
Abstract
Background
Recent studies have highlighted the importance of the complete resection of a brain tumour but the task often remains a challenge for the neurosurgeon. New technologies which add objective information beyond visualisation provided by the traditional operating microscope are required. In this study, we have analysed the cellular density of the tumour/brain interface using three dimensional multi-photon microscopy intensity-images of experimental gliomas and human brain-tumour biopsy samples.
Methods
The density of cellular nuclei was determined in specimens of experimental gliomas in a mouse model and human brain tumour biopsies by analysis of optical tissue sections. Three dimensional multi-photon microscopy image stacks were compared to serial H&E stained sections of conventional histolopathology.
Findings
Both techniques consistently showed a good correlation of cell density values in solid tumour tissue of experimental gliomas versus adjacent brain. The multi-photon microscopy analysis of human biopsy specimens showed that optical analysis of native tissue provided information on the cellular density.
Conclusions
Multi-photon microscopy is an efficient and rapid tool for the study of brain and brain tumour tissue. Multi-photon microscopy allows the detection of individual tumour cells and tumour cell clusters in native tissue biopsies and may therefore provide a tool in the identification of highly cellular lesions during the resection of brain tumours.
Similar content being viewed by others


Introduction
Malignant glioma remains one of the most feared diagnoses. This can partly be attributed to their invasive nature and intrinsic resistance to radiation and chemotherapy [6]. In spite of these difficulties recent developments in local and systemic chemotherapy agents are beginning to improve the prognosis of glioma patients [5, 29, 32]. Furthermore, the extent of resection has been identified as an important prognostic factor in the treatment of gliomas [9, 16]. This has prompted new interest in techniques facilitating intra-operative detection of residual tumour, which facilitates more radical resection of intrinsic brain tumours. The aim of these efforts is however not the resection of individual tumour cells in functionally intact brain-tissue but the identification of highly cellular residual tumour which is not detected by the conventional operating microscope.
Intra-operative detection of 5-aminolevulinic acid-induced porphyrins in malignant brain tumour tissue has shown to delineate tumour and normal brain [1, 28]. Recent prospective clinical trials have demonstrated that fluorescence-guided resection of brain tumours increased the extent of resection and may therefore prolong survival [26, 27]. However, it has also become clear that uptake of 5-aminolevulinic acid or protoporphyrin IX formation may vary within different areas of tumour and that specifically the fluorescence signal of invaded brain and adjacent brain tissue may be difficult to interpret [27, 31]. Nevertheless, these studies highlight that intra-operative detection of residual tumour emerges as a significant factor improving the impact of surgery on survival.
We have recently evaluated optical technologies that do not rely on the metabolic processing of extrinsic markers for analysis of glioma tissue. Optical coherence tomography (OCT) uses the interference of low-coherent light to generate two- and three dimensional images of tissue based on the inherent optical tissue contrast. OCT allows discrimination of solid tumour, tumour invading adjacent brain and normal brain tissue [2]. A clinical pilot study has demonstrated that this technology may detect residual tumour during the resection of malignant brain tumours and that OCT can be integrated into operating microscopes and neuro-endoscopes [3, 17, 24]. However, operating at a resolution of 10–30 µm OCT does not provide subcellular visibility.
Multi-photon excitation microscopy (MPE microscopy) is based on the excitation of endogenous intra- and extracellular fluorophores by two or more near-infrared femtosecond laser pulses, which focus simultaneously into a femtoliter target volume [15]. The two-photon excited auto-fluorescence (TPEAF) of native tissue is recorded by a photomultiplier. Fluorescence intensity images or images of the fluorescence lifetime distribution may be reconstructed at subcellular resolution without the need for tissue labelling or fluorescent markers. MPE microscopy has been applied to dermatological pathologies and normal human skin in vivo [22, 23]. In a neurosurgical setting multi-photon-excitation microscopy may facilitate the analysis of tumour cell density at the resection edge. Recently we have demonstrated that multi-photon microscopy allows structural imaging of human brain tumour and central nervous system histo-architecture at a subcellular level in unprocessed tissue blocks ex vivo. Solid tumour, the tumour–brain interface, and single invasive tumour cells could be visualised [19]. The analysis of the excitation/lifetime profiles of the fluorescent signal within specific excitation volumes discriminated glioma cells and normal brain [8].
