
Abstract
Oxidative phosphorylation (OXPHOS) is the mechanism whereby ATP, the major energy source for the cell, is produced by harnessing cellular respiration in the mitochondrion. This is facilitated by five multi-subunit complexes housed within the inner mitochondrial membrane. These complexes, with the exception of complex II, are of a dual genetic origin, requiring expression from nuclear and mitochondrial genes. Mitochondrially encoded mRNA is translated on the mitochondrial ribosome (mitoribosome) and the recent release of the near atomic resolution structure of the mammalian mitoribosome has highlighted its peculiar features. However, whereas some aspects of mitochondrial translation are understood, much is to be learnt about the presentation of mitochondrial mRNA to the mitoribosome, the biogenesis of the machinery, the exact role of the membrane, the constitution of the translocon/insertion machinery and the regulation of translation in the mitochondrion. This review addresses our current knowledge of mammalian mitochondrial gene expression, highlights key questions and indicates how defects in this process can result in profound mitochondrial disease.
Similar content being viewed by others


Introduction
Mitochondria are organelles that perform several different functions crucial for the homeostasis of most eukaryotic cells. The suggestion has been made that these double-layered organelles derive from the endocytosis of an α-proteobacterium by a pre-eukaryotic cell (Gray 1999; Falkenberg et al. 2007) and that the bacterium was retained as it conferred a selective advantage through the production of ATP. This molecule provides chemical energy for cells and is a product of oxidative phosphorylation (OXPHOS), which takes place at the inner mitochondrial membrane (IMM). Oxidative phosphorylation utilises five multi-subunit complexes, of which four contain a unique combination of both nuclear and mitochondrial (mt) DNA-encoded polypeptides. Human mitochondria house multiple copies of a 16.6-kb circular genome, mtDNA, which encodes 13 proteins translated from 11 mt-mRNA species (9 monocistronic and 2 bicistronic units), plus two mt-rRNAs and 22 mt-tRNAs (Anderson et al. 1981). The 13 polypeptides are all components of the OXPHOS machinery and are synthesised within the organelle by using the mitochondrial translation mechanism, the main component being the mitoribosome (O'Brien 1971). Although all RNA components of this particle are mtDNA-encoded, all the 80 or so protein constituents are derived from nuclear genes, as are all the accessory and biogenesis factors involved in intramitochondrial protein synthesis. The nuclear-encoded proteins are synthesised on cytosolic ribosomes, targeted to the mitochondria and imported into the matrix (for a review, see Neupert 2015). The assembled mitoribosome translates the mt-mRNAs, synthesising proteins that are rapidly inserted into the IMM and integrated into their relevant complexes to form the OXPHOS system.
This review aims to summarise the current knowledge of the mammalian mitochondrial translation system including mitoribosomal biogenesis, pre- and post-translational events involving modification, stabilisation and the fate of mitochondrial transcripts. Finally, a brief overview of known pathogenic mutations related to this process will also be presented.
The mitoribosome
The mitochondrial protein synthesis machinery was first identified by John R. McLean in 1958 in rat liver (McLean et al. 1958) and then isolated from the same organ by Thomas W. O’Brien in 1968 (O'Brien and Kalf 1967). Although mammalian mitoribosomes, like other ribosomes, are composed of a large (mt-LSU) and a small (mt-SSU) subunit, they differ markedly in their density, sedimenting as 55S particles rather than the 80S or 70S species of their eukaryotic cytosolic or bacterial counterparts, respectively (O'Brien 1971). The first cryo-electron-microscopic (cryo-EM) structure of the bovine monosome (the fundamental synthesising particle comprising mt-SSU and mt–LSU) was released in 2003 and showed how significantly the mammalian mitoribosome diverged from its bacterial ancestor (Sharma et al. 2003). The recent release of sub-nanometre resolution structures of porcine (Greber et al. 2014a, 2014b, 2015) and human (Brown et al. 2014; Amunts et al. 2015) mitoribosomes from cryo-EM studies, further highlights these structural and compositional differences (Table 1); an excellent recent review has been published on the structure of the mammalian mitoribosome (Greber and Ban 2016). Whereas bacterial ribosomes are mainly composed of RNA (~70:30% per weight), mammalian mitoribosomes have reversed this ratio and are higher in protein content. This is the result of both the removal of rRNA domains and the increase of the overall protein mass. The increase is effected by the addition of extensions onto many homologous proteins and the acquisition of mitochondrial-specific (mt-specific) proteins, some of which have previously been identified in early proteomics studies (Koc et al. 2001a, 2001b). In a few cases, the new protein mass fills the void generated by the rRNA deletion but, overall, the increase of protein content is mainly found peripherally (Brown et al. 2014) and has been suggested to act as a protective shield for the mt-rRNA, preventing potential damage attributable to the high ROS levels found within the organelle (Lightowlers et al. 2014). The presence of the majority of the mt-specific proteins on the external surface of the subunits suggests that the functional core of the mitoribosome, composed of the mt-mRNA recognition site on the mt-SSU (Amunts et al. 2015; Greber et al. 2015) and peptidyl transferase centre in the mt-LSU (Brown et al. 2014; Greber et al. 2014a, 2014b), has been preserved. This exchange of RNA predominance to protein in the mitoribosome also leads to a different composition of the intersubunit bridges. In bacteria, the two ribosomal subunits interact mainly via RNA:RNA bridges (Liu and Fredrick 2016), whereas in the mammalian 55S particle, a higher proportion of protein-protein and RNA:protein connections is found (Amunts et al. 2015; Greber et al. 2015).
The newly designated ribosomal nomenclature will be used in this review and identifies mt-specific proteins with “m”, universal orthologous proteins with “u” and bacterial-specific orthologues with “b”. This initial letter is followed by “L” for the mt-LSU or “S” for the mt-SSU, which is then followed by an assigned number. In the case of “b” and “u” ribosomal proteins, the letter “m” is also added at the end of the name to distinguish the mitochondrial proteins from the cytosolic ones (Ban et al. 2014).
Mitoribosomal small subunit (28S)
To date, the mammalian mt-SSU is thought to be composed of a 12S mt-rRNA and 30 proteins, of which 15 (porcine) or 14 (human) are mt-specific (Amunts et al. 2015; Greber et al. 2015). The increased protein content of the mitoribosome results in a more elongated subunit compared with the shape of the bacterial counterpart. One of the most remodelled areas is the entrance of the mt-mRNA channel. In the bacterial ribosome (Fig. 1a), uS4 and the C-terminus of uS3 are important for defining the ring-shaped entrance of the channel; both are absent in mt-SSU. The mammalian mitoribosomes (Fig. 1b) compensate for this loss by the extension to uS5m, which now defines the entrance of the channel. The mt-specific protein, mS39 (PTCD3), also lies in close proximity to this channel. PTCD3 is a member of the pentatricopeptide-repeat (PPR)-containing protein family, characterised by their RNA-binding ability (Filipovska and Rackham 2013; Lightowlers and Chrzanowska-Lightowlers 2013). This feature combined with the location of mS39 suggests that it is involved in the recruitment of mt-mRNA to the monosome. One domain of the 12S mt-rRNA that has been deleted is the anti-Shine-Dalgarno sequence, which would conventionally be located close to the exit of the mRNA channel. The absence of this domain is consistent with and perhaps reflects the absence of the corresponding 5’-untranslated region (UTR) on mt-mRNAs (Montoya et al. 1981). The space generated by the lack of this rRNA domain is now occupied by the mitochondrial-specific protein mS37, which takes on the interaction with the 12S mt-rRNA. In contrast to these structural modifications, the central portion of the mRNA channel, which is directly involved in the translation process, is mostly conserved (Greber et al. 2014a, 2014b).

Comparison of structural features of bacterial and human mitochondrial ribosomes. Structures of the Escherichia coli ribosome (PDB 4YBB) and the human mitoribosome (PDB 3J9M) were obtained with Pymol (Open Source, Version 1.8.2.0.). The respective monosomes are depicted left (E. coli) and right (Homo sapiens) with the location of the entrance to the mRNA tunnel (a, b) and the exit site of the polypeptide tunnel (c, d) circled to indicate the region expanded in the main part of the figure (green small subunit structures, blue large subunit structures). Comparison of the entrance to the mRNA tunnel indicates that uS3 (dark pink) and uS5 (yellow) are present in the ribosomes from both human (b) and bacterial a) mitochondria. The bacterial entrance site is, in part, defined by uS4 (red), whereas in mitoribosomes, an additional RNA-binding protein, mS39 (orange), lies close to the entrance and is involved in mRNA recruitment. The bacterial polypeptide exit site (c) is defined by uL22 (orange), uL23 (pink), uL24 (yellow) and uL29 (dark pink). All of these are present in human mitoribosomes (d), with a further ring of proteins surrounding the exit site composed of mL39 (cyan), mL44 (brown) and mL45 (red). Amongst these, mL45 might be involved in anchoring the structure to the inner mitochondrial membrane
Mitoribosomal large subunit (39S)
As with the mt-SSU, several differences can be seen between mt-LSU and its bacterial counterpart. The mammalian mt-LSU is composed of 16S mt-rRNA and the recent ribosomal nomenclature lists 52 proteins (53 in humans), of which 22 are mt-specific proteins (Brown et al. 2014; Greber et al. 2014a, 2014b). With respect to the RNA content, the bacterial LSU contains the 23S and the 5S rRNA, located in the central protuberance (Ban et al. 2000). Intriguingly, the cryo-EM structure of the mammalian mitoribosome reported in 2014 has revealed an RNA density that resembles a domain of bacterial 5S rRNA (Greber et al. 2014a, 2014b) but the authors have been unable to identify its nature. More recent cryo-EM studies of porcine and human mt-LSU particles have confirmed the presence of an additional RNA species and identified this as mt-tRNAPhe (Greber et al. 2014a, 2014b) and mt-tRNAVal (Brown et al. 2014), respectively. Intriguingly, these two mt-tRNA genes are found in close apposition to the 12S and 16S mt-rRNA genes and are transcribed as a single polycistronic RNA unit in mammalian mitochondria. Whereas the mt-LSU from all mammalian species analysed contain only one or other of these mt-tRNA species (and not the other 20 species), no evidence has been obtained for tissue-specific variation. Further, under conditions of mt-tRNAVal depletion caused by a destabilising pathogenic mutation, the human mitoribosome has recently been shown to be able to switch to the incorporation of mt-tRNAPhe and is still functional (J. Rorbach et al., in preparation). One of the areas of the mitoribosome that has changed the most throughout evolution is the polypeptide exit site (PES; Fig. 1c, d). The exit region of the tunnel is defined by a universally conserved ring of proteins (uL22m, uL23m, uL24m) but, in mammals, a second layer of additional mt-specific proteins (mL39, mL44, mL45) are found extending the conserved ring (Fig. 1d). The recruitment of these additional proteins around the exit site might be linked to the synthesis of highly hydrophobic mt-encoded proteins (see Localisation of mitochondrial translation).
