
Abstract
The antioxidant activity of ortho- and meta-substituted indolin-2-one derivatives was investigated in the gas phase and water. The reaction enthalpies of the individual steps of three antioxidant mechanisms of the studied derivatives were calculated and compared with the corresponding values of indolin-2-one. The results show that electron-withdrawing substituents increase the bond dissociation enthalpy and ionization potential, whereas electron-donating substituents increase the proton affinity. The indolin-2-one derivatives with the lowest bond dissociation enthalpy, ionization potential, and proton affinity values were identified as the compounds with high antioxidant activity. The results show that indolin-2-one derivatives with substituents in the ortho position are promising potential novel antioxidants. The results also show that the protective role of indolin-2-one derivatives occurs via hydrogen atom transfer and a sequential proton loss electron transfer mechanism in the gas phase and water, respectively. The calculated reaction enthalpies of the substituted indolin-2-ones are linearly dependent on the Hammett constants and E HOMO such that the latter can be utilized in the selection of suitable substituents for the synthesis of novel antioxidants based on indolin-2-one.
Graphical abstract

Similar content being viewed by others

Introduction
Excess formation of reactive oxygen species (ROS) by various enzymatic and nonenzymatic processes in living organisms is associated with the oxidation of DNA, proteins, and lipids. ROS play an important role in the initiation and progression of various diseases such as atherosclerosis, cancer, cardiovascular disease, neurodegenerative disease, and aging [1]. Antioxidants help cells to cope with oxidative stress by effectively quenching free radicals. The indole framework is a medicinally relevant scaffold and has become widely identified as a target structure or pharmacophore. The indole scaffold is present in thousands of isolated natural products and also medicinal (synthetic) compounds with diverse therapeutic activity. In animals, the most significant indolic compound is indolin-2-one (Fig. 1) [2]. Although indolin-2-one can be added to the diet without limitation, an indication of the required amount is necessary [3]. Indolin-2-one often produces an initial burst of pro-oxidant activity. Some oxidation products of indolin-2-one are shown to be powerful antioxidants as determined from measurements by the Warburg manometric technique [4]. Indolin-2-one has been shown to impose protective effects in oxidant stress-mediated tissue injury in vivo. Indolin-2-one as a hydrophilic antioxidant possesses a high capacity to modulate the inflammatory cascade triggered by oxidative stress and to improve altered angiogenesis which is central to the management of diabetic foot ulcer [4]. Indolin-2-one has been reported to act by different modes such as protection and inhibition of low density lipoprotein (LDL) lipid peroxidation, reduction of myocardial infarction as a promising drug for cardiac ischemia and reperfusion, stimulation of wound healing in genetically diabetic mice, reduction of the glutamate-induced oxidative effects, and antiproliferative effects in rat aortic smooth muscle cells in vivo [5].

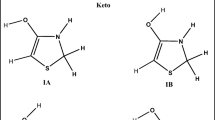
Structure of indolin-2-one and studied ortho- and meta-substituted indolin-2-ones: X = Br, Et, CH=CH2, CCH, CF3, Cl, CN, COMe, COH, COOH, F, NMe2, NHMe, NH2, NO2, OMe, OH, Ph, t-Bu
The indolic antioxidants (ArNH) inhibit oxidation by transferring their indolic H atom to a chain-carrying peroxyl radical (ROO·) at a rate much faster than that of chain propagation [6]. This yields a non-radical product (ROOH) that cannot propagate the chain reaction. It is proposed that chain-breaking antioxidants exert their protective role via two major mechanisms. The first involves a direct hydrogen atom transfer (HAT) from the antioxidant to the free radical (ROO·) and yields a non-radical product (ROOH) that cannot propagate the chain reaction:
For HAT, the N–H bond dissociation enthalpy (BDE) represents one of the important parameters in the evaluation of antioxidant action [7]. The second mechanism, single electron transfer followed by proton transfer (SET–PT), takes place in two steps:
In the first step, ArNH·+ radical cation is formed (Eq. 2.1). In the second step, deprotonation of ArNH+· occurs (Eq. 2.2). Here, indolin-2-one acts as an electron donor and yields an indolyl radical cation. The ionization potential (IP) and proton dissociation enthalpy (PDE) describe the energetics of the SET–PT mechanism [8].