In this study, we have analysed the cellular density of the tumour/brain interface using three dimensional multi-photon microscopy intensity-images of experimental gliomas and human brain-tumour biopsies. We demonstrate that automated-analysis of multi-photon microscopy intensity-images may be used to analyse the cellular density of unprocessed biopsy specimens taken from the edge of resection during the resection of malignant gliomas.
Material and methods
Multi-photon microscopy imaging
Multi-photon excitation images were obtained using a DermaInspect in-vivo imaging system (JenLab, Jena, Germany). The system contains a solid-state, mode-locked 80 MHz titanium:sapphire laser (MaiTai, Spectra Physics, Darmstadt, Germany) with a tuning range of 710–920 nm, a mean laser output of >900 mW at 800 nm, and a 75 fs pulse width. All measurements were performed at 750 nm, which has been determined optimal for imaging of native brain and brain-tumour tissue in previous studies [8, 19]. The scanning module contains a motorised beam attenuator, a shutter, and a two axis galvoscanner. A piezodriven 40× focussing optic (z-axis) with NA 1.3 and 140 µm working distance (Plan Neofluar 40X, 1.3, Zeiss, Göttingen, Germany) was used. Motions of the sample in x- and y-directions were performed by computer-controlled stepper-motors (Owis, Staufen, Germany). The signal of the fluorophores was detected by a photo-multiplier tube module (H7732-01, Hamamatsu, Herrsching, Germany), after passing a beam splitter (Chroma 725DCSPXR, AHF analysentechnik AG, Tübingen, Germany) and a short-pass filter (BG39, Schott, Mainz, Germany).
Orthotopic glioma mouse model, tumour specimen and histopathology
The human glioblastoma derived cell lines G-112 and U87 were grown in minimal essentail medium (MEM) containing 10% fetal calf serum (FCS). For intracranial implantation in nude Naval Medical research Institute (NMRI) mice cells were expanded and harvested in log phase growth by trypanisation. Cells were washed in phosphate buffered saline (PBS) three times and were resuspended at a concentration of 2 × 104 /µl. All procedures were performed in accordance with regulations of the Animal Care and Use Committee of the University Hospital of Schleswig-Holstein under permit No. 30/o/03. Prior to the implantation animals were anaesthetised by peritoneal injection of ketamine/xylazine solution (200 mg ketamine and 20 mg xylazine in 17 ml of saline) at 0.15 mg/10 g of body weight. For the procedure, the cranium was fixed in a stereotactic frame (TSE Systems, Bad Homburg, Germany). A 1 mm burr hole was placed 3 mm lateral to the bregma and a stereotactic implantation of 3 µl cell suspension injected over 3 min was placed in an area corresponding to the internal capsule 0.5 mm below the fibre tracts of the corpus callosum. Following implantation, 50 mg/kg novaminsulfone was administered subcutaneously and 1 mg/ml novaminsulfone was added to the drinking water for 3 days. In the following 4 weeks the highly infiltrative cell line (G-112) and a less infiltrative cell line (U-87) grew low angiogenic tumours to the size convenient for imaging studies (approximately 5 mm in diameter).
Tumour bearing brains were explanted following a lethal intraperitoneal injection of 50 mg/kg xylazine and 350 mg/kg ketamine. Coronal sections of the mouse brains were cut immediately and the tissue samples were placed in a humidified chamber (MiniCeM, JenLab, Jena Germany) adherent to a 0.17 µm cover glass for imaging.
Human biopsy specimens were taken during neurosurgical procedures for routine diagnostic procedures. The biopsy-sites of solid tumour, edge of the tumour, or adjacent brain were registered using a VectorVision 2 neuronavigation system (Brain Lab, Hettenheim, Germany). Biopsy specimens were immediately subjected to multi-photon microscopy. Following the imaging studies all specimens were formalin fixed, paraffin embedded and 5 µm histolopathological sections were cut parallel to the optical plane of the multi-photon microscopy imaging. Sections were processed for H&E and CD-68 staining.
