
Abstract
Regorafenib is an oral multikinase inhibitor that has shown antitumor activity in a range of solid tumors. Based on data from phase III clinical trials, regorafenib is indicated for the treatment of adult patients with metastatic colorectal cancer who have previously been treated with, or are not considered candidates for, other available therapies, and in patients with advanced gastrointestinal stromal tumors that cannot be surgically removed and no longer respond to other appropriate treatments. A panel of oncology nurses, research coordinators, and other medical oncology experts, experienced in the care of patients treated with regorafenib, met to discuss the best practice for the management of regorafenib-associated adverse events (AEs). The panel agreed that, in clinical trials and daily practice with regorafenib, AEs are common but mostly manageable. The most common and/or important AEs associated with regorafenib were considered to be hand–foot skin reaction, rash or desquamation, stomatitis, diarrhea, hypertension, liver abnormalities, and fatigue. This manuscript describes the experience and recommendations of the panel for managing these AEs in everyday clinical practice. Appropriate education, monitoring, and management are considered essential for reducing the incidence, duration, and severity of regorafenib-associated AEs.
Similar content being viewed by others

Introduction
Regorafenib (Stivarga, Bayer HealthCare Pharmaceuticals) is a novel, orally administered multikinase inhibitor [1]. Preclinical studies demonstrated that regorafenib targets cell-signaling pathways involved in tumor formation and progression, including inhibition of protein kinases associated with angiogenesis (e.g., VEGFR1–3 and TIE2), oncogenesis (e.g., KIT and RET), and maintenance of the tumor microenvironment (e.g., PDGFR and FGFR) [1]. The potential value of regorafenib in humans has been studied in a program of clinical trials, following phase I evidence of tolerability and preliminary antitumor activity in patients with a range of advanced solid tumors [2].
Regorafenib is indicated (pending marketing authorization in some countries) for the treatment of adult patients with metastatic colorectal cancer (mCRC) who have previously been treated with, or are not considered candidates for, other available therapies, and in patients with advanced gastrointestinal stromal tumors (GISTs) that cannot be surgically removed and no longer respond to other appropriate treatments [3–6]. The recommended dose of regorafenib is 160 mg to be taken once daily for 3 weeks, followed by 1 week off therapy. This 4-week period is considered a treatment cycle. Treatment should be continued for as long as benefit occurs or until unacceptable toxicity is experienced. Of note, regorafenib is metabolized by CYP3A4 and UGT1A9 [7]. Co-administration of strong CYP3A4 inducers (e.g., rifampin and phenytoin) and strong CYP3A4 inhibitors (e.g., clarithromycin, grapefruit juice, and itraconazole) should be avoided. Co-administration of regorafenib may increase systemic exposure to UGT1A1 and UGT1A9 substrates (e.g., SN-38, an active metabolite of irinotecan and a substrate of UGT1A1).
The applications for marketing authorization of regorafenib in mCRC and GIST were based on data from two international, randomized, phase III trials of regorafenib, named CORRECT (a double-blind, placebo-controlled study of regorafenib plus best supportive care [BSC] versus placebo plus BSC in patients with mCRC that had progressed after standard therapy) and GRID (a double-blind, placebo-controlled study of regorafenib plus BSC versus placebo plus BSC for patients with metastatic and/or unresectable GIST whose disease had progressed despite prior treatment with at least imatinib and sunitinib) [8–11].
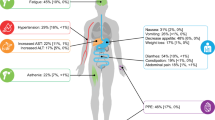
In the CORRECT study, 760 patients were randomized in a 2:1 ratio to receive regorafenib or placebo [9]. The primary study endpoint was overall survival—this endpoint was met at a planned interim analysis, with a hazard ratio (HR) of 0.77 (95 % confidence interval [CI] 0.64–0.94; one-sided p = 0.0052) for overall survival with regorafenib versus placebo (median, 6.4 versus 5.0 months, respectively). Patients assigned to the regorafenib group received 78.9 % of the planned dose during the study, and dose modifications were required in 378 of 500 patients (76 %). The placebo group received 90.1 % of the planned dose, and 97 of 253 patients (38 %) required dose modification. The most common reason for dose modification was adverse events (AEs), with 67 % of regorafenib patients and 23 % of placebo patients experiencing AEs leading to dose modification. The most common grade 3 or higher regorafenib-related AEs were hand–foot skin reaction (HFSR; 46.6 %, all grades; 16.6 %, grade 3; and grade 4, not applicable), fatigue (47.4 %, all grades; 9.2 %, grade 3; 0.4 %, grade 4), hypertension (27.8 %, all grades; 7.2 %, grade 3; and 0 %, grade 4), and diarrhea (33.8 %, all grades; 7.0 %, grade 3; and 0.2 %, grade 4).