Recently, a third mechanism of primary antioxidant action has been discovered. This two-step mechanism was named sequential proton loss electron transfer (SPLET):
The reaction enthalpy of the first step (Eq. 3.1) corresponds to the proton affinity (PA) of the ArN− anion [9]. In the second step (Eq. 3.2) electron transfer from the ArN− anion to ROO· occurs and the indolyl radical is formed. From the antioxidant action viewpoint, the net result of the SPLET mechanism is the same as in the two previously mentioned mechanisms—the transfer of the hydrogen atom to the free radicals. Although reaction enthalpies (BDE, IP, PA) related to three mechanisms are of importance in evaluating the antioxidant action, other criteria, including solubility, bioavailability, and nontoxicity must also be considered when designing an effective and safe antioxidant.
The biological implications and the great potential of indolin-2-ones as an antioxidant aroused our interest in elucidating its antioxidant activity by means of density functional theory (DFT)/B3LYP calculations, which have been successfully used for a variety of antioxidants [10, 11]. The substituent effect is one of the important structural effects influencing the chemical, physicochemical, and biochemical properties of chemical species [12, 13]. In recent years many experimental and theoretical investigations have been carried out on phenol and mono-substituted phenols in the gas phase and the solvent environment [14–17]. Theoretical studies of the substituent effect on the antioxidant activity of indolin-2-one can be utilized in the synthesis of substances with enhanced antioxidant properties. Various substituents such as electron-withdrawing groups (EWG) and electron-donating groups (EDG) can be located in the ortho and meta positions on the aromatic ring of indolin-2-one (Fig. 1). In the present paper the substituent effect on reaction enthalpies of homolytic (HAT mechanism) and heterolytic two-step (SPLET and SET–PT) mechanisms of N–H bond cleavage for mono-substituted indolin-2-ones were investigated in the gas phase and water. Indolin-2-one is a powerful water-soluble antioxidant; therefore, in this study water was chosen as the main cell environment in order to assess the substituent effect on the aforementioned enthalpies in solution. Also the correlations of calculating enthalpies with corresponding E HOMO values and Hammett constants of substituents were investigated.
Thus, the main aims of this work are (1) to find novel indolin-2-one derivatives with high antioxidant activity from a theoretical point of view; (2) to identify the substituents that are the most effective in reducing the BDE, IP, and PA; (3) to compare the effects of the same substituent in ortho and meta positions on reaction enthalpies; (4) to explore how the polar solvents alter the reaction enthalpies of the three mechanisms of the studied derivatives; (5) to find which mechanism is preferred from a thermodynamic point of view in the gas phase and solvent; (6) to identify the dependences between reaction enthalpies and structural parameters for the studied derivatives.
Results and discussion
The total enthalpies of the studied species X, H(X), at the temperature T are usually estimated from Eq. (4):
From the calculated total enthalpies we determined the following quantities:
The calculated gas phase enthalpy of the proton, H(H+), and electron, H(e−), is 6.197 and 3.145 kJ mol−1, respectively. The enthalpy of H+ hydration is −1,090 kJ mol−1. The B3LYP/6-311++G** computed electron hydration enthalpy, Δhydr H(e−) = −105 kJ mol−1, has been employed in this paper [22–24]. It is known that potential inaccuracies related to computed solvation enthalpies of electrons, protons, and hydrogen atoms will be cancelled when the substituent effect is studied as the difference in the reaction enthalpies of substituted and unsubstituted indolin-2-one, i.e., in terms of ΔBDE, ΔPA, and ΔIP. On the other hand, BDE, PA, and IP values allow the determination of the preferred reaction pathway in the studied solvent and gas phase [25–30].
Ionization potentials in gas phase and water
This paper represents the first theoretical systematic study of substituted indolin-2-one IP values. In previous studies [17, 26] the substituent effect on IPs of para- and meta-substituted phenols and pyridinethiols were investigated in the gas phase employing the B3LYP approach. No systematic study of the solvent effect of substituted indolin-2-ones on IPs and ∆IPs has been published yet. In this paper, the calculated IP for indolin-2-one in the gas phase reached 651 kJ mol−1. The computed gas phase IPs and ∆IP = IP(X−ArNH) − IP(ArNH) for substituents in ortho and meta positions are reported in Table 1.