Evaluation of cell density in experimental gliomas and human tumour biopsies
The cellular density in tissue specimens was evaluated based on multi-photon microscopy images and corresponding conventional histolopathology. Multi-photon microscopy image scans of 30 × 30 µm (900 µm2) were acquired as stacks of 10 consecutive images at a gap width of 5 µm along the z-axis (Fig. 1). The number of cells/scan was determined using ImageJ 1.36 (http://rsb.info.nih.gov/ij), by adjusting a threshold according to the brightness and contrast of the tissue, only identifying nuclei as objects. These objects were than automatically counted. For a computer based cell count of H&E stained tissue sections light-microscopy images at a magnification of 100x were digitised using a CCD camera (DFC 480 Photoscanner in combination with the DM IRB microscope, Leica, Wetzlar, Germany). Images were saved as RGB images in jpeg format for analysis using ImageJ. An RGB-split of the colour graphics was performed and an intensity based threshold was manually adjusted according to the tissue contrast in the red channel. An automated cell count was performed for a 30 × 30 µm area of the image. The analysis was performed for 10 consecutive 5 µm sections.

a Schematic presentation of multi-photon microscopy of native brain and brain tumour tissue. b Coronal section of a tumour bearing mouse brain illustrating the analysis of consecutive image stacks from microscopic fields along an axis from the tumour center to normal brain. The upper image shows normal brain, the lower image shows a highly cellular region of the tumour centre. Multi-photon microscopy images were acquired as stacks of 10 consecutive images. The cell number was determined by a computer based cell count. The overlay shows the segmented nuclei of a selected area
3-Dimensional reconstruction of the tumour–brain interface
Three-dimensional reconstruction of the multi-photon microscopy image stacks was done using software developed by the OpenQVis project [25], a collaborative research project of the Computer Graphics Group at the University of Erlangen-Nuernberg and the VIS Group at the University of Stuttgart, Germany, available under GNU public license (http://openqvis.sourceforge.net). The OpenQVis project implements methods for interactive high-quality volume visualisation for general purpose hardware achieving high image quality comparable to traditional ray-casting solutions. In order to use this software, the stacks of 2D images recorded in greyscale TIF format were converted to ASCII format PMG using freeware graphics IrfanView. The stacks of PNG files were converted into the proprietary binary 3D data format required by the OpenQVis. After creation of the corresponding annotation files that contain the pixel sizes the data can be directly loaded into the OpenQVis software. This software allows the definition of transfer functions used to assign visual properties (colour and opacity) to the 3D pixels (voxels) of the volume data set. These parameters were adjusted manually. The tumour tissue, appearing brighter on the MPE-images, remained visible while the surrounding brain tissue became transparent. Thus the 3-dimensional topography of the tumour–brain barrier in an intact tissue volume block could be visualised.
Results
Analysis of the cell density at the tumour/brain interface in experimental gliomas
The cellular density was analysed at the tumour/brain interface of two human glioblastoma derived cell lines (G-112 and U87) which served as an orthotopic glioma mouse model. In mouse brain these cell lines show different patterns of invasive growth. The non-invasive cell line U-87 has been demonstrated to rapidly form large tumours with a well defined tumour/brain interface. The highly invasive cell line G-112 shows single cell invasion following myelinated fibre tracts and vascular structures but also invasive cell clusters projecting into adjacent brain [8, 19].
Multi-photon microscopy discriminated tumour adjacent to brain and brain tumour in specimens implanted with U87 or G-112 tumour cells similar to conventional histolopathology (Fig. 2). Normal brain tissue showed a low density of cellular nuclei and a homogenous intensity image of the brain parenchyma. Compact fibre tracts may produce image signals of varying intensity depending on the orientation of tracts. Multi-photon microscopy of tumours derived from the two cell lines showed a high density of low intensity cellular nuclei surrounded by high intensity cytoplasm. Frequently, the high intensity cytoplasm of tumour cells appeared granular. Morphologically, distinct clusters of cells within the tumours could be identified (Fig. 2 upper panels). Multi-photon microscopy showed the brain/tumour interface of U-87 neoplasms being well defined with solid tumour tissue immediately adjacent to brain tissue, which showed low cellularity. G-112 tumours showed a highly cellular solid tumour and clusters of invasive tumour cells protruding into the brain parenchyma (Fig. 2 lower panels). In order to quantify the cellular density of the tumour/brain interface the number of cellular nuclei was analysed based on MPE-images and corresponding histolopathological sections. According to the growth pattern of the cell lines the analysis of the cellular density of the corresponding tissue volumes showed an abrupt decrease of the cellular density at the tumour/brain interface of the non-invasive U87 tumours, whereas the invasive cell line G-112 showed a continuous decrease from the highly cellular solid tumour to adjacent brain (Fig. 3). The absolute numbers of the cell density determined from MPE-images or histological sections cannot be directly compared, because of a different gap width and overlap of the optical and histolopathological tissue section. The profiles of the cellular density based on multi-photon microscopy images or histolopathological evaluation were not different. Multi-photon microscopy image stacks were acquired from a 50 × 30 × 30 µm tissue volume which allowed the three dimensional reconstruction of the tumour/brain interface (Fig. 3). Assigning low opacity to the low intensity signal of the brain parenchyma allowed a three dimensional display of the high intensity signal of the tumour cells. This analysis showed the tumour/brain interface of U87 tumours to be well defined, whereas G-112 tumours showed a diffuse tumour/brain interface with invasive cell clusters protruding into the tissue volume corresponding to adjacent brain parenchyma.

