
Abstract
Given natural limitations on the length DNA sequences, designing phylogenetic reconstruction methods which are reliable under limited information is a crucial endeavor. There have been two approaches to this problem: reconstructing partial but reliable information about the tree (Mossel in IEEE Comput. Biol. Bioinform. 4:108–116, 2007; Daskalakis et al. in SIAM J. Discrete Math. 25:872–893, 2011; Daskalakis et al. in Proc. of RECOMB 2006, pp. 281–295, 2006; Gronau et al. in Proc. of the 19th Annual SODA 2008, pp. 379–388, 2008), and reaching “deeper” in the tree through reconstruction of ancestral sequences. In the latter category, Daskalakis et al. (Proc. of the 38th Annual STOC, pp. 159–168, 2006) settled an important conjecture of M. Steel (My favourite conjecture. Preprint, 2001), showing that, under the CFN model of evolution, all trees on n leaves with edge lengths bounded by the Ising model phase transition can be recovered with high probability from genomes of length O(logn) with a polynomial time algorithm. Their methods had a running time of O(n 10).
Here we enhance our methods from Daskalakis et al. (Proc. of RECOMB 2006, pp. 281–295, 2006) with the learning of ancestral sequences and provide an algorithm for reconstructing a sub-forest of the tree which is reliable given available data, without requiring a-priori known bounds on the edge lengths of the tree. Our methods are based on an intuitive minimum spanning tree approach and run in O(n 3) time. For the case of full reconstruction of trees with edges under the phase transition, we maintain the same asymptotic sequence length requirements as in Daskalakis et al. (Proc. of the 38th Annual STOC, pp. 159–168, 2006), despite the considerably faster running time.








Similar content being viewed by others

References
Cavender, J.: Taxonomy with confidence. Math. Biosci. 40, 271–280 (1978)
Daskalakis, C., Hill, C., Jaffe, A., Mihaescu, R., Mossel, E., Rao, S.: Maximal accurate forest from distance matrices. In: Proceedings of RECOMB 2006, vol. 3909, pp. 281–295. Springer, Berlin (2006)
Daskalakis, C., Mossel, E., Roch, S.: Optimal phylogenetic reconstruction. In: Proceedings of the Thirty-Eighth Annual ACM Symposium on Theory of Computing (STOC 2006), pp. 159–168 (2006)
Daskalakis, C., Mossel, E., Roch, S.: Phylogenies without branch bounds: contracting the short, pruning the deep. SIAM J. Discrete Math. 25(2), 872–893 (2011)
Erdos, P.L., Steel, M., Szekely, L., Warnow, T.: A few logs suffice to build (almost) all trees (I). Random Struct. Algorithms 14, 153–184 (1997)
Erdos, P.L., Steel, M.A., Szekely, L.A., Warnow, T.J.: A few logs suffice to build (almost) all trees (II). Theor. Comput. Sci. 221(1–2), 77–118 (1999)
Farris, J.S.: A probability model for inferring evolutionary trees. Syst. Zool. 22, 250–256 (1973)
Fischer, M., Steel, M.: Sequence length bounds for resolving a deep phylogenetic divergence. J. Theor. Biol. 256, 247–252 (2008)
Gronau, I., Moran, S., Snir, S.: Fast and reliable reconstruction of phylogenetic trees with very short edges. In: Proceedings of the Nineteenth Annual ACM-SIAM Symposium on Discrete Algorithms (SODA 2008), pp. 379–388 (2008)
Kimura, M.: Estimation of evolutionary distances between homologous nucleotide sequences. Proc. Natl. Acad. Sci. 78(1), 454–458 (1981)
Mossel, E.: On the impossibility of reconstructing ancestral data and phylogenies. J. Comput. Biol. 10(5), 669–678 (2003)
Mossel, E.: Phase transitions in phylogeny. Trans. Am. Math. Soc. 356(6), 2379–2404 (2004)
Mossel, E.: Distorted metrics on trees and phylogenetic forests. IEEE Comput. Biol. Bioinform. 4, 108–116 (2007)
Roch, S.: Sequence length requirement of distance-based phylogeny reconstruction: breaking the polynomial barrier. In: Proceedings of the 49th IEEE Symposium on Foundations of Computer Science (FOCS 2008), pp. 729–738 (2008)
Semple, C., Steel, M.: Phylogenetics. Mathematics and Its Applications, vol. 22. Oxford University Press, Oxford (2003)
Steel, M.: My favourite conjecture. Preprint (2001)
Acknowledgements
We thank Elchanan Mossel for invaluable discussions regarding reconstruction of ancestral sequences. Thanks to Costis Daskalakis, Elchanan Mossel and Sebastien Roch for pointing out errors in a preliminary version of the paper.
We also thank the an anonymous reviewer of an earlier version for pointing out most of the analysis in Appendix C.
Radu Mihaescu was supported by a National Science Foundation Graduate Fellowship, by the Fannie an John Hertz Foundation graduate fellowship and by the CIPRES project. All other authors were supported by CIPRES.
Author information
Authors and Affiliations
Corresponding author
Appendices
Appendix A: The Triangle Inequality for the CFN Model
The main purpose of this appendix is to provide a proof of Lemma 5.4:
Lemma 5.4
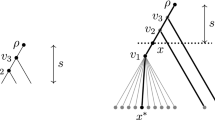
Let T′ be an induced subtree of T rooted at ρ and let v∈(V(T)∩T′)∖δ(T′) (Fig. 9). Then
with \(\tilde{\rho}(T')\) a “learned” character value, where the learning occurs by any bottom-up recursive majority algorithm on T′, as outlined in Sect. 3.