In ortho and meta positions, the highest IP values were found for strong EWG substituents (NO2, CF3, and CN); the lowest IPs were obtained for strong EDG substituents (NMe2, NH2, and NHMe). For F, Cl, and Br in ortho and meta positions, the IP values are higher in comparison to indolin-2-one. For meta- and ortho-substituted indolin-2-ones with OH and OMe, IP values are lower in comparison to indolin-2-one. For meta- and ortho-substituted indolin-2-ones with COH, COOH, and COMe, IP values are higher in comparison to indolin-2-one. The difference between the highest and lowest IP values for meta and ortho positions was 92 and 141 kJ mol−1, respectively.
EWG substituents are known to stabilize the parent molecule and destabilize the radical and radical cation, resulting in an increase in IP; however, EDG substituents have the opposite effect, i.e., their presence in the molecule leads to a decrease in IP. The decreases in IPs (negative ∆IPs) of EDG-substituted indolin-2-ones are the combined result of the cation radical stabilization and the parent molecule destabilization. However, the increased IPs (positive ∆IPs) of EWG-substituted indolin-2-ones may stem from the combination of both the parent and the cation radical destabilization. These results are in accordance with data published for substituent phenols [17, 24].
The computed IPs using the aforementioned calculated Δhydr H(e−) value of water for substituted indolin-2-ones in the meta and ortho positions are reported in Table 1. Table 1 summarizes ΔIP values, too. In water, the determination of IP requires the value of the electron hydration enthalpy, Δhydr H(e−). The B3LYP/6-311++G** computed electron hydration enthalpy, Δhydr H(e−) = −105 kJ mol−1, has been employed in this paper [24]. Potential inaccuracies related to the employed electron hydration enthalpy value will be cancelled when the substituent effect is expressed in terms of ΔIPs. Water causes considerable changes in the enthalpies of the molecule and cation radical of the studied indolin-2-ones. In this paper, the calculated IP for indolin-2-one in water reached 423 kJ mol−1.
For strong EDGs, i.e., NMe2, NH2, and NHMe, in meta and ortho positions, we found a drop in IP values. For meta and ortho indolin-2-ones substituted with an NO2 group the IP value is higher than the corresponding value of indolin-2-one. In water, substituent-induced changes are lower than those observed in the gas phase. The EWG substituents stabilize the parent molecule and destabilize the radical cation. On the other hand, EDG substituents have the opposite effect. Water causes attenuation of substituent effects in terms of a narrower ΔIP range. Again, substituents in ortho positions exert a stronger influence upon IP than the same substituents in the meta position. Calculated IPs related to substituted indolin-2-ones in water are lower than the corresponding values in the gas phase. Mainly, owing to the negative enthalpy of electron hydration in water, the IP is significantly lower than that in the gas phase.
For substituted indolin-2-ones in the ortho position the substituents exert a significantly stronger influence upon IP than in the meta position in the gas phase and water. These results stem from the fact that the radical cation derived from the electron abstraction from any of the ortho-substituted indolin-2-ones can be stabilized by the electron donation of the ortho hydroxyl and intramolecular hydrogen bond formation (e.g., with NH2, NHMe, halogen groups). Alternatively, if the substituent can act as a hydrogen bond donor to the indolic nitrogen atom the corresponding indolyl radical cation may be stabilized relative to the parent structure with a consequent decrease in its IP. Therefore, owing to the potential formation of hydrogen bonds in the ortho position, substituents in the ortho position have a stronger influence upon IP (ca. 3–40 kJ mol−1) in comparison to the same substituent in the meta position. The overall results of the calculations of IP can be summarized by the fact that EWG-substituted indolin-2-ones with higher IPs may exhibit weaker antioxidant activity compared to EDG ones in the gas phase and water. The obtained results in this work are in agreement with previous studies on substituted phenols and chromans [17, 18, 24].
Proton affinities in gas phase and water
PA represents the reaction enthalpy of the first step in the SPLET mechanism. PAs of substituted indolin-2-ones have not been obtained by theoretical calculations previously. In previous studies [25, 26] the substituent effect on PAs of para- and meta-substituted phenols and pyridinethiols were investigated by DFT using B3LYP functional in the gas phase.