Coronal sections of U87 and G-112 tumour bearing mouse brains were scanned by multi-photon microscopy along an axis from tumour centre to adjacent brain. The multi-photon microscopy images of native tissue are shown above with corresponding H&E stained histolopathological images of the specimen below. The tumour/brain interface in U-87 neoplasms appeared well defined, whereas in G-112 derived tumours clusters of invasive cells and single invasive tumour cells were found in tumour adjacent brain

Three-dimensional reconstruction of the multi-photon microscopy image stacks obtained from the tumour/brain interface of U87 and G-112 experimental gliomas in mouse brain. Three dimensional images were reconstructed by assigning visual properties (colour and opacity) to the 3D pixels (voxels) of the volume data set (left panel). The graphs show a plot of the density of cellular nuclei at the tumour/brain interface from consecutive microscopic fields obtained by multi-photon microscopy or based on the analysis of H&E stained histolopathological sections. While the absolute values of the nuclear density determined on MPE images or by conventional histopathology cannot be compared (see “Discussion”) the relative numbers of nuclei reflect a well defined tumour–brain interface for U87 and the presence of an invasion zone for G112 lesions
Evaluation of human glioma biopsy specimens by multi-photon microscopy
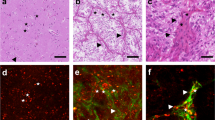
Biopsy specimens of tumour tissue and from the edge of the resection were taken during neurosurgical procedures for diagnostic purposes. A tissue volume of 1 mm3 of each biopsy was subjected to TPEAF imaging and then submitted to processing for routine diagnostic histolopathology. Pre-operative MRI data was used for intra-operative neuronavigation, which also served to register and document the biopsy sites. Similar to our findings in experimental gliomas multi-photon microscopy showed solid tumour as a high density of low intensity cellular nuclei and high intensity cytoplasmic areas (Fig. 4). No biopsies of normal human brain were available for this analysis, but tumour adjacent brain from the edge of the resection showed similar multi-photon imaging characteristics to normal murine brain. However, in human specimens of solid tumour, tumour necrosis as well as tumour adjacent brain, cells with a large number of cytoplasmic granules of very high autofluorescence intensity were a frequent finding. These cells were also present in specimens, for example, taken from the resection edge that histolopathologically showed few or no tumour cells. The specific morphology of these comparably large cells with multiple granula led to the assumption that these might be monocytes. Cells of this type were a common finding in human tumour samples and adjacent brain tissue. In order to confirm this, staining with a surface-marker specific for monocytes (CD-68) was performed. Histolopathologically, CD-68 positive cells were found in a similar distribution and frequency and showed similar histo-architecture. Another difference between human and murine brain adjacent to tumour was the presence of round low intensity amorphous structures up to 8 µm observed in human biopsies on the MPE-images (Fig. 4, pannel 2). Whether these correlate to brain oedema remains undecided in this study. Multi-photon microscopy intensity images of 10 consecutive optical sections were used to determine the number of cellular nuclei in biopsy specimens from solid tumour and the edge of resection (Fig. 5). Following the optical tissue analysis the specimen were processed for routine histolopathology and H&E staining. Consecutive sections of a corresponding tissue volume were used to quantify cellular nuclei. Multi-photon microscopy and routine histolopathology resulted in similar cell counts and similar standard deviations for determination of cell numbers in consecutive sections (Fig. 5). Table 1 shows the determinations of cell number by multi-photon microscopy from specimens taken during surgery for two metastatic brain tumours and three malignant gliomas. Tumour specimens taken from areas corresponding to gadolinium contrast enhancing regions demonstrated a high cellular density (2.8–4.8/900 µm2) by multi-photon microscopy. Based on multi-photon intensity images tumour cells from metastatic and primary brain tumours showed similar imaging characteristics. Specimens analysed from the central necrosis of both metastatic and primary brain tumours showed none or few cells (with the exeption of cells with granules of high autofluorescence). Specimens of tumour adjacent brain showed a low cellular density corresponding to tumour-free margins confirmed by routine histopathology (0.7–2.4/900 µm2). A single specimen taken from a region of suspected residual tumour showed a high cellular density similar to specimens taken from contrast enhancing tumour. Both frozen section and routine histolopathology confirmed solid tumour at the resection edge of this specimen.