Mitoribosomal A-, P- and E-sites
As described earlier, the functional core of the mitoribosome has been conserved throughout evolution. Similarly to most ribosomes, mitoribosomal A-, P- and E-sites can be identified (Wettstein and Noll 1965). The A-site residues contributed by the mt-SSU are important for decoding, and the mt-LSU has maintained the ability to mediate the main contact between mt-tRNA and 16S mt-rRNA, although the A-site finger structure is missing as a consequence of a loss during evolution of the specific rRNA domain (Greber et al. 2014a, 2014b, 2015). This loss might be related to the different characteristics of mammalian mt-tRNA, which do not all adopt the typical cloverleaf structure (Suzuki et al. 2011). The P-site is also relatively preserved but the interactions made with the mt-tRNA are stronger than those observed for the bacterial ribosome (Schuwirth et al. 2005), as the mt-LSU has a P-site finger that interacts with the T-loop of mt-tRNA while it is bound to the site. Since this T-loop of mammalian mt-tRNAs is smaller than those in bacterial tRNA, the finger-like structure is probably necessary to maintain the required orientation of the RNA species during peptide bond formation. Finally, the presence of an E-site is controversial, as most of the contact points that exist between the bacterial ribosome and tRNA seem to be lost as a consequence of the shortened mt-rRNA. However, the existence of this site has been confirmed in recent cryo-EM studies that have highlighted the presence of a modified binding pocket (Greber et al. 2014a, 2014b).
Additional roles of mt-specific proteins
Some of the mt-specific mitoribosomal proteins have been suggested to have additional roles. Examples include mS29 (DAP3; Kissil et al. 1999), mL37 (Levshenkova et al. 2004), mL41 (Yoo et al. 2005) and mL65 (previously named mS30; Sun et al. 1998), all of which have been linked to the control of apoptosis, whereas bL12m has been implicated in POLRMT function and stability (Surovtseva et al. 2011; Nouws et al. 2016). In addition to a potential role in apoptosis, mS29 has an intrinsic GTP-binding site and is phosphorylated, which may affect monosome formation since its location is at the interface (Amunts et al. 2015; Greber et al. 2015). The cryo-EM studies performed on human and porcine samples have also confirmed the presence of a ribosome-dependent peptidyl-tRNA hydrolase, ICT1, as a structural component of the mt-LSU (Richter et al. 2010). Although levels of free ICT1 in the mitochondrion are reported to be extremely low (Richter et al. 2010; Chrzanowska-Lightowlers and Lightowlers 2015), ICT1 might function to help release stalled mitoribosomes (Akabane et al. 2014; Feaga et al. 2016). One particularly striking additional role for a mitoribosomal protein concerns uL18m (Zhang et al. 2015). Under conditions of heat shock, a cytosolic isoform of this protein is generated through translational initiation at an internal CUG codon of its cognate transcript. This isoform is integrated into cytosolic 80S ribosomes, which selectively translate mRNAs encoding heat shock proteins. This is a unique example of the way that a mitoribosomal protein can influence stress adaptation in the eukaryotic cell. A role for the same protein, uL18M, has been suggested for 5S RNA import into mitochondria (Smirnov et al. 2011) but no potential function of 5S RNA in mitochondria has yet been determined.
Pre-translation events
Mitoribosomal biogenesis and processing and the maturation of the mt-transcripts are all prerequisites for intraorganellar protein synthesis. The assembly pathway of the mitoribosomal subunits, however, and the mechanism of recruitment of mt-mRNAs to the mitoribosome remain largely uncharacterised.
Mt-mRNA processing and stabilisation
All the mt-mRNA transcripts, with the exception of MTND6, are matured by a poly(A) polymerase (mtPAP; Tomecki et al. 2004; Slomovic et al. 2005). This process introduces a poly- or oligo(A) extension that serves to complete the UAA stop codon in 7 transcripts (Ojala et al. 1981). Unlike the bacterial or cytosolic counterpart, the role that this modification exerts on mt-mRNA stability does not follow a conventional pattern. In the absence of polyadenylation, a consistent effect is seen on the steady state levels of most transcript species, albeit that this may be an increase or decrease. This pattern is observed irrespective of the mechanism by which the poly(A) tail had been removed, although the manner in which this regulation is effected is still unclear (Tomecki et al. 2004; Nagaike et al. 2005; Wilson et al. 2014; Bratic et al. 2016).
The half-lives of mt-mRNAs can be regulated by cis-acting elements, as described above, and by trans-acting factors. The best characterised mt-mRNA-specific proteins involved in transcript stability are the LRPPRC/SLIRP complex (Sasarman et al. 2010) and FASTKD4, which prevents their degradation (Wolf and Mootha 2014). The only protein-coding transcript derived from the light strand, MTND6, interacts with the mitochondrial isoform of FASTK (Jourdain et al. 2015) and GRSF1, which modulates its stability (Antonicka et al. 2013; Jourdain et al. 2013).
Maturation of mt-tRNAs
Modifications of bacterial tRNAs are introduced to mediate stability and functionality. This is equally true of mammalian mt-tRNA species (Nagaike et al. 2005; Suzuki et al. 2011). The many different modifications have been comprehensively described by Suzuki (2014) and Salinas-Giege et al. (2015). Once the mt-tRNAs have been modified and the CCA added to the 3’ terminus (Nagaike et al. 2001), the transcripts can be charged with their cognate amino acid by the relevant mitochondrial aminoacyl-tRNA synthetase (Diodato et al. 2014) and can participate in the synthesis of mt-encoded proteins.
Mitoribosomal assembly
Several studies support the hypothesis that the assembly of the mitoribosome takes place in two mitochondrial subcompartments, defined as nucleoids and RNA granules. Nucleoids are centres of mtDNA maintenance, replication and transcription, whereas post-transcriptional RNA processing and maturation occur in the RNA granules. Both the compartments contain mitoribosome assembly factors and proteins involved in RNA stability, plus a number of mitoribosomal proteins (Bogenhagen et al. 2014; Antonicka and Shoubridge 2015). As a consequence, early mitoribosome biogenesis has been suggested to be initiated in the nucleoids and to be completed in the RNA granules. Irrespective, RNA processing and at least the early stages of mitoribosome assembly clearly occur in close proximity to mtDNA, consistent with co-transcriptional processing and even mitoribosomal assembly.
The biogenesis of ribosomes involves several key players that act on rRNA and ribosomal proteins to assemble complete subunits. Relatively few factors have been identified that are necessary for mammalian mitoribosomal assembly compared with the generation of 80S ribosomal particles (Hage and Tollervey 2004) and, although the steps in the process are mostly undefined, an increasing number of crucial factors are emerging. After transcription, the 12S mt-rRNA is processed from the major polycistronic RNA unit and, at some stage in the assembly pathway, is bound at its 3’ terminal helix by an RNA-binding GTPase, ERAL1, which stabilises the mt-rRNA prior to the final maturation of the mt-SSU (Dennerlein et al. 2010; Uchiumi et al. 2010). Both the 12S and 16S mt-rRNA species are known to be modified. The extent of mt-rRNA modification was originally assessed in hamster RNA, where nine modifications were detected (Dubin and Taylor 1978). These included pseudouridylation, base methylation and 2’-O-ribose methylation at conserved sites. A uracil methylation site was also identified on the 12S mt-rRNA but has not yet been confirmed to be present on the human orthologue. Of the five base methylations confirmed in 12S mt-rRNA (Metodiev et al. 2009), the data are consistent with the modifications being performed by TFB1M (adenine; Seidel-Rogol et al. 2002; Metodiev et al. 2009) and NSUN4 (cytosine; Metodiev et al. 2014). The 16S mt-rRNA is also a substrate for modifications including three 2’-O-ribose methylations performed by methyltransferases (MRM1, MRM2, MRM3; Rorbach et al. 2014; Lee and Bogenhagen 2014) and, potentially, one pseudouridylation (Ofengand and Bakin 1997). The latter was not detected in the hamster-derived 16S and the enzyme that could be responsible is still unknown.
The formation of many ribonucleoprotein particles including ribosomes requires GTPases and ATP-dependent RNA helicases, although, to date, fewer than expected have been identified in mammalian mitochondria (for a review, see De Silva et al. 2015). The energy derived from GTP hydrolysis is used to regulate the association or dissociation of proteins or to promote conformational changes. These proteins might also act as placeholders for proteins that will join the immature ribosome at a later stage in the assembly process. Finally, because GTPases are usually active in their GTP-bound state, these proteins might act as sensors of the GTP/GDP ratio and respond under starvation conditions, when the GTP/GDP ratio is lower, by reducing mitoribosome assembly to match the reduced nutrient availability. At present, two mitochondrial GTPases (Mtg1, Mtg2) are associated with the IMM and have been reported to interact with the immature mt-LSU (Kotani et al. 2013), whereas, thus far, only one (C4orf14, or NOA1) has been shown to be involved in mt-SSU assembly (He et al. 2012).