The present calculated PA for indolin-2-one reached 1,422 kJ mol−1 in the gas phase. The computed PAs and ∆PAs for the various substituents in the ortho and meta positions of indolin-2-one in the gas phase are reported in Table 2. The highest values of PA for ortho and meta positions were found for NMe2, NH2, and NHMe groups. The lowest PA values of these positions were found in the case of NO2, CF3, and CN groups. The F, Cl, and Br groups in ortho and meta positions cause decrease the PA in comparison to indolin-2-one. For indolin-2-ones with COH, COOH, and COMe groups in the meta and ortho positions, PA values are lower than the PA value of indolin-2-one.
The differences between the highest and lowest gas phase PA values for ortho and meta substituents were 129 and 71 kJ mol−1, respectively. In agreement with previous studies on substituted phenols [15–17] it can be concluded that EDG substituents increase PA, whereas EWG substituents decrease PA. It is known that a charged molecule is more sensitive to the effect of a substituent than its neutral counterpart. EWG substituents stabilize ArN− but destabilize the parent structures. EDGs have the opposite effect [27–29].
For PA calculations in water, the proton hydration enthalpy, Δhydr H(H+), is required. Therefore, we have utilized Δhydr H(H+) = −1,022 kJ mol−1 [24]. The present calculated PA for indolin-2-one reached 241 kJ mol−1 in water. For indolin-2-ones substituted in meta and ortho positions, computed PAs and ∆PAs in water are reported in Table 2.
Again, EWG substituents decrease PAs, whereas EDG groups increase PAs in agreement with results from substituted phenols in water [17]. Again, strong electron-donating NMe2, NH2, and NHMe cause an increase in PA. The presence of COH, COOH, and COMe groups in ortho and meta positions results in decreased PA. Halogens in ortho and meta positions cause a drop in PAs. The largest decrease in PA is for indolin-2-one with an NO2 group in ortho or meta positions. Differences between the highest and lowest PA values for the two studied positions were 89 (ortho) and 60 kJ mol−1 (meta).
Water causes considerable changes in the enthalpies of anions. Calculated PAs related to substituted indolin-2-ones in water are lower than the corresponding values in the gas phase. Mainly, owing to the large negative enthalpy of proton hydration, PAs in water are significantly lower than gas phase values.
It confirms that water attenuates substituent-induced changes. For substituted indolin-2-ones in the ortho position substituents exert significantly a stronger influence upon PA than in the meta position in the gas phase and water. These results stem from this fact that the anion derived from the proton abstraction from any of the ortho-substituted indolin-2-ones can be stabilized by the EWG substituent. Alternatively, if the substituent can act as a hydrogen bond donor to the indolic nitrogen atom the corresponding indolyl anion may be stabilized relative to the parent structure with a consequent decrease in its PA.
Therefore owing to the potential formation of hydrogen bonds in the ortho position, substituents in the ortho position have a stronger influence upon PA (ca. 5–35 kJ mol−1) in comparison to the same substituent in the meta position. The obtained results of this work are in agreement with previous studies on substituted phenols and chromans [17, 28, 29].
Bond dissociation enthalpies in gas phase and water
The bond dissociation enthalpy is a very important characteristic of a molecule that usually refers to the standard thermodynamic state in the gas phase. When these thermodynamic quantities are known they become an invaluable tool for the calculation of activation energies and rate constants of homolytic reactions. In many of the previous studies, experimental and theoretical methods were used to investigate the effect of various substituents on the BDE values of phenols and other natural phenolic antioxidants. In previous studies [15–17] the solvent and substituent effects on O–H BDEs substituted phenols were investigated.
No systematic study of the solvent effect of substituted indolin-2-ones on BDEs and ∆BDEs has been published yet. The N–H BDE value of indolin-2-one is ca. 371 and 355 kJ mol−1 in the gas phase and water, respectively. In this section we investigated the substituent and solvent effects of indolin-2-one on BDE. The computed gas phase BDE and ∆BDE values, where ∆BDE = BDE (X−ArNH) − BDE (ArNH), for substituents placed in the ortho and meta positions (Fig. 1) are reported in Table 3. The EDGs decrease BDE values, whereas EWGs increase the BDE values. The N–H BDE of structures with NO2, COH, COOH, and COMe substituents in ortho and meta positions was higher than the BDE value of indolin-2-one. For ortho- and meta-substituted indolin-2-ones with NMe2, NH2, and NHMe the BDE values are lower than the BDE value of indolin-2-one; for those with halogen substituents the BDE values are higher than that of indolin-2-one; for those with OMe and OH substituents the BDE values are lower than that of indolin-2-one. The difference between the highest and lowest BDE values for ortho- and meta-substituted indolin-2-ones was 89 and 51 kJ mol−1, respectively. The obtained results can be interpreted in light of the fact that EWGs in ortho and the meta positions stabilize the parent molecule and destabilize the radical; hence, they increase the N–H BDE. However, EDGs in ortho and meta positions have the opposite effect; therefore, their presence leads to a decrease in the N–H BDE.