In vitro imaging of a brain metastasis from a bronchial carcinoma. The images in the upper panel show the neuronavigation registered biopsy sites on MRI-scans (tumour outlined white). One biopsy was taken from areas of contrast enhancing solid tumour and the others were obtained from adjacent brain. The panel below shows the corresponding TPEAF images. The cell density of the tumour biopsy exceeds the values for adjacent brain. In contrast to images of tumour bearing mouse brain, all TPEAF images of human tumour samples showed a large number of cells with highly autofluorescent intracellular cytoplasmic granula. The corresponding conventional H&E staining is shown below. The lowest panel shows the immuno-histochemistry for the monocyte marker CD68, demonstrating the presence of CD68 positive cells in human biopsies, suggesting that the nature of the cells with highly autofluorescent granula may be monocytes

Quantitative analysis of the density of cellular nuclei in tumour samples and biopsies obtained from the edge of the resection cavity. Cell counts were based on H&E stained histological sections and on multi-photon images of 900 µm2 microscopic fields. The mean number of nuclei and standard deviation was determined from 30 fields of consecutive images
Discussion
MPE microscopy offers the unique possibility to image three-dimensional tissue structures at the limits of optical microscopy and allows structural and photochemical imaging of native tissue at subcellular resolution [8, 13–15, 19, 22]. Both the structural imaging of the histo-architecture and the biochemical sensitive imaging of the fluorescence lifetime of endogenous fluorophores may be useful in the in vivo detection of brain tumours. Beyond exogenous fluorophores, the auto-fluorescence of native tissue can produce a sufficient fluorescence signal. The restriction of the optical excitation to a femtoliter region provides three-dimensional optical sectioning and avoids out of focus photo damage. The fluorescence is recorded by a photomultiplier and fluorescence intensity images or images of the fluorescence lifetime distribution may be reconstructed [15]. We have recently shown that multi-photon microscopy allows detailed imaging of the normal brain histo-anatomy in native mouse brain [19]. Based on autofluorescence intensity images the histo-architecture of cortex, white matter and myelinated fibre tracts within the basal ganglia or the ependyma can be readily identified [8]. Experimental gliomas on multi-photon intensity images can be identified because of their high cellularity and high autofluorescence of the tumour cell cytoplasm contrasting with the low signal intensity nuclei. This allowed identification of single invasive tumour cells in invasive experimental gliomas in mouse brain [19]. Here we demonstrate that a systematic and quantitative analysis of the tumour/brain interface in experimental gliomas allowed conclusions on the degree of invasion of tumour adjacent brain based on MPE microscopy visualisation of the density of cellular nuclei. Based on the intensity images and nuclear counts per tissue volume the non-invasive U87 tumour could be delineated from the highly invasive G-112 tumours. Strictly speaking, the cell count analysis of MPE images was not performed on a volume data set, but on a stack of adjacent images. A real 3D reconstruction of the volumes, based on a stack width of 5 µm between individual images of 1.5 µm excitation depth, would have added arithmetical errors without providing additional information. In this respect, the cell number determined from MPE images cannot be identical to results obtained by conventional histolopathology, In biopsies taken from human malignant gliomas the density of tumour cells i.e. the degree of tumour infiltration could be estimated based on the same parameters [19]. In this study, we have extended these experiments to a quantitative analysis of the cellular density in specimens taken from human brain tumours and biopsies from the edge of the resection cavity. Our data show that multi-photon microscopy allowed the determination of cell density in native tissue biopsies similar to cell counts obtained from formalin fixed and H&E stained tissue sections. However, while in biopsy specimens obtained from human brain tumours the density of nuclei could be determined, the identification of individual tumour cells remained difficult, because of cellular elements of different histotypes within the tissue volume. For example, in several specimens including tumour adjacent brain, cells of high autofluorescence with prominent cytoplasmic granules were found. These cell types increased the cell count per tissue volume, but were unlikely to represent tumour cells. The signal intense subcellular structures may also result from lysosomal degradation within ex vivo biopsy material but the morphology of these cells made this unlikely. CD-68 staining of these tissue-samples identified cells in a similar distribution to the highly fluorescing cells in the multi-photon intensity images. This supports the hypothesis that these cells were monocytes, which is also suggested by their morphology. Further, the human tumour samples underwent similar processing compared to the experimental tissue obtained from immuno-deficient NMRI mice, which lacked these cell types. The problem of unidentified cell types within specimens may potentially be overcome by analysis of fluorescence lifetimes of endogenous fluorophores. The analysis of the excitation wavelength to fluorescence lifetime showed characteristic autofluoresence lifetime profiles for cells derived from different tissues, suggesting that fluorescence lifetime spectroscopy may differentiate histotypes of cells based on the excitability of cell type specific endogenous chromophores, their chemical states, or their interaction with other biomolecules [13, 14, 19, 30].
