molybdenum and tungsten, and the 6-311G(d,p) basis, including polarization functions [69, 70] for a better description of the sulfur and oxygen atoms, was used for the remaining atoms.
Solvation energies were estimated by single-point energy calculations using the integral equation formalism polarized continuum model in Gaussian 03 [71]. The calculations used water as the solvent (ε = 78.39 and probe radius 1.385 Å) and the default UA0 radii. Reduction potentials were calculated from these energies using a solvation energy for the electron of 4.28 eV [72].
The stereochemistry of the mpt (C9H7N6O2S2 2−) and prz (C7H6N2OS2 2−) ligands (Fig. 2) was the same as that found in the enzymes. In particular, we used the structures of dimethyl sulfoxide reductase [73] and aldehyde oxidoreductase [74]. The quality and accuracy of the DFT calculations were estimated by comparison with experimental structural data. Quite a number of X-ray structures of enzyme model complexes of the general composition [MO(dithiolene)2]n− with n = 1 or 2 and M is Mo or W have been published [28–46] and are therefore available for evaluation of the quality of our computations, all showing the same general geometry as our computed compounds. In particular, X-ray structures of compounds with the edt ligand ([MoO(edt)2]1/2−) [47] and the pyranedithiolene (pdt) ligand ([MoO(pdt)2]2−) [48] are known, i.e., the exact models that are used in this study. The latter allows a comparison of the largest quantity of structural parameters. The computed structure of this compound in oxidation state +4 is almost identical to the experimental data obtained by crystallography. The only large difference is that the calculated Mo–S bond lengths are approximately 0.1 Å too long (2.47 vs. 2.37 Å). This has frequently been observed before in theoretical studies [75]. With respect to the overall geometry, no differences were found.

The ligand systems investigated and the resulting complexes with the numbering scheme of the compounds and the relevant atoms
Results
Complexes investigated
The complexes investigated consist of a metal (either molybdenum or tungsten) with two identical equatorial dithiolene ligands and one apical oxo ligand in a (distorted) square-pyramidal coordination geometry. The emphasis of this study lies on the dithiolene ligands. Four different ligands as increasingly accurate models for the natural molybdopterin ligand, as shown in Fig. 2, were investigated. For each of the four ligands, we studied the metal in each of its three biologically relevant oxidation states, +4, +5, and +6. We therefore introduce the systematic numbering depicted in Fig. 2.
When the metal is in oxidation state +6, synthetic models usually have a second oxo ligand [42–46, 49–52]. However, to simplify the calculations and to evaluate the interaction of the dithiolene with the metal center, we have abstained from including this second oxo ligand and kept the same coordination number and ligand set throughout all three oxidation states. Thereby, we may understand even subtle influences of the different functional groups of molybdopterin on electronic and geometric properties of the central metals.
Structural analysis
One interesting structural feature is the folding angle, which was introduced into the discussion about dithiolene ligands by Lauher and Hoffmann [76]. It defines the angle between the M–S–S plane and the S–C=C–S plane, as shown in Fig. 3. Upon oxidation, one of the dithiolene ligands bends towards the metal and the apical oxygen, while the other bends slightly away from them. This is illustrated in Fig. 4 for the molybdenum compounds with the pdt ligand. This behavior is a consequence of the changing π-electron-density distribution in response to the change of oxidation state of the metal. To be more precise, the “dithiolate folding effect” [77] depends on the occupation of the d orbitals of the metal. In high oxidation states (+5/d 1 and +6/d 0), the need for a flow of electron density towards the metal to stabilize these high oxidation states is greater. A folding of the dithiolene ligand towards the metal causes a much better π overlap of the metal d orbitals with the sulfur p orbitals and therefore provides a better electron-transfer pathway, resulting in a stabilization of the compound. In Fig. 5, the folding angle values are depicted for all compounds.

Definition of the folding angle

Bending of both ligands upon oxidation of the molybdenum pdt compounds 2a

The folding angles (towards M=O, left; away from it, right) in all oxidation states for all molybdenum (top) and tungsten (bottom) compounds
The folding angles of the tungsten compounds are larger than those of the molybdenum compounds. Tungsten therefore is able to achieve a better π overlap with the coordinated sulfur atoms than molybdenum. The differences in the folding angles between the four ligand systems are more pronounced for the molybdenum complexes than for the tungsten complexes. Interestingly, the largest differences were found for the ligand that bends away from the metal.