Klein et al. [17] indicated that the PCM method could describe the substituent effect in very good agreement with experimental data for O–H BDEs of substituted phenols in water. The calculated BDE for indolin-2-one in water is 16 kJ mol−1 lower than the corresponding value in the gas phase. The computed BDEs and ΔBDEs in water for ortho- and meta-substituted indolin-2-ones are reported in Table 3. For ortho- and meta-substituted indolin-2-ones with NMe2, NH2, and NHMe the BDE values are lower in comparison to indolin-2-one; for those with halogen substituents the BDE values are higher in comparison to indolin-2-one; for those with COH, COOH, and COMe the BDE values are higher than the BDE value of indolin-2-one. The difference between the highest and lowest BDE values in water for ortho and meta positions were 78 and 32 kJ mol−1, respectively. In water, EDGs decrease BDE values, whereas EWGs increase BDE values similar to the results for the gas phase. In comparison to the gas phase, the effects of various substituents including EDGs and EWGs on the BDEs in ortho and meta positions were decreased in the water phase. Therefore, we can conclude that in comparison to the gas phase, the effect of EDG substituents and EWG substituents on BDE in ortho and meta positions decreases in water.
An inspection of the N–H BDE values in Table 3 shows that in water the calculated BDE values were lower than gas phase ones. Overall the results reveal a weak dependence of BDEs values on the solvent polarity. Water causes changes in the enthalpies of the molecule and radical of the studied structures. Since water causes unequal stabilization/destabilization of the parent molecule and the respective radical this can be a fundamental reason for the obtained results.
Therefore the decrease in BDEs (negative ∆BDEs) for EDG-substituted indolin-2-ones is the combined result of the radical stabilization and the parent molecule destabilization. However, increased BDEs (positive ∆BDEs) for EWG-substituted indolin-2-ones seem to be due to the combined destabilization of both the parent and the radical.
In the present study computed results for substituted indolin-2-ones show that substituents in ortho positions exert a significantly stronger influence on N–H BDE than substituents in the meta position in the gas phase and water. It can be concluded that some of the ortho-substituted indolin-2-ones can form hydrogen bonds and this can have a marked effect on the reaction enthalpies of ortho-substituted indolin-2-ones. If the substituent can act as a hydrogen bond donor to the indolic nitrogen atom (e.g., o-NH2 group) the corresponding indolyl radical may be stabilized relative to the parent structure with a consequent decrease in its N–H BDE. Therefore, owing to the potential formation of hydrogen bonds in the ortho position, substituents in the ortho position have a stronger influence upon BDE (ca. 3–30 kJ mol−1) in comparison to the same substituent in the meta position. The overall results of the calculations of N–H BDE can be summarized by the fact that EWG-substituted indolin-2-ones with higher BDEs may exhibit weaker antioxidant activity in comparison to EDG ones in the gas phase and water. The obtained results in this work are in agreement with previous studies on substituted phenols, chromans, and other natural phenolic antioxidants [16, 25].
Thermodynamically preferred mechanism
In general, a negative Gibbs free energy represents the main criterion for a thermodynamically preferred process. However, in the case of the studied reactions the absolute values of the entropic term −TΔr S reach few a tens of kilojoules per mole and all free energies, Δr G = Δr H − TΔr S, are only shifted in comparison to corresponding enthalpies [24]. Therefore, comparison of BDEs, PAs, and IPs can indicate which mechanism is thermodynamically preferred. Calculated gas phase IPs and PAs of mono-substituted indolin-2-ones are significantly higher, by 281 and 1,048 kJ mol−1, than BDEs, respectively. Therefore, the HAT mechanism represents the most likely process in the gas phase from a thermodynamic point of view. In water, the PA values are lower than the BDE and the IP values by 112 and 187 kJ mol−1, respectively. In water, the IP values remain still higher than the BDEs by ca. 60 kJ mol−1. Significantly lower PAs indicate that SPLET represents the thermodynamically preferred reaction pathway in water.