Furthermore, experimental glioma tissue showed significantly longer fluorescence lifetimes than normal cortex or white matter [8]. Characterisation of the fluorescence lifetime versus excitation profiles of biopsies from human gliomas demonstrated that the degree of malignancy tended to correlate positively with fluorescence lifetimes [8]. This suggests that the photochemical analysis may offer means for specific detection of residual tumour in biopsy specimens, which may increase the diagnostic value of this technique. We have further demonstrated that the specificity of tumour cell detection by multi-photon microscopy may be significantly increased by detection of 5-ALA induced PpIX fluorescence in experimental gliomas [7]. The fluorescence lifetime analysis of induced PpIX fluorescence by multi-photon microscopy demonstrated that PpIX fluorescence was significantly prolonged compared to fluorescence of endogenous fluorophores. In contrast to variable fluorescence lifetimes of endogenous fluorophores in different tumour specimens, the long lifetime of 5-ALA induced PpIX fluorescence allowed identification of the tumour/brain interface using constant detection parameters, which in the future may facilitate automated detection of tumour and normal brain. Therefore, intra-operative in vivo multi-photon microscopy of brain tissue conceptually provides a high resolution non-invasive diagnostic tool, which in addition to structural information may also offer functional information. The imaging setup used for this study has been developed for dermatological applications and allows subcellular imaging of human skin in vivo [14]. However, for neurosurgical intracerebral applications such a device may not be useful because the movement of the target tissue which follows the respiratory and arterial cycles does not allow high numerical aperture non-contact imaging. This limitation may be overcome by technical developments of multi-photon microscopy scanner probes connected to optic fibres, which have recently facilitated in vivo imaging of the mouse central nervous system over extended periods of time [10, 11, 18]. König et al. have further demonstrated that multi-photon microscopy can be performed through optical rods [12]. However, the device used for this study does allow fast optical serial biopsies during the operation. This might provide additional important information to the operating neurosurgeon to enhance the advantages of the present state of technical development. The analysis of samples produces images with a resolution comparable to conventional H&E staining without the time delay for tissue processing.
Such probes may be placed in direct contact to the target tissue and may offer solutions to the specific requirements of intra-operative high resolution optical brain imaging in the future. TFEAF investigations in animal models and biopsy tissues may also be valuable in the development and verification of optical tumour diagnosis based on large field autofluorescence imaging or spectral resolved detection of backscattered light [4, 20, 21].
Conclusion
Multi-photon microscopy is an efficient and rapid tool for the study of brain and brain tumour tissue. Multi-photon microscopy allows the detection of individual tumour cells and tumour cell clusters in native tissue biopsies and may therefore provide a tool in the identification of highly cellular neoplasms during the resection of brain tumours.