ATP-dependent RNA helicases bind and remodel RNA possibly promoting the unwinding of RNA to initiate ribonucleoprotein (RNP) assembly or the displacement of RNA from RNP particles (Linder and Jankowsky 2011). At present, few such helicases have been identified in human mitochondria and, of these, SUPV3L1 is important in mt-RNA metabolism and for the stable maintenance of mtDNA (Borowski et al. 2013), whereas DDX28 (Tu and Barrientos 2015) and DHX30 are both involved in mitoribosome assembly (Antonicka and Shoubridge 2015).
A number of other factors have been identified as being involved in the assembly of mammalian mitoribosomes. Knockout of the gene encoding mTERF3 in mice and flies causes an increase in mtDNA transcription. Intriguingly, biogenesis of mt-LSU is also affected. RNA immunoprecipitation assays have revealed an association with 16S rRNA, consistent with mTERF3 playing a role in the assembly of the mt-LSU (Wredenberg et al. 2013). Other non-mitoribosomal proteins have been shown by RNA immunoprecipitation to bind 16S rRNA, such as a member of the Fas-activated Serine Threonine Kinase family FASTKD2 and the aforementioned helicase DDX28, both of which have been shown to be required for mt-LSU assembly (Antonicka and Shoubridge 2015; Popow et al. 2015; Tu and Barrientos 2015). Interestingly, the mAAA-protease, a membrane-spanning homo- or hetero-oligomer of two proteins AFG3L2 and paraplegin, also appears to affect mt-LSU assembly. Prior to insertion into the mt-LSU, cleavage of the subunit bL32m has been shown to be important in certain species but this does not happen in the conventional fashion. Rather than co-translocational cleavage by the matrix metalloendopeptidase, maturation occurs following complete import and is effected by the mAAA-protease (Nolden et al. 2005). The autosomal recessive spastic paraplegia type 7 can be caused by pathogenic mutations in paraplegin, the loss of which has been shown, in yeast and certain mouse tissues, to reduce the cleavage of bL32 leading to a mitoribosomal assembly defect. Similarly, defects of AFG3L2 can cause a rare form of spinocerebellar ataxia (SCA28) and tissue-specific mouse knockouts result in decreased steady state levels of bL3m (Almajan et al. 2012). C7orf30 (MALSU1) promotes the incorporation of uL14m into the mt-LSU, an event that is necessary for subunit stability (Rorbach et al. 2012; Fung et al. 2013). Depletion of the IMM protein MPV17L2, which is known to interact with the mt-LSU, leads to a reduction of both subunits, highlighting a possible role for this protein in their assembly (Dalla Rosa et al. 2014). Finally, a small proportion of GRSF1 has been suggested to be involved in the assembly of the mt-SSU, as its depletion leads to the accumulation of incomplete mt-SSU; however, this has not been experimentally determined (Antonicka et al. 2013; Jourdain et al. 2013).
Molecular mechanisms of mitochondrial translation
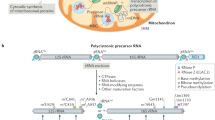
Transcription of human mtDNA is driven from within the noncoding region by promoters on both strands to form polycistronic transcription units (Gustafsson et al. 2016). Processing of these units is driven in part by the natural positioning and folding of mt-tRNA structures, which are excised by the tRNase Z ELAC2 mitochondrial isoform (Rossmanith 2011) and the three subunit protein-only mitochondrial RNase P (Holzmann et al. 2008). Maturation of all light-strand protein-encoding RNAs is facilitated by the addition of a poly(A) tail of approximately 50 nucleotides by the mitochondrial poly (A) polymerase (Tomecki et al. 2004, Temperley et al. 2010a, 2010b). The resultant nine monocistronic and two dicistronic mt-mRNA species are translated by the mitoribosome in a process that can be divided into initiation, elongation, termination and recycling (Fig. 2; for reviews, see Christian and Spremulli 2012; Ott et al. 2016). These mt-mRNA species have a modified codon usage. Mitochondria from different organisms vary in which codon is reassigned but an almost universal change is for the UGA stop codon to be recognised as tryptophan. Other changes include the recognition of AUA as methionine and AGA or AGG becoming unassigned codons that are not recognised by any mt-tRNA or protein factor (Chrzanowska-Lightowlers et al. 2011; Suzuki et al. 2011).

Representation of protein synthesis in human mitochondria showing the four phases of initiation, elongation, termination and recycling that comprise mitochondrial translation. Following ribosome recycling (top), the mitochondrial small subunit (mt-SSU in blue) remains bound to initiation factor mtIF3 (dark green). Initiation commences as mtIF2 (light green) bound to a GTP molecule (red) joins this complex. Once successful recruitment of mRNA has been achieved and fMet-tRNAMet in the P-site anchors to the start codon, GTP is hydrolysed to GDP (orange), the initiation factors are released and the mitochondrial small subunit (mt-LSU in darker blue) can associate, forming the monosome. During elongation (centre), the nascent polypeptide chain is bound to a P-site tRNA, while the A-site is sampled by charged mt-tRNAs delivered by mitochondrial elongation factor-Tu (mtEF-Tu in yellow), until the correct codon-anticodon pair forms. GTP hydrolysis and mtEF-Tu release follows together with exchange of the GDP (light orange) for a new GTP molecule mediated by mtEF-Ts (orange). The charged A-site mt-tRNA changes its conformation juxtaposing its amino acid to that of the extending nascent chain within the peptidyl-transferase centre. This facilitates peptide bond formation transferring the polypeptide chain onto the A-site mt-tRNA. The elongation factor mtEF-G1 (dark orange) promotes the ribosome movement that repositions the mt-mRNA within the 55S and the mt-tRNAs from the A- and P-sites to the P- and E-sites. The E-site mt-tRNA leaves the monosome in anticipation of a new round of elongation. This cycle continues until the polypeptide is complete and a stop codon is presented in the A-site. Termination (bottom) described the recognition of the stop codon by a release factor protein (mtRF1a in pink), which then adopts a modified conformation that promotes hydrolysis of the ester bond anchoring the nascent chain to the final mt-tRNA. Once the polypeptide chain is released, the two recycling factors, mtRRF1 (dark red) and mtRRF2 (red), promote the dissociation of the ribosomal subunits and premature re-association is prevented by the formation of an mtIF3/mt-SSU complex
The first step in the initiation of protein synthesis is the recruitment of the mt-mRNA to the mt-SSU, which is bound by the initiation factor mtIF3 to inhibit premature re-association with the mt-LSU. The PPR protein, mS39, found at the entrance of the mt-mRNA channel has been reported to aid recruitment of the mt-mRNA (Amunts et al. 2015; Greber et al. 2015). The codons recognised as initiating triplets by the mitoribosome are AUG, AUA and AUU to which f-Met-tRNAMet (a subset of met-tRNAMet is formylated by mitochondrial methionyl-tRNA formyltransferase; Tucker et al. 2011) is recruited by mtIF2:GTP. Charged mt-tRNAMet and mtIF2 can bind the mt-SSU in the absence of the mt-mRNA; however, the association is considerably weaker. If a positive codon:anticodon interaction occurs, then a stable complex is created and the interaction with the mt-LSU follows. This formation of the monosome triggers hydrolysis of the mtIF2-bound GTP to GDP concomitantly with the release of initiation factors 2/3 from the mt-SSU. If f-Met-tRNAMet is not available or if the start codon is not present in the P-site, the inspection fails and the mRNA is released.
In mt-mRNAs, the start codon is found at or very near the 5’ terminus in monocistronic transcripts, with efficiency of translation initiation being predicted to be reduced the more distal the start codon (Christian and Spremulli 2010). The two bicistronic RNA units, RNA7 (MTND4/MTND4L) and RNA14 (MTATP8/MTATP6), present an enigma with respect to translation initiation. Since these transcripts contain overlapping open reading frames (ORF), the downstream ORF has, by default, a 5’-UTR that consists of the upstream coding sequence. How the mitoribosome locates this internal initiating codon is still unclear.
Once the monosome is formed, elongation of the nascent chain can start. In this step, a ternary complex forms composed of the mitochondrial elongation factor mtEF-Tu, GTP and a charged mt-tRNA, which can enter the A-site. If the correct codon:anticodon interaction takes place, the mitoribosome stimulates the hydrolysis of GTP leading to the release of GDP:mtEF-Tu. The GTP:mtEF-Tu complex is restored by the direct interaction of mtEF-Tu with the nucleotide exchange factor mtEF-Ts (Cai et al. 2000). After the release of mtEF-Tu, the formation of the peptide bond is catalysed at the peptidyl transferase centre (PTC) in the mt-LSU. Once the bond is formed, the P-site of the mitoribosome is occupied by a deacylated mt-tRNA, and the dipeptidyl-tRNA is found in the A-site. The interaction of the elongation factor mtEF-G1 with the mitoribosome alters its structural conformation, which leads to the release of the mt-tRNA from the P-site and the movement of the dipeptidyl-tRNA by three nucleotides into the P-site. Recently, the presence of the E-site in mammalian mitoribosome has been confirmed (Amunts et al. 2015; Greber et al. 2015). Although different from the characteristic bacterial E-site, the deacylated mt-tRNA moves to this site before exiting the mt-monosome. The elongation process is then reiterated until a stop codon is positioned in the A-site.