Dependence of calculated reaction enthalpies of indolin-2-one derivatives on the Hammett constants
The Hammett equation (and its extended forms) has been one of the most widely used means for the study and interpretation of organic reactions and their mechanisms. Hammett constants σ m (for a substituent in the meta position) and σ p (for a substituent in the para position) obtained from the ionization of organic acids in solutions can frequently successfully predict equilibrium and rate constants for a variety of families of reactions [12, 13]. Hammett constants correlate very well with the changes in the BDE, IP, and PA values in the case of anilines, phenols, chromans, and thiophenols [17, 27]. In the present paper we investigated the dependences between reaction enthalpies of meta-substituted indolin-2-ones and corresponding Hammett constant values. The presence of linear dependences can be utilized in the synthesis of novel indolin-2-one derivatives with high antioxidant activity. The BDE, IP, and PA values computed for the meta-substituted indolin-2-ones in the gas phase and water are corrected against Hammett constants. The equations obtained from the linear regressions and the correlation coefficients in the gas and water are reported in Table 4. We can conclude that the DFT method describes the expected linear dependence of BDE, IP, and PA values on Hammett constants satisfactorily. The obtained equations enable the rapid estimation of BDE, IP, and PA for meta-substituted indolin-2-ones from the corresponding Hammett constant values. This can be useful in the selection of suitable candidates for the synthesis of novel indolin-2-one derivatives with enhanced antioxidant properties.
Dependence of calculated reaction enthalpies of indolin-2-one derivatives on corresponding E HOMO
To accelerate the discovery of novel antioxidants, considerable effort has been devoted to investigating the structure–activity relationships (SARs) for antioxidants. Furthermore, rational design strategies for antioxidants have been proposed and applied in research. It was shown that IPs determined using the DFT computational approach are sufficiently accurate to characterize the electron-donating ability of antioxidants [30]. The energy of the highest occupied molecular orbital (E HOMO) represents an alternative parameter to assess the electron-donating ability of antioxidants. This is widely used in antioxidant studies because of the simple calculation procedure, where only the calculation for the parent molecule is required. In this paper, the E HOMO values found for indolin-2-one in the gas phase and water are −5.17 and −5.72 eV, respectively. As a general rule, the higher the E HOMO, the more active the compound is as an antioxidant. The computed E HOMO values of the investigated indolin-2-ones in the gas phase and water are summarized in Table 5. These reveal that in the case of EWG substituents, E HOMO values become more negative, whereas the presence of EDG substituents results in less negative E HOMO values. Therefore, indolin-2-ones with strong EDGs are better electron donors, i.e., they participate in the SET–PT mechanism more easily. A previous study showed that the B3LYP/6-311+G (2d, 2p) method significantly underestimates vertical gas phase ionization potentials obtained from E HOMO for mono-substituted anilines, phenols, and thiophenols [30]. However, the trends in ionization potentials, in terms of ΔIPs, are described reliably. Therefore, we expected to find linear dependence between the calculated IPs and the corresponding E HOMO values. The equations obtained from the linear regressions and the correlation coefficients in the gas and water are reported in Table 4. From the obtained equations we can conclude that E HOMO can be employed for the rapid estimation of reaction enthalpies for the first step of the SET–PT mechanism. These linear dependences can be utilized in the selection of suitable substituents for the synthesis of novel indolin-2-one derivatives with enhanced antioxidant activity.
Conclusions
In this article, the reaction enthalpies of the individual steps of three antioxidant action mechanisms, HAT, SET–PT, and SPLET, for various ortho- and meta-substituted indolin-2-ones were calculated in the gas phase and water. The obtained results indicate that electron-withdrawing substituents increase the bond dissociation enthalpy (BDE) and ionization potential (IP), whereas electron-donating ones cause a rise in the proton affinity (PA). Water attenuates the substituent effect on all reaction enthalpies. The results show that the indolin-2-ones with substituents in the ortho position are promising potential novel antioxidants. In the gas phase, BDEs are lower than PAs and IPs, i.e., HAT represents the thermodynamically preferred pathway. On the other hand, the SPLET mechanism represents the thermodynamically favored process in water. The results show that calculated enthalpies can be successfully correlated with Hammett constants (σ m) of the substituted indolin-2-ones. It has been also found that IP values for substituted indolin-2-ones can be estimated from their E HOMO values. This fact may be useful for the development of novel indolin-2-one-based antioxidants.