The GRID study randomized 199 patients in a 2:1 ratio to receive regorafenib or placebo [11]. According to an independent, blinded central review, median progression-free survival (primary endpoint) was 4.8 months with regorafenib and 0.9 months with placebo (HR, 0.27; 95 % CI, 0.19–0.39; p < 0.0001). Because of the high rate of crossover (85 % of the placebo group crossed over on disease progression to receive open-label regorafenib), the overall survival results from GRID did not reach statistical significance. Patients in the regorafenib group received 78.0 % of the planned dose and patients in the placebo group received 83.8 % of the planned dose. Drug-related AEs were reported for 98 % of patients receiving regorafenib and 68 % of patients receiving placebo, the most common being HFSR (56.1 %, all grades; 19.7 %, grade 3; grade 4, not applicable), hypertension (48.5 %, all grades; 22.7 %, grade 3; and 0.8 %, grade 4), diarrhea (40.2 %, all grades; 5.3 %, grade 3; and 0 %, grade 4), and fatigue (38.6 %, all grades; 2.3 %, grade 3; and 0 %, grade 4).
Following these trials, an open-label phase IIIb study of regorafenib in patients with mCRC that had progressed after standard therapy was established [12]. This prospective, multicenter, single-arm, interventional, expanded-access study (named CONSIGN) will provide regorafenib to patients with mCRC that has failed standard therapies, for whom no treatment alternatives are available, in the time between the positive phase III results and approval or availability of regorafenib locally. The study aims to collect additional safety data on regorafenib. The primary endpoint is safety, and patients will continue on treatment until reaching one of the main criteria—death, unacceptable toxicity, patient's withdrawal of consent, treating physician's decision that discontinuation of treatment is in the patient's best interest, or substantial noncompliance with the protocol. The study is ongoing.
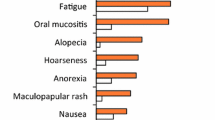
Experience to date shows that regorafenib can provide survival benefits in patients with mCRC or advanced GIST, but that AEs are common and can necessitate dose modification. The AE profile of regorafenib was consistent across both phase III studies—HFSR, fatigue, diarrhea, and hypertension were the most common drug-related AEs [9, 11]. In our experience, while other AEs (e.g., nausea and anorexia) may also occur, they do not have an impact on patients' quality of life to the extent that those covered in this manuscript may do, and do not tend to require treatment modification.
We noticed that the AEs of targeted therapies are often compared with AEs of chemo- and radiation therapy, rather than with the AEs of hormonal therapy. However, chemotherapy-associated AEs are cumulative, while most AEs associated with hormonal therapy appear and, if adequately managed, may resolve within the first 12 weeks of treatment—similar to most regorafenib-associated AEs.
Sound knowledge of the AE profile associated with regorafenib and proactive AE monitoring and management can help patients to better tolerate the drug and optimize treatment outcome.
Objective
A panel of medical oncology experts from Europe and Canada, experienced in the care of patients treated with regorafenib, met to discuss the best practice for the management of regorafenib-associated AEs. The agenda for the day was developed by the meeting chair (Mieke De Wit) and included a detailed discussion of the most common AEs among patients receiving regorafenib and the personal experience of the expert panel in the management of these events. The meeting was supported by Bayer HealthCare Pharmaceuticals, which funded Succinct Healthcare Communications to manage logistics and provide medical-writing support. This manuscript presents the experience and recommendations of the group and communicates the AE management considerations that we believe to be the most important for maintaining quality of life and adherence to treatment in patients receiving regorafenib.
Management strategy for regorafenib-related AEs
HFSR
The three grades of HFSR, according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 4.03, are described in Table 1 [13]. Reported in around half of the patients receiving regorafenib, HFSR was the most common AE in the CORRECT and GRID studies [9, 11].