In the +4 oxidation state, the folding angle is larger for the compounds with the smaller and less accurate molybdopterin models edt (1) and pdt (2), while the compounds with the prz ligand (3) are in good agreement with the complexes of the most complete molybdopterin model (mpt, 4). In the +5 oxidation state, the folding angles are larger for the smaller models than for the mpt complexes (4), while in oxidation state +6, the folding angle of the prz (3) compounds is smaller and that of the edt (1) and pdt (2) compounds is again larger.
Naturally, besides the folding angle, the bond lengths of the most prominent connections are of interest. In all cases, a decrease of the M–S bond lengths and an increase of the C=C bond lengths are observed (Fig. 6) in response to the oxidation of the compounds from oxidation state +4 to oxidation state +6. This is in accordance with the findings of a recent study by Lim et al. [78] of planar nickel bisdithiolene. The higher charge of the metal and its decreased size upon oxidation are responsible for the decreased M–S bond lengths. Moreover, the lack of electrons close to the metal center after removal of two electrons when going from oxidation state +4 to oxidation state +6 causes the C=C bond to support the complex by donating π-electron density towards the center. This results in an increase of the C=C bond length.

Dependence of the M–S and C=C bond lengths on oxidation states for the molybdenum (top) and tungsten (bottom) complexes
It is notable that the M–S bond lengths are shorter for tungsten than for molybdenum, owing to the higher charge of the nucleus, relativistic effects, and stronger contractions. The decrease of the M–S bond lengths (approximately 0.044 Å for W and 0.052 Å for Mo) and the increase of the C=C bond lengths (approximately 0.022 Å for W and 0.026 Å for Mo) are also less pronounced for tungsten than for molybdenum. This is caused by the fact that the W–S bonds are already comparatively short in oxidation state +4 and that the stronger ligand folding, resulting in a stronger π–overlap between tungsten and sulfur, provides more electron density directly from the sulfur atoms and therefore less electron density is needed from the C=C bond.
The four Mo–S bonds of each compound are not identical, as is shown in detail in Table 1. In all cases, the decrease of the M–S bond lengths is stronger for the ligand that bends towards the metal center than for the ligand that bends away. This is caused by the fact that the ligand bending towards the metal shares more π-electron density with the metal owing to the better π overlap of the metal d orbitals with the sulfur p orbitals.
The four M–S bonds are most similar for complexes in oxidation state +4. The variation is smallest for the smallest ligand (edt; 1) and largest for the pdt ligand (2). In this oxidation state, the differences occur between the sulfur atoms of the same dithiolene ligand, while the M–S bond lengths for the sulfur atoms in trans position to each other are equal (edt 1, prz 3, mpt 4) or almost equal (pdt 2) (a trans trend).
In the +5 and +6 oxidation states, however, the M–S bond lengths of the sulfur atoms of the same dithiolene ligand are more or less identical (a cis trend), while there is a difference for the sulfur atoms in trans position to each other. This cis trend is caused by the ligand folding. The ligand that bends away from the metal and the ligand that bends towards the metal are different with respect to the possible overlap between metal d orbitals and sulfur p orbitals. The much better overlap between the metal and the ligand that bends towards the metal results in much shorter M–S distances for this ligand. The difference between the two ligands of each compound with respect to the M–S bonds is most evident in oxidation state +6: The differences are approximately 0.01 Å for the +5 oxidation state and approximately 0.03 Å for the +6 oxidation state. The differences are most pronounced for the smallest ligand (edt). For molybdenum, the average M–S bond lengths vary between 2.41 and 2.48 Å, whereas they show a slightly smaller range of 2.41 and 2.46 Å for tungsten (Fig. 6).
The smallest models (with edt) show the most pronounced differences from the mpt (4) compounds with respect to the M–S and C=C bond lengths (Fig. 6). The M–S bonds are longer and the C=C bonds are shorter owing to the fact that there is no possibility of a distribution of π-electron density to additional atoms of the ligand. The pdt compounds (2) mimic the behavior of the mpt compounds (4) quite well in oxidation states +4 and +5. However, in oxidation state +6, the difference from the mpt complexes is almost as large as that of the edt compounds (1). Accordingly, the most pronounced geometric difference between the different ligand systems is found in oxidation state +6 of the tungsten series.