Protein synthesis is complete once a stop codon enters the A-site but the nascent peptide then needs to be released from the mitoribosome. The termination codon is recognised by a protein rather than by a tRNA species and, in human mitochondria, mitochondrial release factor 1a (mtRF1a) is believed to be sufficient to terminate all 13 ORFs (Soleimanpour-Lichaei et al. 2007). This factor is a class I RF, which, unlike class II, demonstrates sequence-specific recognition of the A-site codon (UAA, UAG). This first step in termination is mediated by the interaction of two protein domains with the RNA, the tripeptide motif and the tip of the α-5 helix. Once these two regions have recognised the A-site triplet as a stop signal, the RF structure alters to position a second conserved domain, the GGQ motif (Frolova et al. 1999) into the PTC. This facilitates the hydrolysis of the ester bond between mt-tRNA and the final amino acid. Hence, mtRF1a in the presence of GTP promotes the release of the polypeptide from the mt-LSU (Schmeing et al. 2005). In human mitochondria, UAA and UAG are used as stop codons to terminate nine monocistronic and two bicistronic ORFs, respectively. The sequencing of human mtDNA in 1981 logically suggested that, since the triplets following the coding sequence for the remaining two ORFs, MTCO1 and MTND6, were AGA and AGG, respectively, these must have been recoded as alternative stop codons, as no corresponding tRNA was present in the mitochondrial genome (Anderson et al. 1981). Fine mapping of the termination codons of transcripts positioned at the A-site of the human mitoribosome in intact cells, however, showed that these two species terminated with the classical UAG codon (Temperley et al. 2010a, 2010b). An explanation for this observation is that a −1 frameshift occurs, potentially driven by structured RNA immediately downstream of the termination codons within the transcripts. Although this mechanism is plausible for humans, the frameshift alone may not create a stop codon in other vertebrates. More recently, another member of the mitochondrial translation release factor family, ICT1, has been suggested to be involved in the termination of the synthesis of COXI and ND6. As discussed above, ICT1 is a structural component of mt-LSU but is not located in the proximity of the A-site. Despite this, a limited free pool of ICT1 might have peptidyl hydrolase activity, analogous to the bacterial ArfB (YaeJ; Akabane et al. 2014). Recent studies of isolated ICT1 have confirmed its ability to hydrolyse peptidyl-tRNA on stalled ribosomes but, intriguingly, not when the RNA template extends more than 14 nucleotides past the A-site (Feaga et al. 2016). This has led to the conclusion that ICT1 is unlikely to act as a natural translation terminator in vivo, as MTCO1 and MTND6 mt-mRNA have 3’ extensions longer than 14 nucleotides. As four members of the mammalian mitochondrial translation release factor family have been found (Chrzanowska-Lightowlers et al. 2011), other release factors might be involved in terminating the translation of these two mt-mRNAs, perhaps in other mammalian species (Young et al. 2010).
After the release of the polypeptide, two ribosomal recycling factors, mtRRF1 and mtEF-G2, promote the dissociation of the ribosomal subunits and the release of mt-mRNA and deacylated mt-tRNA (Rorbach et al. 2008; Tsuboi et al. 2009). These two factors are finally released and the translation cycle can reinitiate.
Regulation of mitochondrial translation
As the components of the OXPHOS complexes are synthesised both in the cytosol and in the mitochondrial matrix, their synthesis must be coordinated in order to lead to an efficient assembly of the complexes. Translational activators abound in yeast mitochondria. These proteins regulate the synthesis of various proteins and associate selectively with the (mainly 5’) UTRs of all yeast mt-mRNA species (for a review, see Herrmann et al. 2013). Their exact mode of function remains unknown but these activators establish a feedback loop whereby the absence of available partners to produce a complete OXPHOS complex can inhibit further translation of the associated transcript. The absence of UTRs in the majority of human mitochondrial transcripts would appear to preclude the functioning of such proteins. However, one translational activator has been identified in human mitochondria, namely TACO1 (Weraarpachai et al. 2009). The absence of this activator in patients with pathogenic mutations of TACO1 results in the selective loss of MTCO1 translation, which encodes COXI of complex IV. The mechanism of action of TACO1 is not known but cannot be mediated via a 5’-UTR as no such sequence exists on MTCO1. It has been postulated to promote the recognition of the start codon of MTCO1 or to stabilise the polypeptide during its synthesis. TACO1 might also interact with the translation termination factor to ensure that the nascent polypeptide is not released prior to its completion (Weraarpachai et al. 2009).
As translational activators are unlikely to work in a similar fashion to those in yeast, how can the level of mitochondrial translation be modulated in response to the import of cytosolic components of the OXPHOS complexes? An important insight into this process has been advanced by the identification of MITRAC (the mitochondrial translation regulation assembly intermediate of cytochrome c oxidase (Mick et al. 2012). This dynamic complex appears to connect the assembly of cytochrome c oxidase (COX) with the synthesis of the mitochondrially encoded COXI. The molecular mechanisms underlying this link are unclear. However, pathogenic mutations have been reported in two MITRAC components C12orf62 and MITRAC12, which function early in COX assembly, and their loss results in the inhibition of COXI synthesis (Szklarczyk et al. 2012; Weraarpachai et al. 2012). No similar complex has been reported to coordinate complex I assembly with the synthesis of mitochondrial components, although a link for the assembly of complex III has been suggested (Tucker et al. 2013).
The cellular environment has also been suggested to have an effect on mitochondrial translation. In the cytosol, microRNAs interact with the proteins AGO2 and GW182, creating a complex that is able to reduce the cytosolic translation of mRNAs (Czech and Hannon 2011). MicroRNAs and AGO2 have also been postulated to form a complex within mitochondria, where, conversely, they have been reported to enhance the translation of certain transcripts during muscle differentiation (Zhang et al. 2014). The existence of microRNAs in mitochondria, however, is still controversial, particularly as no evidence has been found for the enrichment of microRNAs reported in the first detailed analysis of the transcriptome from purified human mitochondria (Mercer et al. 2011).
Mitochondrial protein synthesis can be regulated by post-translational modifications of mitoribosomal components. These can be phosphorylated (Miller et al. 2009) or acetylated as a result of the levels of ATP, acetyl-CoA and NADH within mitochondria. A role in the association of the mt-LSU and mt-SSU might be played by the mt-specific protein mS29 (see above). This protein is able to bind GTP and is found bound to GDP in the mammalian 55S structures (Amunts et al. 2015; Greber et al. 2015). The binding affinity is higher for the mt-SSU than for the monosome (O’Brien et al. 1990; Denslow et al. 1991), suggesting a possible regulatory effect of GTP-hydrolysis on mS29 (Amunts et al. 2015). In addition, mS29 contains phosphorylation sites on its intersubunit face (Miller et al. 2008) possibly further influencing the formation of the monosome (Miller et al. 2009). In addition to this example, several other mitoribosomal proteins are reported to be modified and are found in the proximity of the subunit interface or in domains crucial for translation, such as the mRNA channel, the PTC or the PES (Miller et al. 2009). The assembly of the monosome is also promoted by the complex mTERF4-NSUN4, which interacts with the mt-SSU and supports the recruitment of the mt-LSU (Cámara et al. 2011; Metodiev et al. 2014).
Localisation of the mitoribosome and mitochondrial translation
Mitochondrial ribosomes have evolved to translate highly hydrophobic components of the OXPHOS chain that need to be inserted into the membrane to prevent aggregation and precipitation in the matrix. As a consequence, the mitoribosome is probably anchored to the membrane in close proximity to the PES to align with the insertion machinery. This interaction has been shown in yeast in recent cryo-EM tomography studies (Pfeffer et al. 2015). Biochemical studies with the bovine mitoribosome have demonstrated that approximately 40% of mitoribosomes interact with the IMM (Li and Spremulli 2000) but the way that this interaction is mediated is still unclear. This same study has suggested that the association is mediated by electrostatic interactions between mitoribosomal proteins and the IMM and by interaction between the mitoribosome and IMM proteins.
Few proteins have been reported to interact with both the IMM and the mitoribosome. One of these is OXA1L, a polytopic membrane insertase and member of the YidC/Oxa/Alb3 family, which mediates the insertion of proteins within the IMM (Hennon et al. 2015). Its homologue in yeast has been suggested to interact with the mitoribosome (Jia et al. 2003) and the C-terminus of the human protein has been cross-linked to components of the mt-LSU, namely uL13m, bL20m, bL28m, mL48, mL49 and mL51 (Haque et al. 2010), although these are not located in close proximity to the PES. MPV17L2 is an integral membrane protein that has been co-localised with the mt-LSU on sucrose gradients, suggesting its involvement in anchoring the subunit to the IMM (Dalla Rosa et al. 2014). LetM1, another IMM protein, contains a large matrix domain and its homologue in yeast (Mdm38) has been reported to interact with the mitoribosome (Lupo et al. 2011). In vitro studies performed on LetM1 have shown its interaction with the mitoribosomal protein bL36m (Piao et al. 2009) and led the authors to propose that LetM1 acts as a membrane locator for the mitoribosome. This is an interesting proposal, although the recent cryo-EM data suggest its location is not ideal for localising the mitoribosome to the membrane or for assuring the close proximity of the PES to the membrane.
Indeed, the release of the mammalian mt-LSU structure (Greber et al. 2014a, 2010b) implicates the mt-LSU component, mL45, in membrane association. This protein is localised in close proximity to the PES, a position that is ideal to allow the rapid insertion of the newly synthesised hydrophobic proteins into the membrane. The hypothesis of the involvement of mL45 in the interaction with the membrane is strengthened by its structural homology with the IMM-interacting protein Tim44 (Handa et al. 2007) and by the confirmation of the ability of its yeast homologue Mba1 to mediate the interaction between mitoribosomes and the IMM (Ott et al. 2006; Pfeffer et al. 2015). The putative role of mL45 in anchoring the mitoribosome to the membrane is intriguing but, if it does indeed play this role, it is highly likely to share it with other anchoring sites that lie within the mitoribosome and that may interact directly with the membrane or via an integral membrane protein(s) that function as a receptor(s) for the mitoribosome.
How is the newly synthesised polypeptide correctly inserted into the IMM? Two systems have been evolutionarily conserved to mediate the integration of nascent peptides into membranes: the Sec complex and the YidC/Oxa/Alb3 insertases. To date, the major candidate that has been implicated in this process in mammals is OXA1L (see above), a member of the YidC/Oxa/Alb3 insertase family, which may play a pivotal role in this process, at least for a subset of the mitochondrially encoded proteins. OXA1L is the homologue of yeast Oxa1, which is the key component of the yeast cytochrome oxidase assembly (OXA) translocase and is involved not only in the integration of mitochondrially encoded proteins, but also in the biogenesis of some nuclear-encoded mitochondrial products (Stiller et al. 2016). Intriguingly, knockdown studies of humans cells have shown that OXA1L is necessary to obtain only a functional complex I and V and does not appear to be involved in cytochrome c oxidase or complex III assembly in man (Stiburek et al. 2007). MITRAC and the UQCC (ubiquinol-cytochrome c reductase complex chaperone) members (see above) possibly function without the aid of OXA1L to assemble complexes III and IV. One other member of the YidC/Oxa/Alb3 family that has been reported to be present in human mitochondria is the homologue of the yeast Cox18p. This promotes the insertion of the C-terminal of Cox2p in yeast but its function in mammals has not been fully characterised.