HFSR is characterized by localized hyperkeratotic lesions that may be surrounded by erythematous regions within the skin. The areas of the skin under the most pressure or flexure are most likely to be affected, such as the palms of the hand, fingertips, finger webs, distal phalanges, soles of the feet, and toes [14]. HFSR usually occurs early (within 4 weeks of initiation of regorafenib treatment; most commonly within the first 2 weeks) [14, 15], and, although it is not life threatening and usually resolves with appropriate management, it can negatively affect patients' quality of life physically, psychosocially, and socially [16].
As with many potential AEs, early detection and prompt initiation of therapeutic management can reduce the severity and duration of HFSR [14]. Close monitoring is crucial—during the first two cycles of treatment, we recommend that patients should be assessed for HFSR weekly by a healthcare provider; from cycle 3 onwards every 4 weeks. It is important to encourage patients and caregivers to monitor for potential symptoms of HFSR and to maintain frequent communication between the patient and healthcare provider to ensure that symptoms are detected and managed at the earliest possible stage.
Preventive measures can decrease the likelihood of patients experiencing HFSR [14]. A full-body skin examination should be carried out before treatment initiation to ensure that any changes can be accurately noted, and any calluses should be softened and removed before and during treatment.
It is important that patients understand the best practices for managing HFSR and what actions to take at home to prevent or manage the occurrence. Care should be taken to avoid pressure and friction on the skin. The patient should be advised to avoid traumatic activity to minimize stress on the skin. The pressure points and pressure-sensitive areas on the feet can be protected with cotton socks, well-padded and well-fitting shoes, foam-padded soles, and insole cushions. Padded gloves should be worn when using tools that might damage the skin on the hands. Effective use of creams and moisturizers can help to maintain the condition of the skin. Soaps and hand sanitizers containing alcohol and the use of hot water can dry the skin and should be avoided. If HFSR is painful, patients should be advised to cool the skin with cold packs and to treat with analgesics such as nonsteroidal anti-inflammatory drugs.
If active management does not alleviate HFSR symptoms, dose modification may be required and usually leads to rapid alleviation of symptoms (within 1–2 weeks) [14]. We believe that the regorafenib trial protocols contain effective advice on the management of HFSR, including details of appropriate dose modifications when necessary [9, 11]. These recommendations, along with the typical manifestations of symptoms, preventive measures, and suggested topical treatments are shown in Table 2. With these measures, HFSR is usually resolved or effectively controlled—Fig. 1 shows examples of HFSR in patients being treated with regorafenib and how the reaction can be resolved with appropriate management and dose modification.

Hand–foot skin reaction: a occurring 2 weeks after initiation of regorafenib therapy; b 2 weeks later, following interruption of regorafenib and reintroduction at a reduced dose (120 mg) according to protocol-specified guidelines; c and d localized hyperkeratotic lesions on the feet of a patient receiving regorafenib. Pictures courtesy of Leiden University Medical Center, Netherlands, and University Hospitals Leuven, Belgium
Rash or desquamation
Maculopapular rash is a disorder characterized by the presence of macules (flat, discolored areas of skin) and papules (solid elevations of skin), with symptoms including photosensitivity, erythema, dry or peeling skin, blistering, and pruritus [17]. The grading of rash, as described in Table 1, is according to body-surface coverage—macules or papules covering less than 10 % of body surface are grade 1, covering 10–30 % are grade 2, and covering more than 30 % are grade 3 [13]. In our experience, grades 1–2 rash is most common among patients receiving regorafenib, whereas grade 3 is less common. Regorafenib-related rash or desquamation of any grade was experienced by 26 % of the patients receiving regorafenib in the CORRECT trial (incidence of grade 3 was 6 %) and by 18 % of the patients in the GRID trial (grade 3, 2 %) [9, 11]. Regorafenib-associated rash predominantly occurs within the first cycle and the incidence is considerably reduced in subsequent cycles.
With appropriate management, and dose modification where necessary, rash is normally resolved. We recommend that, during the first two cycles, patients should be monitored for rash every week, in combination with the HFSR assessment, with the frequency decreasing to every 4 weeks from cycle 3. Preventive measures, such as regular use of emollients (not containing alcohol) from initiation of treatment, the use of mild soaps, and avoidance of extreme temperatures and direct sun exposure, can help to prevent or reduce the severity of rash. Rash, in all three grades, can be effectively managed with antihistamines and a short treatment with dermatology ointments containing class IV corticosteroids (e.g., clobetasol propionate 0.05 % in cold cream)—a dermatologist should be consulted to determine the most appropriate treatment.