The dependence of the M–S bond lengths on a change in the oxidation state of the metal for the mpt (4) compounds is best reproduced by the prz (3) compounds. The same applies for the C=C bond, for which the prz compounds reproduce not only the trends, but also the absolute values of the mpt ligand.
Electronic structure and bond analysis
In this section, we study various aspects of the electronic structure, e.g., the charges obtained by natural population analysis, the binding orbitals (natural bond analysis), and the redox-active orbitals (the highest occupied molecular orbitals, HOMOs, and the lowest unoccupied molecular orbitals, LUMOs). The natural population analysis charges for the metal, the apical oxo ligand, the two sulfur atoms of each of the two dithiolene ligands, and the doubly bonded carbon atoms of the two ligands are shown for the molybdenum compounds in Fig. 7. The quite similar graphs for the analogous tungsten compounds can be found in Fig. S1. The charge on the tungsten atom is approximately 0.02e higher than that on the molybdenum atom in all complexes. Again this is the result of the higher charge of its nucleus.

The natural population analysis (NPA) charges on molybdenum and the apical oxo ligand, as well as the average charge on the two sulfur atoms and the two ethene carbon atoms of each ligand
When the compounds are oxidized from oxidation state +4 to oxidation state +5, the charge on the metal increases as expected. But the oxidation to +6 causes the charge to decrease slightly, as a consequence of a quite effective electron density distribution from the ligands towards the metal. This behavior was found throughout all the complexes investigated. For example, the molybdenum charge is 0.55 for 1a IV, 0.67 for 1a V, and 0.57 for 1a VI with the edt ligand. In all complexes investigated, formally about 3.5, 4.5, and 5.5 electrons are transferred from the ligands onto the metal in oxidation states +4, +5, and +6, respectively.
The negative charges on the directly coordinated ligand atoms decrease regularly during the two oxidations. In fact, the charge on the four sulfur atoms even becomes positive in oxidation state +6. The total charge transfer from the four sulfur atoms onto the metal is therefore large. For the MoO(edt)2 complexes, the charge on the apical oxo ligand is −0.60 for 1a IV, −0.54 for 1a V, and −0.46 for 1a VI, and for the sulfur atoms of the ligand that bends away from the metal, the values are −0.24 for 1a IV, −0.10 for 1a V, and +0.11 for 1a VI. The charges on the sulfur atoms in trans position to each other, belonging to different ligands, are of course not equal upon oxidation. The ligand that achieves a better π overlap with the metal by bending towards it in oxidation states +5 and +6 donates more electron density onto the metal than the other ligand. Therefore, the increase in charge is larger for the sulfur atoms of this ligand than for the sulfur atoms of the opposite ligand. This corresponds to the M–S bond lengths with the trans trend in oxidation state +4 and the cis trend in oxidation states +5 and +6.
The results with the edt ligand are quite different from those for the other three ligand systems, especially for the carbon atoms. On the other hand, all data of the pdt (2), prz (3), and mpt (4) ligands are very similar, not only with respect to the trends, but also with respect to the absolute values. This is because the charge can be distributed throughout a much larger molecule for the latter three ligands, whereas in edt it has to stay at the carbon atoms of the double bond.
For the other three ligands, the charge on the oxygen atom in the pyrane ring is more or less identical (not shown in Fig. 7). The same is true for the nitrogen atoms of the pyrazine ring for prz (3) and mpt (4). Since the values of the relevant atoms for the three ligand systems with the pyrane ring are more or less equal, the influence of the pterin functional group on the natural population analysis charges is rather insignificant. In conclusion, the small edt ligand system is clearly not a proper model of the molybdopterin ligand.