Post-translation events
As previously mentioned, at the end of translation, the mitoribosomal subunits are separated and are recycled. However, what happens to the mt-mRNA?
After translation, the mt-mRNAs are possibly protected by interaction with RNA chaperones (e.g. LRPPRC/SLIRP, FASTKD2, FASTKD4) and reused to programme a new round of translation, although these species might be degraded in order to eliminate aberrant or damaged transcripts. The identity of the RNA-degrading apparatus in mitochondria is still under investigation. Whereas the helicase involved in RNA metabolism has been identified as SUPV3L1, the key component(s) with ribonucleolytic activity may not be completely resolved. SUPV3L1 (Minczuk et al. 2002) has been found to interact with the human polynucleotide phosphorylase (PNPase; Borowski et al. 2010), which, on depletion, causes the accumulation of mt-RNA decay intermediates and the increased stability of mitochondrial transcripts. PNPase clearly is an important factor in mitochondrial RNA degradation (Chujo et al. 2012) but most of this protein is localised in the mitochondrial intermembrane space and a role in RNA import has been suggested (Wang et al. 2010). The degradation of RNA species in mitochondria is likely to be aided by REXO2, a protein found both in the cytosol and in mitochondria. This protein is an exonuclease able to digest very short oligo RNA and might help PNPase or other enzymes to degrade and recycle unwanted RNA (Bruni et al. 2013). More recently, an endoribonuclease, LACTB2, has been identified in mammalian mitochondria. This protein has been thoroughly characterised in vitro, including high resolution structural data, but its physiological function remains unclear (Levy et al. 2016).
Mitochondrial translation and pathologies
Mitochondrial diseases are a class of heterogeneous disorders that can be characterised by mutations of either mtDNA or nuclear DNA and that cause severe defects of OXPHOS function. Symptoms can vary from mild to profound, from highly tissue selective to multi-systemic, making clinical diagnosis challenging. Many pathogenic mtDNA deletions or point mutations in genes encoding mt-tRNA and mt-mRNA have been well documented since the first reports in 1988 (Holt et al. 1988; Wallace et al. 1988). Additionally, because of the advances in whole exome sequencing, a dramatic recent increase has been seen in the identification of novel mutations in nuclear-encoded mitochondrial proteins. As mitochondrial translation has a key role in OXPHOS, mutations in most of the components involved in the process can lead to pathologies, as shown in Table 2; a comprehensive list appears in several recent reviews (Lightowlers et al. 2015; Mayr et al. 2015; Shen et al. 2016).
Regarding the protein synthesis machinery, whereas several mutations of the mt-rRNAs have been associated with disease (see http://www.mitomap.org/MITOMAP), only a few mutations of mitoribosomal proteins have been identified to date. In particular, a mutation in bS16m has been linked to hypotonia and fatal neonatal lactic acidosis (Miller et al. 2004), whereas two different mutations in mS22 result in cardiomyopathy and tubulopathy (Saada et al. 2007; Smits et al. 2011). Concerning components of the mt-LSU, a mutation in uL3m has been shown to cause cardiomyopathy (Galmiche et al. 2011), a defect also reported for a mutation of the mitochondrial-specific protein mL44 (Carroll et al. 2013). Growth retardation and neurological deterioration have been diagnosed as a consequence of a mutation in the mitoribosomal protein bL12m (Serre et al. 2013). Recently, mutations in uS7m have been discovered and linked to deafness with renal and hepatic failure (Menezes et al. 2015). Further to the role of mitochondrial ribosome protein (MRP) mutations in disease, Auwerx and colleagues have reported that genetic variation in genes encoding MRPs are linked to longevity in certain mice strains. Moreover, by depleting Mrps5 in Caenorhabditis elegans, they have obtained evidence suggesting that a mito-nuclear protein imbalance caused by a relative decrease in mitochondrial translation can lead to activation of a mitochondrial unfolded protein response underlying this increased longevity (Houtkooper et al. 2013).
Concluding remarks
Progress in understanding the mechanisms underlying mammalian mitochondrial gene expression in recent years has been impressive. This has been attributable, in large part, to the production of high resolution structures of the mammalian mitoribosome and the identification of pathogenic mutations in key players via whole exome sequencing. Establishing a reconstituted in vitro translation system remains an enormous challenge (accepting the impressive initial efforts by Suzuki and colleagues; Takemoto et al. 2009). Major limitations include the isolation of sufficient quantities of mammalian mitochondria that are devoid of template mt-mRNA and mt-tRNAs to use as a substrate and also the central role played by associations with the IMM and translocon, which has yet to be characterised. Structural biology and genetics will undoubtedly further our detailed understanding, particularly regarding the assembly process of the mitoribosome, but the next step will also require the establishment of a faithful in vitro reconstituted mitochondrial translation system. We look forward to this possibility with great excitement.
References
Akabane S, Ueda T, Nierhaus KH, Takeuchi N (2014) Ribosome rescue and translation termination at non-standard stop codons by ICT1 in mammalian mitochondria. PLoS Genet 10:e1004616
Almajan ER, Richter R, Paeger L, Martinelli P, Barth E, Decker T, Larsson NG, Kloppenburg P, Langer T, Rugarli EI (2012) AFG3L2 supports mitochondrial protein synthesis and Purkinje cell survival. J Clin Invest 122:4048–4058
Amunts A, Brown A, Toots J, Scheres SH, Ramakrishnan V (2015) Ribosome. The structure of the human mitochondrial mitoribosome. Science 348:95–98
Anderson S, Bankier AT, Barrell BG, De Bruijn MHL, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJH, Staden R, Young IG (1981) Sequence and organization of the human mitochondrial genome. Nature 290:457–465
Antonicka H, Shoubridge EA (2015) Mitochondrial RNA granules are centers for posttranscriptional RNA processing and ribosome biogenesis. Cell Rep 10:920–932. doi:10.1016/j.celrep.2015.01.030
Antonicka H, Sasarman F, Nishimura T, Paupe V, Shoubridge EA (2013) The mitochondrial RNA-binding protein GRSF1 localizes to RNA granules and is required for posttranscriptional mitochondrial gene expression. Cell Metab 17:386–398
Ban N, Nissen P, Hansen J, Moore PB, Steitz TA (2000) The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science 289:905–920
Ban N, Beckmann R, Cate JH, Dinman JD, Dragon F, Ellis SR, Lafontaine DL, Lindahl L, Liljas A, Lipton JM, McAlear M, Moore PB, Noller HF, Ortega J, Panse VG, Ramakrishnan V, Spahn CM, Steitz TA, Tchorzewski M, Tollervey D, Warren AJ, Williamson JR, Wilson D, Yonath A, Yusupov M (2014) A new system for naming ribosomal proteins. Curr Opin Struct Biol 24:165–169
Bogenhagen DF, Martin DW, Koller A (2014) Initial steps in RNA processing and ribosome assembly occur at mitochondrial DNA nucleoids. Cell Metab 19:618–629
Borowski LS, Szczesny RJ, Brzezniak LK, Stepien PP (2010) RNA turnover in human mitochondria: more questions than answers? Biochim Biophys Acta 1797:1066–1070
Borowski LS, Dziembowski A, Hejnowicz MS, Stepien PP, Szczesny RJ (2013) Human mitochondrial RNA decay mediated by PNPase-hSuv3 complex takes place in distinct foci. Nucleic Acids Res 41:1223–1240
Bratic A, Clemente P, Calvo-Garrido J, Maffezzini C, Felser A, Wibom R, Wedell A, Freyer C, Wredenberg A (2016) Mitochondrial polyadenylation is a one-step process required for mRNA integrity and tRNA maturation. PLoS Genet 12:e1006028
Brown A, Amunts A, Bai XC, Sugimoto Y, Edwards PC, Murshudov G, Scheres SH, Ramakrishnan V (2014) Structure of the large ribosomal subunit from human mitochondria. Science 346:718–722
Bruni F, Gramegna P, Oliveira JM, Lightowlers RN, Chrzanowska-Lightowlers ZM (2013) REXO2 is an oligoribonuclease active in human mitochondria. PLoS ONE 8:e64670
Cai YC, Bullard JM, Thompson NL, Spremulli LL (2000) Interaction of mitochondrial elongation factor Tu with aminoacyl-tRNA and elongation factor Ts. J Biol Chem 275:20308–20314
Cámara Y, Asin-Cayuela J, Park CB, Metodiev MD, Shi Y, Ruzzenente B, Kukat C, Habermann B, Wibom R, Hultenby K, Franz T, Erdjument-Bromage H, Tempst P, Hallberg BM, Gustafsson CM, Larsson NG (2011) MTERF4 regulates translation by targeting the methyltransferase NSUN4 to the mammalian mitochondrial ribosome. Cell Metab 13:527–539