Stomatitis
Stomatitis, also referred to as oral mucositis, is the inflammation of the mucous lining of any of the structures in the mouth and oropharynx. The CTCAE version 4.03 describe five grades (see Table 1) [13]. Regorafenib-associated stomatitis is a common AE [9, 11] and tends to occur between 5 and 14 days after the start of a treatment cycle. In our experience, the effect may occur in cycle 1 only or may be recurrent, and dose reduction may be necessary. While the development of stomatitis is thought to be a drug-related effect in patients receiving regorafenib, other patient-related factors can increase the intensity of the effect. These include old age, dental hygiene, nutritional status, associated infections, and dental pathology [18].
Preventive measures are important in reducing the risk of stomatitis. Good oral hygiene is crucial and it is important that patients are appropriately educated about effective dental care before treatment. Teeth and gums should be cleaned with fluoride toothpaste and a soft toothbrush or swab (as tolerated) after meals and before sleep; the mouth should be regularly rinsed with an alcohol-free mouthwash, saline solution, or bicarbonate; dentures should be removed and cleaned daily; and painful stimuli such as hot foods and drinks and spicy foods should be avoided.
If stomatitis occurs, patients should increase the frequency of mouthwash (e.g., every 1–2 h). In addition to saline-containing mouthwashes, solutions such as 60 ml mycostatin (100,000 IU/ml nystatin), 250 ml hexetidine, 10 ml mepivacaine hydrochloride (10 mg/ml), and 1 g bicarbonate can be considered. If patients find the mouthwash painful, they should be advised to use pain medication beforehand (e.g., viscous lidocaine 2 %, coating agents, and, when needed, systemic approaches following the World Health Organization pain management ladder). If the patient reports symptoms in the throat, and fungal infection is suspected, then nystatin 5 ml four times daily (swallowed once daily) for 10 days should be recommended. Oral intake of food and drink should be assessed because pain, dry mouth, and difficulty swallowing can occur. At grades 3–4, the mouthwash protocol should be intensified to 8–12 rinses per day, or every hour, with the addition of analgesics if considered appropriate. If no improvement is seen after 7 days, then regorafenib should be interrupted until the stomatitis is reduced to grade 1, at which point treatment can be reintroduced. Ideally, regorafenib should be reintroduced at the same dose. Other treatments, such as coating agents, topical analgesic or anti-inflammatory agents, topical anesthetics, and alternative mouthwashes are also available and may be considered.
Diarrhea
Diarrhea is common among patients receiving regorafenib [9, 11]. Five grades of diarrhea are described in the CTCAE version 4.03 (Table 1) [13]. Although grade 1 diarrhea (an increase of <4 stools per day over baseline) may not present a significant problem for patients, grade 2 diarrhea (increase of 4–6 stools per day over baseline) can have a moderate effect on quality of life, limiting the patient's ability to leave the house, and grade 3 diarrhea (increase of ≥7 stools per day over baseline and incontinence) severely affects self-care activities of daily living (ADL). That said, in our experience, the prolonged occurrence of grade 2 diarrhea may negatively affect a patient's quality of life and ADL more than a short-lasting grade 3 event, and so it is important that patients are educated about the need to report and manage any occurrence of diarrhea.
Dietary advice that may minimize the likelihood of diarrhea (e.g., ensuring a low intake of fiber) should be given to patients on initiation of treatment. In order to effectively manage diarrhea and prevent symptoms from increasing in severity, patients should be advised to contact their healthcare team if they have an increase of more than three stools per day. Patients should also be educated about the variation in stool form and advised that changes to stool number or consistency do not always indicate diarrhea that requires treatment. The Bristol stool chart is a useful tool to help explain the variation to patients—stools are classified into seven types, with types 5–7 tending towards diarrhea [19].
We recommend that patients experiencing diarrhea of grades 1 and 2 should be advised to take loperamide (two 2-mg tablets) after the first stool and one additional tablet every 2 h, until 12 h after the last watery stool, for a maximum of 48 h. It is important to confirm that the patient is experiencing diarrhea, as the use of loperamide could otherwise cause constipation. In addition, the risk of developing constipation is increased in a patient already taking opioids for cancer-related pain. If loperamide does not control the diarrhea, for example when diarrhea remains grade 3 or higher after the 48-h loperamide period, the patient is likely to become dehydrated—the patient should be encouraged to increase fluid intake and hospital admission should be considered.