To analyze the bond situation at the metal in more detail, a natural bond analysis of the natural localized molecular orbitals [79] was carried out. In Table 2, the most important parameters (the M–S bonds) are collected. A larger data overview can be found in Table S2. It should be noted that the electronic structure is strongly delocalized, so approximately 2% of the electrons cannot be assigned to a certain Lewis structure. Therefore, the assignment is somewhat ambiguous, especially for the sulfur atoms, for which the default analysis indicates that a lone pair can be converted to a π bond for one or two of the sulfur ligands in the oxidized +6 state. However, a nearly equally good assignment with two lone pairs on all sulfur atoms can always be obtained and this is shown in Table 2 to simplify the comparison.
The Lewis structures based on this analysis for all compounds investigated show a metal–oxygen triple bond. The metal–oxygen triple bond is not uncommon for complexes of this kind [80]. It consists of the usual σ and π bonds for a metal–oxygen double bond, plus another π bond provided by the oxygen through one of its “free” electron pairs.
The four sulfur atoms of the two dithiolene ligands are bound by σ bonds. The contribution of the metal to these σ bonds is for all complexes in all oxidation states in the range 15–27%, while the sulfur atoms contribute between 71 and 85%. The values for the metal–oxygen bond are in the same range (70–80% for oxygen and 20–30% for the metal). The metal is therefore surrounded by five atoms bound in a strongly polarized manner. The polarization decreases slightly in the course of oxidation. For instance, the contribution of the metal increases for the mpt compound (4a) from 20% in oxidation state +4 to 23–26% in oxidation state +5 and stays in this range in oxidation state +6 (18–27%), while the contributions of the sulfur atoms decrease accordingly (77% for oxidation state +4, but 72–73% for oxidation state +5 and 73–82% for oxidation state +6). This behavior was found for all the other complexes as well, although the numbers are slightly different. In general, the contribution of tungsten is marginally smaller (approximately 0.5%) than that of molybdenum and the contributions of the respective sulfur atoms are therefore larger by this same amount. Another difference between the two metals is the degree of metal s-orbital participation, which is slightly larger for tungsten (and the metal d-orbital participation is therefore slightly smaller). This is again related to the larger relativistic effects of tungsten.
In general, the prz ligand (3) gives results that are most similar to those of the mpt ligand (4), with a mean absolute difference in the metal and sulfur contributions to the M–S bonds of only 0.2% and a maximum difference of less than 1%. For the edt (1) and pdt (2) ligands, the differences are appreciably higher, on average 0.7 and 1.4% units. The differences are largest for the oxidized (+6) complexes. In particular, for the oxidized pdt model (2a VI), the molybdenum contribution is only 15–16%, whereas it is 18–27% for the corresponding mpt model (4a VI). This shows that the molybdopterin model has a quite strong influence of the electronic structure of the complex.
The redox behavior of the molecules investigated is to a large extent determined by the redox-active molecular orbitals, i.e., the HOMO in oxidation state +4, the singly occupied molecular orbital (SOMO) in oxidation state +5, and the LUMO in oxidation state +6. These orbitals would be involved in the reaction with a potential redox partner. The shape of the redox-active orbitals is shown in Fig. 8 and their composition in terms of metal and ligand participation is described in Table 3. In all cases, it is predominantly the metal \( d_{{x^{2} - y^{2} }} \) orbital that is involved (i.e., the orbital perpendicular to the oxo ligand and directed between the four M–S bonds). For the sulfur atoms, it is in most cases the p z orbital (directed out of the S–C=C–S plane) that is involved in the redox-active molecular orbital; for oxidation state +4 (i.e., for the HOMO), it is instead the p y orbital (located in the S–C=C–S plane). This is connected to the amount of metal participation: if the metal participation is high (more than 60%), the S p y orbitals are used. Without the bending of the ligand, which only occurs in oxidation states +5 and +6, and with the need for the transfer of electron density onto the metal, the orbital overlap is only sufficient for the in-plane p y orbital. Further participants in the frontier orbitals are the carbon atoms of the C=C bond (p z orbitals) and to a low degree (2%) the apical oxygen (p y orbital).

The highest occupied orbitals of all complexes in oxidation state +4
From Fig. 8 and Table 3, it can be seen that the smaller models have a larger contribution from the metal and the edt and pdt ligands still have a larger difference from the mpt ligand (by 19 and 13% units on average), compared with the prz ligand (8% units difference on average). For the sulfur contribution, the difference is smaller, but the trends are the same.