Carroll CJ, Isohanni P, Pöyhönen R, Euro L, Richter U, Brilhante V, Götz A, Lahtinen T, Paetau A, Pihko H, Battersby BJ, Tyynismaa H, Suomalainen A (2013) Whole-exome sequencing identifies a mutation in the mitochondrial ribosome protein MRPL44 to underlie mitochondrial infantile cardiomyopathy. J Med Genet 50:151–159
Christian BE, Spremulli LL (2010) Preferential selection of the 5'-terminal start codon on leaderless mRNAs by mammalian mitochondrial ribosomes. J Biol Chem 285:28379–28386
Christian BE, Spremulli LL (2012) Mechanism of protein biosynthesis in mammalian mitochondria. Biochim Biophys Acta 1819:1035–1054
Chrzanowska-Lightowlers ZM, Lightowlers RN (2015) Response to "Ribosome rescue and translation termination at non-standard stop codons by ICT1 in mammalian mitochondria". PLoS Genet 11:e1005227
Chrzanowska-Lightowlers ZM, Pajak A, Lightowlers RN (2011) Termination of protein synthesis in mammalian mitochondria. J Biol Chem 286:34479–34485
Chujo T, Ohira T, Sakaguchi Y, Goshima N, Nomura N, Nagao A, Suzuki T (2012) LRPPRC/SLIRP suppresses PNPase-mediated mRNA decay and promotes polyadenylation in human mitochondria. Nucleic Acids Res 40:8033–8047
Czech B, Hannon GJ (2011) Small RNA sorting: matchmaking for argonautes. Nat Rev Genet 12:19–31
Dalla Rosa I, Durigon R, Pearce SF, Rorbach J, Hirst EM, Vidoni S, Reyes A, Brea-Calvo G, Minczuk M, Woellhaf MW, Herrmann JM, Huynen MA, Holt IJ, Spinazzola A (2014) MPV17L2 is required for ribosome assembly in mitochondria. Nucleic Acids Res 42:8500–8515
De Silva D, Tu YT, Amunts A, Fontanesi F, Barrientos A (2015) Mitochondrial ribosome assembly in health and disease. Cell Cycle 14:2226–2250
Dennerlein S, Rozanska A, Wydro M, Chrzanowska-Lightowlers ZM, Lightowlers RN (2010) Human ERAL1 is a mitochondrial RNA chaperone involved in the assembly of the 28S small mitochondrial ribosomal subunit. Biochem J 430:551–558
Denslow ND, Andersg JC, O’Brien TW (1991) Bovine mitochondrial ribosomes possess a high affinity binding site for guanine nucleotide. J Biol Chem 266:9586–9590
Diodato D, Ghezzi D, Tiranti V (2014) The mitochondrial aminoacyl tRNA synthetases: genes and syndromes. Int J Cell Biol 2014:787956
Dubin DT, Taylor RH (1978) Modification of mitochondrial ribosomal RNA from hamster cells: the presence of GmG and late-methylated UmGmU in the large subunit (17S) RNA. J Mol Biol 121:523–540
Falkenberg M, Larsson NG, Gustafsson CM (2007) DNA replication and transcription in mammalian mitochondria. Annu Rev Biochem 76:679–699
Feaga HA, Quickel MD, Hankey-Giblin PA, Keiler KC (2016) Human cells require non-stop ribosome rescue activity in mitochondria. PLoS Genet 12:e1005964
Filipovska A, Rackham O (2013) Modular blocks for building RNA-binding proteins. RNA Biol 10:1426–1432
Frolova LY, Tsivkovskii RY, Sivolobova GF, Oparina NY, Serpinsky OI, Blinov VM, Tatkov SI, Kisselev LL (1999) Mutations in the highly conserved GGQ motif of class 1 polypeptide release factors abolish ability of human eRF1 to trigger peptidyl-tRNA hydrolysis. RNA 5:1014–1020
Fung S, Nishimura T, Sasarman F, Shoubridge EA (2013) The conserved interaction of C7orf30 with MRPL14 promotes biogenesis of the mitochondrial large ribosomal subunit and mitochondrial translation. Mol Biol Cell 24:184–193
Galmiche L, Serre V, Beinat M, Assouline Z, Lebre AS, Chretien D, Nietschke P, Benes V, Boddaert N, Sidi D, Brunelle F, Rio M, Munnich A, Rotig A (2011) Exome sequencing identifies MRPL3 mutation in mitochondrial cardiomyopathy. Hum Mutat 32:1225–1231
Gray MW (1999) Mitochondrial evolution. Science 283:1476–1481
Greber BJ, Ban N (2016) Structure and function of the mitochondrial ribosome. Annu Rev Biochem 85:103–132
Greber BJ, Boehringer D, Leibundgut M, Bieri P, Leitner A, Schmitz N, Aebersold R, Ban N (2014a) The complete structure of the large subunit of the mammalian mitochondrial ribosome. Nature 515:283–286
Greber BJ, Boehringer D, Leitner A, Bieri P, Voigts-Hoffmann F, Erzberger JP, Leibundgut M, Aebersold R, Ban N (2014b) Architecture of the large subunit of the mammalian mitochondrial ribosome. Nature 505:515–519
Greber BJ, Bieri P, Leibundgut M, Leitner A, Aebersold R, Boehringer D, Ban N (2015) The complete structure of the 55S mammalian mitochondrial ribosome. Science 348:303–308
Gustafsson CM, Falkenberg M, Larsson NG (2016) Maintenance and expression of mammalian mitochondrial DNA. Annu Rev Biochem 85:133–160. doi:10.1146/annurev-biochem-060815-014402
Hage AE, Tollervey D (2004) A surfeit of factors: why is ribosome assembly so much more complicated in eukaryotes than bacteria? RNA Biol 1:10–15
Handa N, Kishishita S, Morita S, Akasaka R, Jin Z, Chrzas J, Chen L, Liu ZJ, Wang BC, Sugano S, Tanaka A, Terada T, Shirouzu M, Yokoyama S (2007) Structure of the human Tim44 C-terminal domain in complex with pentaethylene glycol: ligand-bound form. Acta Crystallogr D Biol Crystallogr 63:1225–1234
Haque ME, Elmore KB, Tripathy A, Koc H, Koc EC, Spremulli LL (2010) Properties of the C-terminal tail of human mitochondrial inner membrane protein Oxa1L and its interactions with mammalian mitochondrial ribosomes. J Biol Chem 285:28353–28362
He J, Cooper HM, Reyes A, Di Re M, Kazak L, Wood SR, Mao CC, Fearnley IM, Walker JE, Holt IJ (2012) Human C4orf14 interacts with the mitochondrial nucleoid and is involved in the biogenesis of the small mitochondrial ribosomal subunit. Nucleic Acids Res 40:6097–6108
Hennon SW, Soman R, Zhu L, Dalbey RE (2015) YidC/Alb3/Oxa1 family of insertases. J Biol Chem 290:14866–14874
Herrmann JM, Woellhaf MW, Bonnefoy N (2013) Control of protein synthesis in yeast mitochondria: the concept of translational activators. Biochim Biophys Acta 1833:286–294
Holt IJ, Harding AE, Morgan-Hughes JA (1988) Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 331:717–719
Holzmann J, Frank P, Löffler E, Bennett KL, Gerner C, Rossmanith W (2008) RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell 135:462–474
Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J (2013) Mitonuclear protein imbalance as a conserved longevity mechanism. Nat Rev Genet 497:451–457
Jia L, Dienhart M, Schramp M, McCauley M, Hell K, Stuart RA (2003) Yeast Oxa1 interacts with mitochondrial ribosomes: the importance of the C-terminal region of Oxa1. EMBO J 22:6438–6447
Jourdain AA, Koppen M, Wydro M, Rodley CD, Lightowlers RN, Chrzanowska-Lightowlers ZM, Martinou JC (2013) GRSF1 regulates RNA processing in mitochondrial RNA granules. Cell Metab 17:399–410
Jourdain AA, Koppen M, Rodley CD, Maundrell K, Gueguen N, Reynier P, Guaras AM, Enriquez JA, Anderson P, Simarro M, Martinou JC (2015) A mitochondria-specific isoform of FASTK is present in mitochondrial RNA granules and regulates gene expression and function. Cell Rep 10:1110–1121
Kissil JL, Cohen O, Raveh T, Kimchi A (1999) Structure-function analysis of an evolutionary conserved protein, DAP3, which mediates TNF-alpha- and Fas-induced cell death. EMBO J 18:353–362
Koc EC, Burkhart W, Blackburn K, Moseley A, Spremulli LL (2001a) The small subunit of the mammalian mitochondrial ribosome. Identification of the full complement of ribosomal proteins present. J Biol Chem 276:19363–19374
Koc EC, Burkhart W, Blackburn K, Moyer MB, Schlatzer DM, Moseley A, Spremulli LL (2001b) The large subunit of the mammalian mitochondrial ribosome. Analysis of the complement of ribosomal proteins present. J Biol Chem 276:43958–43969
Kotani T, Akabane S, Takeyasu K, Ueda T, Takeuchi N (2013) Human G-proteins, ObgH1 and Mtg1, associate with the large mitochondrial ribosome subunit and are involved in translation and assembly of respiratory complexes. Nucleic Acids Res 41:3713–3722
Lee KW, Bogenhagen DF (2014) Assignment of 2'-O-methyltransferases to modification sites on the mammalian mitochondrial large subunit 16 S ribosomal RNA (rRNA). J Biol Chem 289:24936–24942
Levshenkova EV, Ukraintsev KE, Orlova VV, Alibaeva RA, Kovriga IE, Zhugdernamzhilyn O, Frolova EI (2004) The structure and specific features of the cDNA expression of the human gene MRPL37. Bioorg Khim 30:499–506
Levy S, Allerston CK, Liveanu V, Habib MR, Gileadi O, Schuster G (2016) Identification of LACTB2, a metallo-β-lactamase protein, as a human mitochondrial endoribonuclease. Nucleic Acids Res 44:1813–1832
Li M, Spremulli LL (2000) Interaction of mammalian mitochondrial ribosomes with the inner membrane. J Biol Chem 275:29400–29406
Lightowlers RN, Chrzanowska-Lightowlers ZM (2013) Human pentatricopeptide proteins: only a few and what do they do? RNA Biol 10:1433–1438