Tree configuration for the proof of Lemma 5.4
We begin by introducing an alternative representation of the CFN model under a percolation framework. This intuitive view lies at the root of the theoretical results regarding information flow on trees in [11] and [12].
Let p(e)<0.5 be the probabilities of mutation along edges e∈E(V) for a CFN model on T. Let α(e) be independent random variables such that
Suppose each edge e in T carries a survival probability θ(e)=1−2p(e), such that the edge e is deleted if α(e)=0. After removing the destroyed edges, each surviving connected component C receives a single character value χ(C), by tossing an independent unbiased coin. We write u↔v for the event that the two nodes u,v are in the same connected component and C v for the component containing v.
It is easy to see that the joint probability distribution on character values at V(T) produced under this alternative model, ℙ T,θ , is the same as the one induced by the original CFN model: ℙ T,L , where L(e)=−log(1−2p(e))/2=−log(θ(e))/2. As before, D is the distance between uniform binary random variables defined in Sect. 2.
Proof or Lemma 5.4
Let \(\tilde{\rho}\) denote \(\tilde{\rho}(T')\) for brevity. The lemma is equivalent to \(\mathbb {E}[v \tilde{\rho}]\geq \mathbb {E}[\rho v] \mathbb {E}[\rho\tilde{\rho}]\). It is an easy exercise to show that, in turn, this is equivalent to \(\mathbb {P}[\rho=\tilde{\rho}|\rho=v] \geq \mathbb {P}[\rho=\tilde{\rho}|\rho\neq v]\). By symmetry, we may assume for the rest of the proof that ρ=1, so χ(C ρ )=1, and our task reduces to showing that
Let E(P)={e 1,…,e s } denote the edges of the path P=P(ρ,v), and let V(P)={v 1,…,v s =v} be the nodes of P, other than ρ. Then 1(ρ↔v)=1(α(E(P))≡1). We proceed by way of a standard coupling argument.
Suppose α(E(T)) is such that such that \(\rho\not \leftrightarrow v\). Given a set of values χ 0 for the characters χ(C), C≠C v , C≠C ρ ,

Now \(\tilde{\rho}\) is a recursive majority function in the character values at δT′, and is therefore coordinate-wise increasing in the values of those characters. Moreover χ(δT′) in the event 1[χ(C v )=1,χ(C ρ )=1,χ(C ≠v,ρ )=χ 0] is coordinate-wise larger than χ(δT′) in the event 1[χ(C v )=−1,χ(C ρ )=1,χ(C ≠v,ρ )=χ 0], while the probabilities of the two events, conditioned on the values α, are the same. Summing over all values χ 0 and all values α such that \(\rho\not\leftrightarrow v\),
For any x∈{±1}s and any b∈{±1}t with t=|E(T)|−s, an identical argument to the one above shows that

Observe that 1[ρ↔v]=1[α(E(P))≡1] implies 1[χ(V(P))≡1], and α(E(T)∖E(P)) and α(E(P)) are independent, thus α(E(T)∖E(P)) and χ(V(P)) are independent. Therefore

The first implication follows from summation over all values of b. The second comes from summation over all values of x such that x s =v=1 and x is compatible with α(E(P))=a. The third implication follows from summing over all values a∈{±1}s, \(a\not\equiv1\).
The last inequality, together with (7), implies

□
Appendix B: Applicability to Other Molecular Models of Evolution
Our method implies similar results for all group based models of evolution, where character alphabet is a group G admitting a non-trivial morphism ϕ:G→ℤ2. In a group-based model of evolution, the probability of transformation of the character χ from state a to state b along any edge e of the tree only depends on a −1 b. In other words, for an edge e=(u,v)∈E(T),
By the definition of a morphism,
Thus