The size of the energetic gap in oxidation state +6 between the LUMO (former HOMO/SOMO) and the new HOMO (former HOMO-1) depends on the molybdopterin ligand model as well. For instance, it is 1.90 eV (Mo) and 2.23 eV (W) for the edt compounds (1) and is slightly smaller for the pdt compounds (2), 1.74 eV (Mo) and 2.07 eV (W). However, for the other two ligands, prz (3) and mpt (4), it is significantly smaller and the values are very similar to each other [0.61 and 0.65 eV (Mo) and 0.60 and 0.73 eV (W) for prz and mpt, respectively]. Again, the prz ligand (3) is a sufficient model for the mpt ligand (4), whereas the smaller ligands (1, 2) show considerable deviations.
The smaller gap between the HOMO and the LUMO for the complexes with the larger ligands means that they are easier to reduce than the smaller models. The reduction would be the relevant catalytic step for those enzymes that are oxidases and the regeneration step for those that are reductases. It is therefore an important part of the enzymatic catalysis. However, if a sixth CH3O− ligand is added to the oxidized complexes (cis to the oxy group), this difference almost disappears: then, the HOMO/LUMO gap is 1.93–2.05 eV for the molybdenum complexes, and 2.37–2.44 eV for the tungsten complexes (Table S3). These findings indicate that the molybdopterin ligand has in this respect an important influence on the active site of the arsenite oxidase [81] but, interestingly, does not have as much influence on the active sites of the majority of the members of the dimethyl sulfoxide reductase family of enzymes.
The impact on the metal without an additional ligand is nicely illustrated in Table 4, which shows the calculated redox potentials of the transition between oxidation states +4 and +5, which are those that are experimentally available and directly relevant for the enzymatic reactions in nature. The transition between oxidation states +5 and +6 (Table S4) can usually not be observed since the complexes and enzymes in oxidation state +6 contain a second oxo ligand. The MIV ↔ MV potentials calculated in water are negative and in the range from −0.10 to −0.14 V for the molybdenum compounds and are more negative for tungsten (−0.33 to −0.38 V). Experimental redox potentials in acetonitrile have been measured for [MoO(edt)2]2−/− [47] and [MoO(pdt)2]2−/− [48], −0.85 and −0.80 V. This is a reasonable deviation, considering the difference in solvents (the calculated potentials in diethyl ether are −1.38 and −1.35 V, respectively; Table S4). In particular, the sign and size of the difference between the two ligands is accurately reproduced. For both metals, the mpt ligand gives the most negative values, which differ by approximately 0.04–0.05 V from those for pdt and prz, and by approximately 0.02–0.03 V from those for edt. In the case of the redox potentials, the influence of the four different ligand systems on the complex properties is not very pronounced (up to 0.05 V), even though there are examples in the literature where different substituents on the dithiolene cause differences in redox potential as large as 1 V [18, 19]. From the calculation of the redox potentials and the HOMO–LUMO/SOMO gaps, it can be concluded that the differences in redox behavior that are to be expected for the four complexes with the same metal are more kinetic in nature (gap sizes between redox-active orbitals) than of thermodynamic origin since the redox potentials were calculated from the energetic differences of the reduced state and the oxidized state.
Conclusion
In this paper, we have studied how the model used for the molybdopterin ligand affects the properties of the active site. Our results show that there are quite extensive differences between the properties obtained with different models, especially with the smaller edt and pdt models. For the M–S bond lengths, the average difference between the edt and mpt ligands is 0.012 Å, whereas the difference is only 0.002 and 0.001 Å for the pdt and prz ligands, respectively. For the C=C bond lengths, the average differences (compared with mpt) are 0.004, 0.001, and 0.002 Å for the edt, pdt, and prz models, respectively. However, for the folding angles, all three ligand models give similar average differences of 0.8°, 1.5°, and 1.4°, respectively. The natural population analysis charges differ in the order edt > pdt > prz, with 0.034, 0.018, and 0.016 for the metals, 0.020, 0.017, and 0.013 for oxygen, 0.019, 0.012, and 0.006 for the carbon atoms, and 0.022, 0.003, and 0.002 for the sulfur atoms, showing the greatest differences for the edt compounds and the smallest for the prz compounds. The natural bond orbital analysis shows the largest differences for the edt and pdt ligands, especially for the oxidized state, with differences in the composition of the M–S orbitals of up to 11% units. For the HOMO–LUMO gap, the edt and pdt ligand models give a larger difference from the mpt model (0.04 a.u.) compared with the prz ligand (0.02 a.u.). Finally, for redox potentials, the differences are rather small and similar for all the three simplified models, approximately 0.03 V on average.