References
Bogaards A, Varma A, Collens SP, Lin A, Giles A, Yang VX, Bilbao JM, Lilge LD, Muller PJ, Wilson BC (2004) Increased brain tumour resection using fluorescence image guidance in a preclinical model. Lasers Surg Med 35(3):181–190
Böhringer HJ, Boller D, Leppert J, Knopp U, Lankenau E, Reusche E, Hüttmann G, Giese A (2006) Time-domain and spectral-domain optical coherence tomography in the analysis of brain tumour tissue. Lasers Surg Med 38(6):588–597
Böhringer HJ, Lankenau E, Rohde V, Hüttmann G, Giese A (2006) Optical coherence tomography for experimental neuroendoscopy. Minim Invasive Neurosurg 49(5):269–275
Bottiroli G, Croce AC, Locatelli D, Nano R, Giombelli E, Messina A, Benericetti E (1998) Brain tissue autofluorescence: an aid for intra-operative delineation of tumour resection margins. Cancer Detect Prev 22:330–339
Giese A, Kucinski T, Knopp U, Goldbrunner R, Hamel W, Mehdorn HM, Tonn JC, Hilt D, Westphal M (2004) Pattern of recurrence following local chemotherapy with biodegradable carmustine (BCNU) implants in patients with glioblastoma. J Neurooncol 66(3):351–360
Giese A, Bjerkvig R, Berens ME, Westphal M (2003) Cost of migration: invasion of malignant gliomas and implications for treatment. J Clin Oncol 21(8):1624–1636
Kantelhardt SR, Diddens H, Leppert J, Pettkus N, Rohde V, Hüttmann G, Giese A (2008) Multi-photon excitation fluorescence microscopy of 5-aminolevulinic acid induced fluorescence in experimental gliomas. Laser Surg Med 40:273–281
Kantelhardt SR, Leppert J, Krajewski J, Petkus N, Reusche E, Tronnier VM, Huttmann G, Giese A (2007) Imaging of brain and brain tumour specimens by time-resolved multi-photon excitation microscopy ex vivo. Neuro-oncol 2:103–112
Keles GE, Lamborn KR, Berger MS (2001) Low-grade hemispheric gliomas in adults: a critical review of extent of resection as a factor influencing outcome. J Neurosurg 95:735–745
Kim D, Kim KH, Yazdanfar S, So PTC (2004) High-speed handheld multi-photon multifoci microscopy. Multi-photon microscopy in the biomedical science IV, at Biomedical Optics 2001, San Jose, CA. SPIE Proc 5323:267–272
Kim KH, Buehler C, So PTC (1999) High-speed, two-photon scanning microscope. Appl Opt 38:6004–6009
König K, Ehlers A, Riemann I, Schenkl S, Buckle R, Kaatz M (2007) Clinical two-photon microendoscopy. Microsc Res Tech 70:398–402
König K, Schenke-Layland K, Riemann I, Stock UA (2005) Multi-photon autofluorescence imaging of intratissue elastic fibres. Biomaterials 26:495–500
König K, Rieman I (2003) High-resolution multi-photon tomography of human skin with subcellular spatial resolution and picosecond time resolution. J Biomed Optics 8:432–439
König K (2000) Multi-photon microscopy in life science. J Microsc 200:83–104
Lacroix M, Abi-Said D, Fourney DR, Gokaslan ZL, Shi W, DeMonte F, Lang FF, McCutcheon IE, Hassenbusch SJ, Holland E, Hess K, Michael C, Miller D, Sawaya R (2001) A multivariate analysis of 416 patients with glioblastoma multiforme: prognosis, extent of resection, and survival. J Neurosurg 95:190–198
Lankenau E, Klinger D, Winter C, Malik A, Müller HH, Oelckers S, Pau HW, Just T, Hüttmann G (2007) Combining Optical Coherence Tomography (OCT) with an operating microscope. In: Buzug TM et al (ed) Advances in medical engineering. Springer, Berlin, pp 343–348
Lee WC, Huang H, Feng G, Sanes JR, Brown EN, So PT, Nedivi E (2006) Dynamic remodeling of dendritic arbors in GABAergic interneurons of adult visual cortex. PLoS Biol 4(2):e29
Leppert J, Krajewski J, Kantelhardt SR, Schlaffer S, Petkus N, Reusche E, Huttmann G, Giese A (2006) Multi-photon excitation of autofluorescence for microscopy of glioma tissue. Neurosurgery 58(4):759–767
Lin WC, Toms SA, Motamedi M, Jansen ED, Mahadevan-Jansen A (2000) Brain tumour demarcation using optical spectroscopy; an in vitro study. J Biomed Opt 5:214–220
Majumder SK, Gebhart S, Johnson MD, Thompson R, Lin WC, Mahadevan-Jansen A (2007) A probability-based spectroscopic diagnostic algorithm for simultaneous discrimination of brain tumour and tumour margins from normal brain tissue. Appl Spectrosc 61:548–557
Masters BR, So PT, Gratton E (1998) Multi-photon excitation microscopy of in vivo human skin. Functional and morphological optical biopsy based on three-dimensional imaging, lifetime measurements and fluorescence spectroscopy. Ann N Y Acad Sci 838:58–67
Masters BR, So PT, Gratton E (1997) Multi-photon excitation fluorescence microscopy and spectroscopy of in vivo human skin. Biophys J 72:2405–2412