Lightowlers RN, Rozanska A, Chrzanowska-Lightowlers ZM (2014) Mitochondrial protein synthesis: figuring the fundamentals, complexities and complications, of mammalian mitochondrial translation. FEBS Lett 588:2496–2503
Lightowlers RN, Taylor RW, Turnbull DM (2015) Mutations causing mitochondrial disease: What is new and what challenges remain? Science 349:1494–1499
Linder P, Jankowsky E (2011) From unwinding to clamping—the DEAD box RNA helicase family. Nat Rev Mol Cell Biol 12:505–516
Liu Q, Fredrick K (2016) Intersubunit bridges of the bacterial ribosome. J Mol Biol 428:2146–2164. doi:10.1016/j.jmb.2016.02.009
Lupo D, Vollmer C, Deckers M, Mick DU, Tews I, Sinning I, Rehling P (2011) Mdm38 is a 14-3-3-like receptor and associates with the protein synthesis machinery at the inner mitochondrial membrane. Traffic 12:1457–1466
Mayr JA, Haack TB, Freisinger P, Karall D, Makowski C, Koch J, Feichtinger RG, Zimmermann FA, Rolinski B, Ahting U, Meitinger T, Prokisch H, Sperl W (2015) Spectrum of combined respiratory chain defects. J Inherit Metab Dis 38:629–640
McLean J, Cohn GL, Brandt IK, Simpson MV (1958) Incorporation of labeled amino acids into the protein of muscle and liver mitochondria. J Biol Chem 233:657–663
Menezes MJ, Guo Y, Zhang J, Riley LG, Cooper ST, Thorburn DR, Li J, Dong D, Li Z, Glessner J, Davis RL, Sue CM, Alexander SI, Arbuckle S, Kirwan P, Keating BJ, Xu X, Hakonarson H, Christodoulou J (2015) Mutation in mitochondrial ribosomal protein S7 (MRPS7) causes congenital sensorineural deafness, progressive hepatic and renal failure and lactic acidemia. Hum Mol Genet 24:2297–2307
Mercer TR, Neph S, Dinger ME, Crawford J, Smith MA, Shearwood AM, Haugen E, Bracken CP, Rackham O, Stamatoyannopoulos JA, Filipovska A, Mattick JS (2011) The human mitochondrial transcriptome. Cell 146:645–658
Metodiev MD, Lesko N, Park CB, Cámara Y, Shi Y, Wibom R, Hultenby K, Gustafsson CM, Larsson NG (2009) Methylation of 12S rRNA is necessary for in vivo stability of the small subunit of the mammalian mitochondrial ribosome. Cell Metab 9:386–397
Metodiev MD, Spåhr H, Loguercio Polosa P, Meharg C, Becker C, Altmueller J, Habermann B, Larsson NG, Ruzzenente B (2014) NSUN4 is a dual function mitochondrial protein required for both methylation of 12S rRNA and coordination of mitoribosomal assembly. PLoS Genet 10:e1004110
Mick DU, Dennerlein S, Wiese H, Reinhold R, Pacheu-Grau D, Lorenzi I, Sasarman F, Weraarpachai W, Shoubridge EA, Warscheid B, Rehling P (2012)MITRAC links mitochondrial protein translocation to respiratory-chain assembly and translational regulation.Cell 151:1528–1541
Miller C, Saada A, Shaul N, Shabtai N, Ben-Shalom E, Shaag A, Hershkovitz E, Elpeleg O (2004) Defective mitochondrial translation caused by a ribosomal protein (MRPS16) mutation. Ann Neurol 56:734–738
Miller JL, Koc H, Koc EC (2008) Identification of phosphorylation sites in mammalian mitochondrial ribosomal protein DAP3. Protein Sci 17:251–260
Miller JL, Cimen H, Koc EC (2009) Phosphorylated proteins of the mammalian mitochondrial ribosome: implications in protein synthesis. J Proteome Res 8:4789–4798
Minczuk M, Piwowarski J, Papworth MA, Awiszus K, Schalinski S, Dziembowski A, Dmochowska A, Bartnik E, Tokatlidis K, Stepien PP, Borowski P (2002) Localisation of the human hSuv3p helicase in the mitochondrial matrix and its preferential unwinding of dsDNA. Nucleic Acids Res 30:5074–5086
Montoya J, Ojala D, Attardi G (1981) Distinctive features of the 5'-terminal sequences of the human mitochondrial mRNAs. Nature 290:465–470
Nagaike T, Suzuki T, Tomari Y, Takemoto-Hori C, Negayama F, Watanabe K, Ueda T (2001) Identification and characterization of mammalian mitochondrial tRNA nucleotidyltransferases. J Biol Chem 276:40041–40049
Nagaike T, Suzuki T, Katoh T, Ueda T (2005) Human mitochondrial mRNAs are stabilized with polyadenylation regulated by mitochondria-specific poly(A) polymerase and polynucleotide phosphorylase. J Biol Chem 280:19721–19727
Neupert W (2015) A perspective on transport of proteins into mitochondria: a myriad of open questions. J Mol Biol 427:1135–1158
Nolden M, Ehses S, Koppen M, Bernacchia A, Rugarli EI, Langer T (2005) The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell 123:277–289
Nouws J, Goswami AV, Bestwick M, McCann BJ, Surovtseva YV, Shadel GS (2016) Mitochondrial ribosomal protein L12 is required for POLRMT stability and exists as two forms generated by alternative proteolysis during import. J Biol Chem 291:989–997
O'Brien TW (1971) The general occurrence of 55 S ribosomes in mammalian liver mitochondria. J Biol Chem 246:3409–3417
O'Brien TW, Kalf GF (1967) Ribosomes from rat liver mitochondria. J Biol Chem 242:2172–2179
O’Brien TW, Denslow ND, Anders JC, Courtney BC (1990) The translation system of mammalian mitochondria. Biochim Biophys Acta 1050:174–178
Ofengand J, Bakin A (1997) Mapping to nucleotide resolution of pseudouridine residues in large subunit ribosomal RNAs from representative eukaryotes, prokaryotes, archaebacteria, mitochondria and chloroplasts. J Mol Biol 266:246–268
Ojala D, Montoya J, Attardi G (1981) tRNA punctuation model of RNA processing in human mitochondria. Nature 290:470–474
Ott M, Prestele M, Bauerschmitt H, Funes S, Bonnefoy N, Herrmann JM (2006) Mba1, a membrane-associated ribosome receptor. EMBO J 25:1603–1610
Ott M, Amunts A, Brown A (2016) Organization and regulation of mitochondrial protein synthesis. Annu Rev Biochem 85:77–101. doi:10.1146/annurev-biochem-060815-014334
Pfeffer S, Woellhaf MW, Herrmann JM, Forster F (2015) Organization of the mitochondrial translation machinery studied in situ by cryoelectron tomography. Nat Commun 6:6019
Piao L, Li Y, Kim SJ, Byun HS, Huang SM, Hwang SK, Yang KJ, Park KA, Won M, Hong J, Hur GM, Seok JH, Shong M, Cho MH, Brazil DP, Hemmings BA, Park J (2009) Association of LETM1 and MRPL36 contributes to the regulation of mitochondrial ATP production and necrotic cell death. Cancer Res 69:3397–3404
Popow J, Alleaume AM, Curk T, Schwarzl T, Sauer S, Hentze MW (2015) FASTKD2 is an RNA-binding protein required for mitochondrial RNA processing and translation. RNA 21:1873–1884
Ramakrishnan V, White SW (1998) Ribosomal protein structures: insights into the architecture, machinery and evolution of the ribosome. Trends Biochem Sci 23:208–212
Richter R, Rorbach J, Pajak A, Smith PM, Wessels HJ, Huynen MA, Smeitink JA, Lightowlers RN, Chrzanowska-Lightowlers ZM (2010) A functional peptidyl-tRNA hydrolase, ICT1, has been recruited into the human mitochondrial ribosome. EMBO J 29:1116–1125
Rorbach J, Richter R, Wessels HJ, Wydro M, Pekalski M, Farhoud M, Kuhl I, Gaisne M, Bonnefoy N, Smeitink JA, Lightowlers RN, Chrzanowska-Lightowlers ZM (2008) The human mitochondrial ribosome recycling factor is essential for cell viability. Nucleic Acids Res 36:5787–5799
Rorbach J, Gammage PA, Minczuk M (2012) C7orf30 is necessary for biogenesis of the large subunit of the mitochondrial ribosome. Nucleic Acids Res 40:4097–4109
Rorbach J, Boesch P, Gammage PA, Nicholls TJ, Pearce SF, Patel D, Hauser A, Perocchi F, Minczuk M (2014) MRM2 and MRM3 are involved in biogenesis of the large subunit of the mitochondrial ribosome. Mol Biol Cell 25:2542–2555
Rossmanith W (2011) Localization of human RNase Z isoforms: dual nuclear/mitochondrial targeting of the ELAC2 gene product by alternative translation initiation. PLoS ONE 6: e19152
Saada A, Shaag A, Arnon S, Dolfin T, Miller C, Fuchs-Telem D, Lombes A, Elpeleg O (2007) Antenatal mitochondrial disease caused by mitochondrial ribosomal protein (MRPS22) mutation. J Med Genet 44:784–786
Salinas-Giege T, Giege R, Giege P (2015) tRNA biology in mitochondria. Int J Mol Sci 16:4518–4559
Sasarman F, Brunel-Guitton C, Antonicka H, Wai T, Shoubridge EA (2010) LRPPRC and SLIRP interact in a ribonucleoprotein complex that regulates posttranscriptional gene expression in mitochondria. Mol Biol Cell 21:1315–1323
Schmeing TM, Huang KS, Strobel SA, Steit TA (2005) An induced-fit mechanism to promote peptide bond formation and exclude hydrolysis of peptidyl-tRNA. Nature 438:520–524
Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila-Sanjurjo A, Holton JM, Cate JH (2005) Structures of the bacterial ribosome at 3.5 Å resolution. Science 310:827–834
Seidel-Rogol BL, McCulloch V, Shadel GS (2002) Human mitochondrial transcription factor B1 methylates ribosomal RNA at a conserved stem-loop. Nat Genet 33:23–24
Serre V, Rozanska A, Beinat M, Chretien D, Boddaert N, Munnich A, Rotiq A, Chrzanowska-Lightowlers ZM (2013) Mutations in mitochondrial ribosomal protein MRPL12 leads to growth retardation, neurological deterioration and mitochondrial translation deficiency. Biochim Biophys Acta 1832:1304–1312