Another important observation is that the changes of the bond lengths and folding angles when the oxidation state of the metal is changed are always smallest for the mpt (4) compounds. This could be of significance for the catalytic performance of the active sites of the enzymes for instance resulting in a lower reorganization energy. This ligand also gives smaller differences between molybdenum and tungsten, indicating that the normally observed quite large differences between these two metals may be less significant in the enzymes.
In conclusion, we believe that great care is needed in the selection of the model of the molybdopterin ligand both in theoretical and in experimental investigations if properties depending on this non-innocent ligand should be obtained that accurately represent the native metal site in the proteins and that the slightly smaller pyrazine dithiolene ligand would be an excellent model for most characteristics.
References
Enemark JH, Cooney JJ, Wang J-J, Holm RH (2004) Chem Rev 104:1175–1200. doi:10.1021/cr020608l
Sigel A, Sigel H (eds) (2002) Metal ions in biological systems: molybdenum and tungsten: their roles in biological processes, vol 39. Marcel Dekker, New York
Lorber C, Donahue JP, Goddard CA, Nordlander E, Holm RH (1998) J Am Chem Soc 120:8102–8112. doi:10.1021/ja981015o
Stiefel E I (1997) J Chem Soc Dalton Trans 3915–3923
Hille R (1996) Chem Rev 96:2757–2816. doi:10.1021/cr950061t
Johnson MK, Rees DC, Adams MWW (1996) Chem Rev 96:2817–2839. doi:10.1021/cr950063d
Arion V, Wieghardt K, Weyhermüller T, Bill E, Leovac V, Rufinska A (1997) Inorg Chem 36:661–669. doi:10.1021/ic960802o
Jüstel T, Bendix J, Metzler-Nolte N, Weyhermüller T, Nuber B, Wieghardt K (1998) Inorg Chem 37:35–43. doi:10.1021/ic970850o
Chaudhuri P, Nazari Verani C, Bill E, Bothe E, Weyhermüller T, Wieghardt K (2001) J Am Chem Soc 123:2213–2223. doi:10.1021/ja003831d
Ray K, Weyhermüller T, Neese F, Wieghardt K (2005) Inorg Chem 44:5345–5360. doi:10.1021/ic0507565
Ray K, Begum A, Weyhermüller T, Piligkos S, van Slageren J, Neese F, Wieghardt K (2005) J Am Chem Soc 127:4403–4415. doi:10.1021/ja042803i
Ray K, Bill E, Weyhermüller T, Wieghardt K (2005) J Am Chem Soc 127:5641–5654. doi:10.1021/ja040237l
Bachler V, Olbrich G, Neese F, Wieghardt K (2002) Inorg Chem 41:4179–4193. doi:10.1021/ic0113101
Ray K, Weyhermüller T, Goossens A, Crajé MWJ, Wieghardt K (2003) Inorg Chem 42:4082–4087. doi:10.1021/ic0207092
Petrenko T, Ray K, Wieghardt K, Neese F (2006) J Am Chem Soc 128:4422–4436. doi:10.1021/ja0578451
Patra AK, Bill E, Weyhermüller T, Stobie K, Bell Z, Ward MD, McCleverty JA, Wieghardt K (2006) Inorg Chem 45:6541–6548. doi:10.1021/ic060665n
Patra AK, Bill E, Bothe E, Chlopek K, Neese F, Weyhermüller T, Stobie K, Ward MD, McCleverty JA, Wieghardt K (2006) Inorg Chem 45:7877–7890. doi:10.1021/ic061171t
Lim BS, Donahue JP, Holm RH (2000) Inorg Chem 39:263–273. doi:10.1021/ic9908672
Maiti R, Nagarajan K, Sarkar S (2003) J Mol Struct 656:169–176