Pau HW, Lankenau E, Just T, Behrend D, Hüttmann G (2007) Optical coherence tomography as an orientation guide in cochlear implant surgery? Acta Otolaryngol 127(9):907–913
Rezk-Salama C, Engel K, Bauer M, Greiner G, Ertl T (2000) Interactive Volume Rendering on Standard PC Graphics Hardware Using Multi-Textures and Multi-Stage Rasterisation; in Proc. Eurographics SIGGRAPH Workshop on Graphics Hardware 2000
Stummer W, Pichlmeier U, Meinel T, Wiestler OD, Zanella F, Reulen HJ, ALA-Glioma Study Group (2006) Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: a randomised controlled multicentre phase III trial. Lancet Oncol 7(5):392–401
Stummer W, Novotny A, Stepp H, Goetz C, Bise K, Reulen HJ (2000) Fluorescence-guided resection of glioblastoma multiforme by using 5-aminolevulinic acid-induced porphyrins: a prospective study in 52 consecutive patients. J Neurosurg 93(6):1003–1013
Stummer W, Stepp H, Möller G, Ehrhardt A, Leonhard M, Reulen HJ (1998) Technical principles for protoporphyrin-IX-fluorescence guided microsurgical resection of malignant glioma tissue. Acta Neurochir (Wien) 140(10):995–1000
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO, European Organisation for Research and Treatment of Cancer Brain Tumour and Radiotherapy GroupsNational Cancer Institute of Canada Clinical Trials Group (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352(10):987–996
Teuchner K, Freyer W, Leupold D, Volkmer A, Birch DJ, Altmeyer P, Stucker M, Hoffmann K (1999) Femtosecond two-photon excited fluorescence of melanin. Photochem Photobiol 70:146–151
Utsuki S, Oka H, Sato S, Shimizu S, Suzuki S, Tanizaki Y, Kondo K, Miyajima Y, Fujii K (2007) Histological examination of false positive tissue resection using 5-aminolevulinic acid-induced fluorescence guidance. Neurol Med Chir (Tokyo) 47(5):210–213
Westphal M, Ram Z, Riddle V, Hilt D, Bortey E, Executive Committee of the Gliadel Study Group (2006) Gliadel wafer in initial surgery for malignant glioma: long-term follow-up of a multicenter controlled trial. Acta Neurochir (Wien) 148(3):269–275
Acknowledgment
This study was supported by grants to the University Hospital of Schleswig-Holstein, Campus Lübeck (AG), the Future Investment Program of Schleswig-Holstein (AG and GH) and the Novartis-foundation für therapeutische Forschung, Nürnberg.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Comment
Malignant glioma still remains to have the worst prognosis for survival of the patients. Despite tremendous efforts to find new treatment options the survival time has not increased dramatically. Complete resection of the tumour has been shown to be beneficial for the overall survival of the patient; however, the invasive nature of the tumour creates a big challenge for the neurosurgeon to achieve this.
Multiphoton excitation (ME) microscopy is still a new approach to image a tumour and its surrounding tissue. The authors have already shown the usefulness of this technique on tissue from experimental intracranial gliomas in mouse brain, and biopsies of human gliomas, where they were able to perform structural imaging of tumour and central nervous system histo-architecture at a subcellular level and the ability to distinguish between normal and tumour tissue.
In the present paper the authors measured cell density in native tissue with ME microscopy and compared this with conventional histopathological analysis (H&E staining) of the same tissue. Both techniques consistently allowed them to distinguish between the highly cellular tumour area and the less cellular adjacent normal tissue and even identify tumour cell clusters within less cellular tissue. In these experiments ME microscopy has proven to be a useful tool to quickly analyse the origin of the biopsy tissue (tumour centre or tumour periphery).
The paper is a very valuable contribution to the improvement of treatment option for glioma patients and raises hopes that after further development of the technique it might eventually find use in the operation room.
Verena Amberger-Murphy
Dublin City University
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kantelhardt, S.R., Leppert, J., Kantelhardt, J.W. et al. Multi-photon excitation fluorescence microscopy of brain-tumour tissue and analysis of cell density. Acta Neurochir (Wien) 151, 253–262 (2009). https://doi.org/10.1007/s00701-009-0188-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00701-009-0188-6