Sharma MR, Koc EC, Datta PP, Booth TM, Spremulli LL, Agrawal RK (2003) Structure of the mammalian mitochondrial ribosome reveals an expanded functional role for its component proteins. Cell 115:97–108
Shen L, Diroma MA, Gonzalez M, Navarro-Gomez D, Leipzig J, Lott MT, Oven M van, Wallace DC, Muraresku CC, Zolkipli-Cunningham Z, Chinnery PF, Attimonelli M, Zuchner S, Falk MJ, Gai X (2016) MSeqDR: a centralized knowledge repository and bioinformatics web resource to facilitate genomic investigations in mitochondrial disease. Hum Mutat 37:540–548
Slomovic S, Laufer D, Geiger D, Schuster G (2005) Polyadenylation and degradation of human mitochondrial RNA: the prokaryotic past leaves its mark. Mol Cell Biol 25:6427–6435
Smirnov A, Entelis N, Martin RP, Tarassov I (2011) Biological significance of 5S rRNA import into human mitochondria: role of ribosomal protein MRP-L18. Genes Dev 25:1289–1305
Smits P, Saada A, Wortmann SB, Heister AJ, Brink M, Pfundt R, Miller C, Haas D, Hantschmann R, Rodenburg RJ, Smeitink JA, Heuvel LP van den (2011) Mutation in mitochondrial ribosomal protein MRPS22 leads to Cornelia de Lange-like phenotype, brain abnormalities and hypertrophic cardiomyopathy. Eur J Hum Genet 19:394–399
Soleimanpour-Lichaei HR, Kuhl I, Gaisne M, Passos JF, Wydro M, Rorbach J, Temperley R, Bonnefoy N, Tate W, Lightowlers RN, Chrzanowska-Lightowlers ZM (2007) mtRF1a is a human mitochondrial translation release factor decoding the major termination codons UAA and UAG. Mol Cell 27:745–757
Stiburek L, Fornuskova D, Wenchich L, Pejznochova M, Hansikova H, Zeman J (2007) Knockdown of human Oxa1l impairs the biogenesis of F1Fo-ATP synthase and NADH:ubiquinone oxidoreductase. J Mol Biol 374:506–516
Stiller SB, Höpker J, Oeljeklaus S, Schütze C, Schrempp SG, Vent-Schmidt J, Horvath SE, Frazier AE, Gebert N, Laan M van der, Bohnert M, Warscheid B, Pfanner N, Wiedemann N (2016) Mitochondrial OXA translocase plays a major role in biogenesis of inner-membrane proteins. Cell Metab 23:901–908
Sun L, Liu Y, Frémont M, Schwarz S, Siegmann M, Matthies R, Jost JP (1998) A novel 52 kDa protein induces apoptosis and concurrently activates c-Jun N-terminal kinase 1 (JNK1) in mouse C3H10T1/2 fibroblasts. Gene 208:157–166
Surovtseva YV, Shutta TE, Cotneya J, Cimenb H, Chena SY, Koc EC, Shadela GS (2011) Mitochondrial ribosomal protein L12 selectively associates with human mitochondrial RNA polymerase to activate transcription. Proc Natl Acad Sci U S A 108:17921–17926
Suzuki T (2014) A complete landscape of post-transcriptional modifications in mammalian mitochondrial tRNAs. Nucleic Acids Res 42:7346–7357
Suzuki T, Nagao A, Suzuki T (2011) Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu Rev Genet 45:299–329
Szklarczyk R, Wanschers BF, Cuypers TD, Esseling JJ, Riemersma M, Brand MA van den, Gloerich J, Lasonder E, Heuvel LP van den, Nijtmans LG, Huynen MA (2012) Iterative orthology prediction uncovers new mitochondrial proteins and identifies C12orf62 as the human ortholog of COX14, a protein involved in the assembly of cytochrome c oxidase. Genome Biol 13:R12
Takemoto C, Spremulli LL, Benkowski LA, Ueda T, Yokogawa T, Watanabe K (2009) Unconventional decoding of the AUA codon as methionine by mitochondrial tRNAMet with the anticodon f5CAU as revealed with a mitochondrial in vitro translation system. Nucleic Acids Res 37:1616–1627
Temperley R, Richter R, Dennerlein S, Lightowlers RN, Chrzanowska-Lightowlers ZM (2010a) Hungry codons promote frameshifting in human mitochondrial ribosomes. Science 327:301
Temperley RJ, Wydro M, Lightowlers RN, Chrzanowska-Lightowlers ZM (2010b) Human mitochondrial mRNAs—like members of all families, similar but different. Biochim Biophys Acta 1797:1081–1085
Tomecki R, Dmochowska A, Gewartowski K, Dziembowski A, Stepien PP (2004) Identification of a novel human nuclear-encoded mitochondrial poly(A) polymerase. Nucleic Acids Res 32:6001–6014
Tsuboi M, Morita H, Nozaki Y, Akama K, Ueda T, Ito K, Nierhaus KH, Takeuchi N (2009) EF-G2mt is an exclusive recycling factor in mammalian mitochondrial protein synthesis. Mol Cell 35:502–510
Tu YT, Barrientos A (2015) The human mitochondrial DEAD-box protein DDX28 resides in RNA granules and functions in mitoribosome assembly. Cell Rep 10:854–864
Tucker EJ, Hershman SG, Köhrer C, Belcher-Timme CA, Patel J, Goldberger OA, Christodoulou J, Silberstein JM, McKenzie M, Ryan MT, Compton AG, Jaffe JD, Carr SA, Calvo SE, RajBhandary UL, Thorburn DR, Mootha VK (2011) Mutations in MTFMT underlie a human disorder of formylation causing impaired mitochondrial translation. Cell Metab 14:428–434
Tucker EJ, Wanschers BF, Szklarczyk R, Mountford HS, Wijeyeratne XW, Brand MA van den, Leenders AM, Rodenburg RJ, Reljić B, Compton AG, Frazier AE, Bruno DL, Christodoulou J, Endo H, Ryan MT, Nijtmans LG, Huynen MA, Thorburn DR (2013) Mutations in the UQCC1-interacting protein, UQCC2, cause human complex III deficiency associated with perturbed cytochrome b protein expression. PLoS Genet 9:e1004034
Uchiumi T, Ohgaki K, Yagi M, Aoki Y, Sakai A, Matsumoto S, Kang D (2010) ERAL1 is associated with mitochondrial ribosome and elimination of ERAL1 leads to mitochondrial dysfunction and growth retardation. Nucleic Acids Res 38:5554–5568
Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ 2nd, Nikoskelainen EK (1988) Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science 242:1427–1430
Wang G, Chen HW, Oktay Y, Zhang J, Allen EL, Smith GM, Fan KC, Hong JS, French SW, McCaffery JM, Lightowlers RN, Morse HC 3rd, Koehler CM, Teitell MA (2010) PNPASE regulates RNA import into mitochondria. Cell 142:456–467
Weraarpachai W, Antonicka H, Sasarman F, Seeger J, Schrank B, Kolesar JE, Lochmüller H, Chevrette M, Kaufman BA, Horvath R, Shoubridge EA (2009) Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase deficiency and late-onset Leigh syndrome. Nat Genet 41:833–837
Weraarpachai W, Sasarman F, Nishimura T, Antonicka H, Auré K, Rötig A, Lombès A, Shoubridge EA (2012) Mutations in C12orf62, a factor that couples COX I synthesis with cytochrome c oxidase assembly, cause fatal neonatal lactic acidosis. Am J Hum Genet 90:142–151
Wettstein FO, Noll H (1965) Binding of transfer ribonucleic acid to ribosomes engaged in protein synthesis: number and properties of ribosomal binding sites. J Mol Biol 11:35–53
Wilson WC, Hornig-Do HT, Bruni F, Chang JH, Jourdain AA, Martinou JC, Falkenberg M, Spåhr H, Larsson NG, Lewis RJ, Hewitt L, Baslé A, Cross HE, Tong L, Lebel RR, Crosby AH, Chrzanowska-Lightowlers ZM, Lightowlers RN (2014) A human mitochondrial poly(A) polymerase mutation reveals the complexities of post-transcriptional mitochondrial gene expression. Hum Mol Genet 23:6345–6355
Wittmann HG (1982) Structure and evolution of ribosomes. Proc R Soc Lond B Biol Sci 216:117–135
Wolf AR, Mootha VK (2014) Functional genomic analysis of human mitochondrial RNA processing. Cell Rep 7:918–931
Wredenberg A, Lagouge M, Bratic A, Metodiev MD, Spåhr H, Mourier A, Freyer C, Ruzzenente B, Tain L, Grönke S, Baggio F, Kukat C, Kremmer E, Wibom R, Polosa PL, Habermann B, Partridge L, Park CB, Larsson NG (2013) MTERF3 regulates mitochondrial ribosome biogenesis in invertebrates and mammals. PLoS Genet 9:e1003178
Yoo YA, Kim MJ, Park JK, Chung YM, Lee JH, Chi SG, Kim JS, Yoo YD (2005) Mitochondrial ribosomal protein L41 suppresses cell growth in association with p53 and p27Kip1. Mol Cell Biol 25:6603–6616
Young DJ, Edgar CD, Murphy J, Fredebohm J, Poole ES, Tate WP (2010) Bioinformatic, structural, and functional analyses support release factor-like MTRF1 as a protein able to decode nonstandard stop codons beginning with adenine in vertebrate mitochondria. RNA 16:1146–1155
Zhang X, Gao X, Coots RA, Conn CS, Liu B, Qian SB (2015) Translational control of the cytosolic stress response by mitochondrial ribosomal protein L18. Nat Struct Mol Biol 22:404–410
Zhang X, Zuo X, Yang B, Li Z, Xue Y, Zhou Y, Huang J, Zhao X, Zhou J, Yan Y, Zhang H, Guo P, Sun H, Guo L, Zhang Y, Fu XD (2014) MicroRNA directly enhances mitochondrial translation during muscle differentiation. Cell 158:607–619
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
This study was funded by The Wellcome Trust [096919/Z/11/Z] and The Barbour Foundation.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Mai, N., Chrzanowska-Lightowlers, Z.M.A. & Lightowlers, R.N. The process of mammalian mitochondrial protein synthesis. Cell Tissue Res 367, 5–20 (2017). https://doi.org/10.1007/s00441-016-2456-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-016-2456-0