Amano T, Ochi N, Sato H, Sakaki S (2007) J Am Chem Soc 129:8131–8138. doi:10.1021/ja068584d
Pal K, Chaudhury PK, Sarkar S (2007) Chem Asian J 2:956–964
Waters T, Wang X-B, Yang X, Zhang L, O’Hair RAJ, Wang L-S, Wedd AG (2004) J Am Chem Soc 126:5119–5129. doi:10.1021/ja039652o
Thapper A, Deeth RJ, Nordlander E (2002) Inorg Chem 41:6695–6702. doi:10.1021/ic020385h
Wang J-J, Groysman S, Lee SC, Holm RH (2007) J Am Chem Soc 129:7512–7513. doi:10.1021/ja0724442
Hoffmann M (2007) J Biol Inorg Chem 12:989–1001. doi:10.1007/s00775-007-0271-5
Fritscher M, Hrobárik P, Kaupp M (2007) Inorg Chem 46:8146–8161. doi:10.1021/ic070341e
Tenderholt AL, Szilagyi RK, Holm RH, Hodgson KO, Hedman B, Solomon EI (2007) J Inorg Biochem 101:1594–1600
McNamara JP, Hillier IH, Bhachu TS, Garner CD (2005) Dalton Trans 3572–3579. doi: 10.1039/b507206a
Joshi HK, Enemark JH (2004) J Am Chem Soc 126:11784–11785. doi:10.1021/ja046465x
Baba K, Okamura T, Suzuki C, Yamamoto H, Yamamoto T, Ohama M, Ueyama N (2006) Inorg Chem 45:894-901. doi: 10.1021/ic051493h
Sugimoto H, Tarumizu M, Tanaka K, Miyake H, Tsukube H (2005) Dalton Trans 3558–3565. doi:10.1039/b503828f
Baba K, Okamura T, Yamamoto H, Yamamoto T, Ohama M, Ueyama N (2005) Chem Lett 34:44–45
Wang K, McConnachie JM, Stiefel EI (1999) Inorg Chem 38:4334–4341. doi:10.1021/ic990204k
Oku H, Ueyama N, Nakamura A (1997) Inorg Chem 36:1504–1516. doi:10.1021/ic951277g
Oku H, Ueyama N, Kondo M, Nakamura A (1994) Inorg Chem 33:209–216
Coucouvanis D, Iladjikyriacou A, Toupadakis A, Koo S-M, Ileperuma O, Draganjac M, Salifoglou A (1991) Inorg Chem 30:154–161
Boyde S, Ellis S R, Garner C D, Clegg W (1986) J Chem Soc Chem Commun 1541–1543. doi:10.1039/C39860001541
Davies ES, Aston GM, Beddoes RL, Collison D, Dinsmore A, Docrat A, Joule JA, Wilson CR, Garner CD (1998) J Chem Soc Dalton Trans 3647–3656. doi:10.1039/a805688i
Davies ES, Beddoes RL, Collison D, Dinsmore A, Docrat A, Joule J A, Wilson CR, Garner CD (1997) J Chem Soc Dalton Trans 3985–3996. doi:10.1039/a703682e
Götz B, Knoch F, Kisch H (1996) Chem Ber 129:33–37
Matsubayashi G-E, Nojo T, Tanaka T (1988) Inorg Chim Acta 154:133–135
Ueyama N, Oku H, Nakamura A (1992) J Am Chem Soc 114:7310–7311. doi:10.1021/ja00044a061
Das SK, Biswas D, Maiti R, Sarkar S (1996) J Am Chem Soc 118:1387–1397. doi:10.1021/ja9511580
Baba K, Okamura T, Yamamoto H, Yamamoto T, Ohama M, Ueyama N (2006) Inorg Chem 45:8365–8371. doi:10.1021/ic060719t
Sung K-M, Holm RH (2001) Inorg Chem 40:4518–4525. doi:10.1021/ic010421x
Goddard CA, Holm RH (1999) Inorg Chem 38:5389–5398. doi:10.1021/ic9903329
Donahue JP, Goldsmith CR, Nadiminti U, Holm RH (1998) J Am Chem Soc 120:12869–12881. doi:10.1021/ja982914f
Sugimoto H, Harihara M, Shiro M, Sugimoto K, Tanaka K, Miyake H, Tsukube H (2005) Inorg Chem 44:6386–6392. doi:10.1021/ic050234p
Ueyama N, Oku H, Kondo M, Okamura T, Yoshinaga N, Nakamura A (1996) Inorg Chem 35:643–650. doi:10.1021/ic950204u