Hypertension
In clinical practice, hypertension is common among patients receiving regorafenib, but, when treated appropriately, the effect on quality of life is negligible. Hypertension with regorafenib is not cumulative [15]. In our experience, regorafenib, like other receptor tyrosine kinase inhibitors, can instead be seen as a toxicity intensifier—it may interfere with the recovery of previous cardiovascular damage, while any direct effect on cardiovascular function appears to be reversible [20–22]. Regorafenib-associated hypertension is therefore temporary, and, once regorafenib is discontinued, blood pressure is likely to decrease to levels that are normal for each individual patient.
Before initiation of regorafenib treatment, blood pressure should be checked and controlled if it is found to be elevated. Blood pressure should then be monitored weekly for the first two cycles of treatment, and, if detected, hypertension should be treated. If possible, patients should measure their blood pressure at home on a daily basis—this helps to identify patients whose blood pressure is high when visiting the hospital because they are nervous, rather than because they have persistent hypertension. Table 1 describes, according to CTCAE version 4.03, five grades of hypertension based on the levels of systolic and diastolic blood pressure [13]. Blood pressures exceeding 140/90 mmHg (grade 2 hypertension) are very common and can be treated with antihypertensive treatment such as angiotensin-converting enzyme (ACE) inhibitors. Blood pressures of grade 3 and higher are less common but do occur, and, if they are not controlled by ACE inhibitors, beta blockers can be added as an adjunctive therapy. If hypertension remains a problem, calcium channel blockers can be added to the ACE inhibitors and beta blockers; because of the risk of diarrhea, the use of diuretics is discouraged. Recommendations for the management of treatment-emergent hypertension, as specified in the regorafenib clinical trial protocols [9, 11], are shown in Table 3.
Liver abnormalities
In our experience, abnormalities in liver function tests can be very common among patients receiving regorafenib. One case of fatal liver dysfunction was observed in the CORRECT trial [9]. Treatment with regorafenib may lead to a higher risk of severe liver toxicity, with symptoms that may include yellowish discoloration of the skin and the white of the eye, nausea and vomiting, very dark urine, and changes in sleep patterns. The liver abnormalities generally occur soon after treatment initiation, and it is crucial that aspartate aminotransferase (AST), alanine aminotransferase (ALT), and bilirubin are measured every 2 weeks for the first two cycles. Table 1 gives the grading of the laboratory abnormalities according to CTCAE version 4.03 [13].
We feel that the recommendations for the management of liver toxicities, through dose modification only, contained in the CORRECT and GRID protocols are effective in clinical practice [9, 11]. The recommendations specify that, if AST or ALT levels greater than five times the upper limit of normal (ULN) are detected, then regorafenib dosing should be interrupted until transaminases return to less than three times ULN or to baseline, at which stage reinitiation of regorafenib can be considered. If AST or ALT levels reach more than 20 times ULN, or if AST or ALT levels are more than three times ULN with concurrent bilirubin more than two times ULN, treatment should be discontinued. Patients with Gilbert's syndrome (who have high baseline levels of bilirubin) with elevated bilirubin should be treated according to the guidelines for elevated liver enzymes—elevated transaminases need not be taken into consideration.
Fatigue
In our experience, fatigue is a common and important AE among patients receiving regorafenib. Cancer-related fatigue remains one of the most prevalent, distressing, and activity-limiting symptoms that patients experience, especially in the later stages of their disease [23]. In the CORRECT and GRID clinical trials, fatigue was reported in 47.4 and 38.6 % of patients, respectively [9, 11]. The lower incidence among patients with GIST may reflect the different prior therapies and disease histories of GIST and mCRC patients, as well as the site of disease and any metastases.
Fatigue can affect a patient's quality of life and can be disabling [23]—at grade 2, fatigue is not relieved by rest and can limit instrumental ADL (for example, the ability to do housework or use the telephone); at grade 3, it limits self-care ADL [13]. The three grades of fatigue, according to the CTCAE version 4.03, are described in Table 1. However, grading a subjective AE such as fatigue can be difficult: in our experience, some patients may be hesitant to report the occurrence or severity of fatigue through a belief that experiencing toxicities indicates that the treatment is working or because they expect to experience fatigue due to their age or disease state; others may contact their healthcare team at the first indication. It is important to educate patients about the likelihood of experiencing fatigue, and that reporting it early enables them to receive the appropriate advice and management that may help to prevent it becoming more severe.