Computational details
The geometries of the derivations and respective radicals, radical cations, and anions were optimized using the DFT method with B3LYP functional and the 6-31G (d, p) basis set in the gas phase and solution phase. Single point calculations were performed using the 6-311++G (2d, 2p) basis set [18]. The ground-state geometries of the structures were optimized at the restricted B3LYP level and the geometry of the radicals, radical cations, and anions were optimized at the restricted B3LYP open shell (half electron) level. The optimized structures were confirmed to be real minima by frequency calculation. For the species having more conformers, all conformers were investigated. The conformer with the lowest electronic energy was used in this work. All reported enthalpies were zero point energy (ZPE) corrected with unscaled frequencies. The solvent contributions to the total enthalpies were computed employing the isodensity polarizable continuum model (IPCM) method [19, 20]. All calculations were performed using the Gaussian 98 program package using 10−6 a.u. as the criterion for energy and gradient convergence [21].
References
Thorisson S, Gunstone F, Hardy R (1992) J Am Oil Chem Soc 69:806
Sambasiva M, Narendra D, Thomas K (1984) Cancer Res 44:1072
Benson AM, Cha YN, Bueding E, Heine HS, Talalay P (1996) Cancer Res 39:2971
Cabrai JRP, Neal GE (1983) Cancer Lett 79:125
Chance B, Sies H, Boveris A (1979) Physiol Rev 59:527
Wright JS, Johnson ER, Dilabio GA (2001) J Am Chem Soc 123:1173
Zhang HY, Ji HF (2003) J Mol Struct (Theochem) 663:167
Foti MC, Daquino C, Geraci C (2004) J Org Chem 69:2309
Litwinienko G, Ingold KU (2005) J Org Chem 70:8982
Wang LF, Zhang HY (2005) Bioorg Chem 33:108
Klein E, Lukes V, Ilcin M (2007) Chem Phys 336:51
Krygowski TM, Steupien BT (2005) Chem Rev 105:3482
Hansch C, Leo A, Taft RW (1991) Chem Rev 91:165
Klein E, Lukes V (2006) J Mol Struct (Theochem) 767:43
Denisov ET (1995) Polym Degrad Stab 49:71
Mulder P, Korth H, Pratt DA, Dilabio GA, Valgimigli L, Pedulli GF, Ingold KU (2005) J Phys Chem A 109:2647
Klein E, Rimarcik J, Lukes V (2009) Acta Chim Slov 2:37
Hehre WJ, Radom L, Schleyer PVR, Pople JA (1986) Ab initio molecular orbital theory. Wiley, New York
Miertus S, Scrocco E, Tomasi J (1981) Chem Phys 55:117
Cossi M, Rega N, Scalmani G, Barone VJ (2003) Comput Chem 24:669
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (1998) Gaussian 98. Gaussian, Pittsburgh
Bartmess JE (1994) J Phys Chem 98:6420
Bizarro MM, Costa Cabral BJ, Borges dos Santos RM, Martinho Simões JA (1999) Pure Appl Chem 71:1249
Rimarcik J, Lukes V, Klein E, Ilcin M (2010) J Mol Struct (Theochem) 952:25
Najafi M, Nazarparvar E, Haghighi Mood K, Zahedi M, Klein E (2011) Comput Theoret Chem 965:114
Nam PC, Nguyen MT, Chandra AK (2006) J Phys Chem A 110:10904
Bordwell FG, Zhang XM, Satish AV, Cheng JP (1994) J Am Chem Soc 116:6605
Brinck T, Haeberline M, Jonsson M (1997) J Am Chem Soc 119:4239
Fu Y, Liu L, Guo Q-X (2004) J Phys Org Chem 17:282
DiLabio GA, Pratt DA, Wright JS (2000) J Org Chem 65:2195
Acknowledgments
I would like to thank from Dr. Toraj Miri for valuable discussions about the computational affairs.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Najafi, M. On the antioxidant activity of ortho- and meta-substituted indolin-2-one derivatives. Monatsh Chem 145, 291–299 (2014). https://doi.org/10.1007/s00706-013-1099-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-013-1099-z