Das SK, Chaudhury PK, Biswas D, Sarkar S (1994) J Am Chem Soc 116:9061–9070. doi:10.1021/ja00099a024
Yoshinaga N, Ueyama N, Okamura T, Nakamura A (1990) Chem Lett 1655–1656
Oku H, Ueyama N, Nakamura A (1996) Bull Chem Soc Jpn 69:3139–3150
McNaughton RL, Lim BS, Knottenbell SZ, Holm RH, Kirk ML (2008) J Am Chem Soc 130:4628–4636. doi:10.1021/ja074691b
Sung K-M, Holm RH (2002) J Am Chem Soc 124:4312–4320. doi:10.1021/ja012735p
Lim BS, Holm RH (2001) J Am Chem Soc 123:1920–1930. doi:10.1021/ja003546u
Groysman S, Holm RH (2007) Inorg Chem 46:4090–4102. doi:10.1021/ic062441a
Wang J-J, Tessier C, Holm RH (2006) Inorg Chem 45:2979–2988. doi:10.1021/ic0521630
Jiang J, Holm RH (2005) Inorg Chem 44:1068–1072. doi:10.1021/ic040109y
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2004) Gaussian 03, revision C.02. Gaussian, Wallingford
Glendening ED, Reed AE, Carpenter JE, Weinhold F (1988) J Mol Struct 169:41–62
Carpenter JE, Weinhold F (1988) J Mol Struct 169:41–62
Foster JP, Weinhold F (1980) J Am Chem Soc 102:7211–7218
Reed AE, Weinhold F (1985) J Chem Phys 83:1736–1740
Reed AE, Curtiss LA, Weinhold F (1988) Chem Rev 88:899–926
Becke AD (1993) J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785–789
Hay PJ, Wadt WR (1985) J Chem Phys 82:299–310
Petersson GA, Al-Laham MA (1991) J Chem Phys 94:6081–6090
McLean AD, Chandler GS (1980) J Chem Phys 72:5639–5648
Krishnan R, Binkley JS, Seeger R, Pople JA (1980) J Chem Phys 72:650–654
Tomasi J, Mennucci B, Cancès E (1999) J Mol Struct (Theochem) 464:211–226
Kelly CP, Cramer CJ, Truhlar DG (2006) J Phys Chem 110:16066–16081
Schindelin H, Kisker C, Hilton J, Rajagopalan KV, Rees DC (1996) Science 272:1615–1621
Romao MJ, Archer M, Moura I, Moura JJG, LeGall J, Engh R, Schneider M, Hof P, Huber R (1995) Science 270:1170–1176
Ma B, Lii J-H, Schaefer HFIII, Allinger NL (1996) J Phys Chem 100:8763–8769
Lauher JW, Hoffmann R (1976) J Am Chem Soc 98:1729–1742
Cooney JJA, Cranswick MA, Gruhn NE, Joshi HK, Enemark JH (2004) Inorg Chem 43:8110–8118
Lim BS, Formitchev DV, Holm RH (2001) Inorg Chem 40:4257–4262
Reed AE, Weinhold F (1985) J Chem Phys 83:1736–1740
McNaughton RL, Helton ME, Rubie ND, Kirk ML (2000) Inorg Chem 39:4386–4387
Ellis PJ, Conrads T, Hille R, Kuhn P (2001) Structure 9:125–132
Acknowledgments
We gratefully acknowledge generous financial support by the DFG (International Research Training Group IRTG 1422 “Metal Sites in Biomolecules: Structures, Regulation and Mechanisms” and project SCHU 1480/3-1). The investigation has also been supported by computer resources of Lunarc at Lund University.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Ryde, U., Schulzke, C. & Starke, K. Which functional groups of the molybdopterin ligand should be considered when modeling the active sites of the molybdenum and tungsten cofactors? A density functional theory study. J Biol Inorg Chem 14, 1053–1064 (2009). https://doi.org/10.1007/s00775-009-0548-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-009-0548-y