For the first two cycles of regorafenib treatment, fatigue should be monitored weekly. For subsequent cycles, patients should be monitored for fatigue every 2 weeks and reminded to contact their healthcare team if fatigue develops or worsens between visits. If drug-related fatigue is reported, patients should be advised to take some time out of the day to rest and recuperate their energy levels. Patients should also be educated about the potential benefits of exercise—there is clinical evidence that physical exercise may help to improve cancer-related fatigue [24, 25]. In addition, regorafenib dose modification may be necessary for grade 3 fatigue. Table 3 shows the recommended treatment modifications relevant to fatigue.
Conclusion
Phase III study data have demonstrated that regorafenib can provide survival benefits in patients with mCRC or GIST (overall survival and progression-free survival, respectively) [9, 11]. In our experience using regorafenib in clinical trials and daily practice, AEs are common but mostly manageable. Appropriate education, monitoring, and management are essential for reducing the incidence, duration, and severity of regorafenib-associated AEs, and we have provided a checklist (Table 4) to aid the prompt and appropriate management of these AEs. We recommend attempting to manage the effect before dose reduction or interruption. If dose modification is deemed advisable, the protocols of the phase III regorafenib trials [9, 11] provide clear guidance on approaches for the different AEs, including treatment interruption until the AE has resolved followed by reintroduction of regorafenib at a lower dose (minimum 80 mg daily). If the patient is able to tolerate the reduced dose without recurrence of the AE, we recommend re-escalating the dose if possible.
References
Wilhelm SM, Dumas J, Adnane L, Lynch M, Carter CA, Schütz G, Thierauch K-H, Zopf D (2011) Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer 129:245–255
Mross K, Frost A, Steinbild S, Hedbom S, Büchert M, Fasol U, Unger C, Krätzschmar J, Heinig R, Boix O, Christensen O (2012) A phase I dose-escalation study of regorafenib (BAY 73-4506), an inhibitor of oncogenic, angiogenic, and stromal kinases, in patients with advanced solid tumors. Clin Cancer Res 18:2658–2667
European Medicines Agency (2013) Summary of opinion (initial authorisation): Stivarga regorafenib. EMA, London
US Food and Drug Administration (2012) Drugs: regorafenib. http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm321378.htm. Accessed 17 Sept 2013
Health Canada (2013) Drugs and health products: PrStivarga. http://www.hc-sc.gc.ca/dhp-mps/prodpharma/sbd-smd/drug-med/sbd_smd_2013_stivarga_157970-eng.php. Accessed 17 Sept 2013
US Food and Drug Administration (2013) FDA news release: FDA approves Stivarga for advanced gastrointestinal stromal tumors. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm340958.htm. Accessed 17 Sept 2013
Bayer Pharma AG (2013) Stivarga 40 mg film-coated tablets. Summary of product characteristics. Bayer, Berlin
ClinicalTrials.gov (2013) Patients with metastatic colorectal cancer treated with regorafenib or placebo after failure of standard therapy. http://clinicaltrials.gov/show/NCT01103323. Accessed 17 Sept 2013
Grothey A, Van Cutsem E, Sobrero A, Siena S, Falcone A, Ychou M, Humblet Y, Bouché O, Mineur L, Barone C, Adenis A, Tabernero J, Yoshino T, Lenz H-J, Goldberg RM, Sargent DJ, Cihon F, Cupit L, Wagner A, Laurent D (2013) Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381:303–312, original article and supplementary appendix
ClinicalTrials.gov (2013) Study of regorafenib as a 3rd-line of beyond treatment for gastrointestinal stromal tumors (GIST) (GRID). http://clinicaltrials.gov/show/NCT01271712. Accessed 17 Sept 2013
Demetri GD, Reichardt P, Kang Y-K, Blay J-Y, Rutkowski P, Gelderblom H, Hohenberger P, Leahy M, von Mehren M, Joensuu H, Badalamenti G, Blackstein M, Le Cesne A, Schöffski P, Maki RG, Bauer S, Nguyen BB, Xu J, Nishida T, Chung J, Kappeler C, Kuss I, Laurent D, Casali PG (2013) Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381:295–302, original article and supplementary appendix