which does not depend on a and implicitly does not depend on ϕ(a).
We can then reduce any such model to the binary one by identifying a state g∈G to ϕ(g)∈ℤ2 and applying our analysis mutatis mutandis. The most notable example of group based model of evolution satisfying our requirements is the Kimura 3ST model [10], which is realized by the group ℤ2×ℤ2 [15]. We also note that Kimura 3ST is a generalization of the well known Jukes-Cantor model.
Appendix C: Sequence Length Requirements of the TreeMerge Algorithm
In this appendix we attempt to provide a brief analysis of the sequence length requirements of our algorithm, as well as other techniques using the reconstruction of ancestral characters.
The results listed here unfortunately show that, despite the theoretically optimal sequence length requirement of O ϵ (logn), in the absence of a substantially more involved analysis into the growth rate of the noise of ancestral sequence reconstruction, the current state of the art on learning ancestral sequences does not easily provide a solution for practical applications.
The results listed here show that the constants involved in the O ϵ (logn) sequence length bounds may in fact be prohibitively high, not just for our algorithm, but generally for all algorithms making use of ancestral sequence reconstruction. This is because the constants involved in the O ϵ (logn) notation are exponential in 1/ϵ, where ϵ is, as before, a proxy for the separation between the longest edge in the tree to be reconstructed and the “phase transition” λ 0=log(2)/4.
All of our results below refer to bounds which are immediately available from the analysis in [13]. Most of these bounds are not claimed to be tight except in an asymptotic sense. Thus our analysis here is not meant to be a permanent indictment of this branch of research, as it is in fact likely that a more in depth analysis will provide much tighter bounds, which will lend themselves readily to practical application.
In all the subsequence analysis we preserve the notation of earlier sections.
Proposition C.1
The depth d of the subtree decomposition required for non-decaying recursive accuracy of ancestral reconstruction (see Theorem 3.3 or Theorem 4.1 in [13]) is inversely proportional to the separation ϵ.
Proof
In order to obtain a non-decaying recursive accuracy of ancestral reconstruction, we require a bound of α>1 for the results in Theorem 3.3 or Theorem 4.1 in [13]. This is not a strict requirement and it is in fact likely that its relaxation will provide better bounds, but at a cost to ease of exposition. However, under this analysis, by Stirling approximation we obtain
as mentioned in the prof of Theorem 3.3, and also in Lemma 4.3 of [13]. This implies
□
Proposition C.2
The upper bound on the noise of ancestral reconstruction β(λ max (ϵ)) grows linearly in d, and thus in 1/ϵ.
Proof
We use here the bounds implicit in Lemma 4.6 in [13]. Again, it is in fact possible that better bounds may be obtainable. We restate this lemma in our own notation. □
Lemma 4.6
[13]
If η(v)≤η max , then
As λ max <log(2)/4, we obtain \(e^{-2\lambda_{\mathit{max}}}>1/2\) and thus \(h(e^{-2\lambda_{\mathit{max}}})=1\). Applying the same Stirling approximation as before, namely
one obtains

Note
The penultimate inequality comes from \(h(e^{-2\lambda_{\mathit{max}}} e^{-2\eta_{\mathit{max}}})\leq1\). This is enough to obtain a negative result, but the inequality is in fact strict. Modifying this analysis to obtain a positive result, meaning an upper bound on β(λ max (ϵ)), would in fact require understanding the behavior of \(e^{-2\lambda_{\mathit{max}}} e^{-2\eta_{\mathit{max}}}\), or more precisely bounding the size of the constant C in the proof of Lemma 4.5 in [13]. This quantity seems to us a crucial ingredient for providing upper bounds for the noise of ancestral reconstruction, and a quietly significant open problem in this field.
We tie up this analysis with the bounds of [6], the current standard for inferring sequence lengths required for the correct estimation of distances under the CFN model. From Sect. 2, we infer the sequence length N required for estimating a single phylogenetic distance of length at least M within ϵ/2 error with probability at least 1−ξ, namely
Some rearrangement yields
In our case, M=7λ 0+3β(λ max (ϵ))−5ϵ. The above analysis shows that for small values of ϵ the dominant term in M will be 3β(λ max (ϵ))=Ω(1/ϵ). On the other hand, it is likely that for any algorithm relying on ancestral reconstruction, distances of at least 2β will have to be estimated (for instance the distance between the character sequences learned at two different internal nodes from disjoint clades rooted at said nodes).
Plugging in M=2β(λ max (ϵ)) and β(λ max (ϵ))≥0.1956/ϵ+0.4594 yields

For ϵ=λ 0/2 and ξ=0.1 the above bound yields N≥1.08⋅1013, which is clearly prohibitive from the point of view of practical application.
We thus conclude that, despite the theoretical value of the asymptotic results presented here and in related papers, the underlying mathematical machinery of ancestral sequence reconstruction does not yet readily provide practical alternatives for the “traditional” phylogeny reconstruction algorithms. It is however possible that a substantial tightening of the bounds presented above, together with a relaxation of the “stability” requirements for recursive ancestral sequence learning, may eventually provide such an alternative.
Rights and permissions
About this article
Cite this article
Mihaescu, R., Hill, C. & Rao, S. Fast Phylogeny Reconstruction Through Learning of Ancestral Sequences. Algorithmica 66, 419–449 (2013). https://doi.org/10.1007/s00453-012-9644-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00453-012-9644-4