ClinicalTrials.gov (2013) Regorafenib in subjects with metastatic colorectal cancer (CRC) who have progressed after standard therapy (CONSIGN). http://clinicaltrials.gov/show/NCT01538680. Accessed 17 Sept 2013
US Department of Health and Human Services (2010) Common terminology criteria for adverse events (CTCAE). http://evs.nci.nih.gov/ftp1/CTCAE/. Accessed 17 Sept 2013
Wood LS, Lemont H, Jatoi A, Lacouture ME, Robert C, Keating K, Anderson R (2010) Practical considerations in the management of hand–foot skin reaction caused by multikinase inhibitors. Commun Oncol 7:23–29
Grothey A, Van Cutsem E, Sobrero AF, Siena S, Falcone A, Ychou M, Humblet Y, Bouché O, Mineur L, Barone C, Adenis A, Tabernero J, Yoshino T, Lenz H-J, Goldberg RM, Sargent DJ, Cihon F, Wagner A, Laurent D, Cupit L (2012) Time course of regorafenib-associated adverse events in the phase III CORRECT study. J Clin Oncol 30(Suppl 34):467
Aklilu M, Anderson RT, Keating KN, O'Leary JJ (2008) Patient interviews on hand–foot syndrome: symptom assessment and impact on quality of life. J Clin Oncol 26(Suppl 15):20713
Khayyam KU, Imam F, Sharma M, Pillai KK, Behera D (2010) Pyrazinamide-induced maculopapular rash. Indian J Dermatol 55:384–386
McCarthy GM, Awde JD, Ghandi H, Vincent M, Kocha WI (1998) Risk factors associated with mucositis in cancer patients receiving 5-fluorouracil. Oral Oncol 34:484–490
Lewis SJ, Heaton KW (1997) Stool form scale as a useful guide to intestinal transit time. Scand J Gastroenterol 32:920–924
Ewer MS (2011) Chemotherapy-related cardiac safety. Research review speaker series. http://www.researchreview.co.nz/getmedia/8cc9638a-4d34-4761-a402-32d70b4758bf/ss_ewer.pdf.aspx. Accessed 17 Sept 2013
Ewer MS, Ewer SM (2011) Cardiotoxicity of anticancer treatments: what the cardiologist needs to know. Nat Rev Cardiol 7:564–575
Suter TM, Ewer MS (2013) Cancer drugs and the heart: importance and management. Eur Heart J Manage 34:1102–1111
Hofman M, Ryan JL, Figueroa-Moseley CD, Jean-Pierre P, Morrow GR (2007) Cancer-related fatigue: the scale of the problem. Oncologist 12:4–10
Cheville AL, Kollasch J, Vandenberg J, Shen T, Grothey A, Gamble G, Basford JR (2013) A home-based exercise program to improve function, fatigue, and sleep quality in patients with stage IV lung and colorectal cancer: a randomized controlled trial. J Pain Symptom Manag 45:811–821
Lin KY, Shun SC, Lai YH, Liang JT, Tsauo JY (2013) Comparison of the effects of a supervised exercise program and usual care in patients with colorectal cancer undergoing chemotherapy. Cancer Nurs. doi:10.1097/NCC.0b013e3182791097
Acknowledgments
The panel would like to thank their patients and the nursing staff and physicians at each of their centers. Mieke De Wit and Kristina Vermeersch wish to thank Eric Van Cutsem. Logistic and administrative support in the organization of the focus group meeting and editorial assistance in the preparation of this manuscript were provided by Succinct Healthcare Communications, with financial support from Bayer HealthCare Pharmaceuticals; the authors retained editorial control over the content.
Disclosures
M. De Wit: No disclosures
C.B. Boers-Doets has been a consultant and speaker for Bayer HealthCare Pharmaceuticals
A. Saettini: No disclosures
K. Vermeersch: No disclosures
C. Roncero de Juan: No disclosures
J. Ouwerkerk: No disclosures
S.-S. Raynard: No disclosures
A. Bazin: No disclosures
C. Cremolini has been a consultant and speaker for Bayer HealthCare Pharmaceuticals.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
De Wit, M., Boers-Doets, C.B., Saettini, A. et al. Prevention and management of adverse events related to regorafenib. Support Care Cancer 22, 837–846 (2014). https://doi.org/10.1007/s00520-013-2085-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00520-013-2085-z