
Abstract
Developmental dyslexia is the most common learning disorder in children. Problems in reading and writing are likely due to a complex interaction of genetic and environmental factors, resulting in reduced power of studies of the genetic factors underlying developmental dyslexia. Our approach in the current study was to perform exome sequencing of affected and unaffected individuals within an extended pedigree with a familial form of developmental dyslexia. We identified a two-base mutation, causing a p.R229L amino acid substitution in the centrosomal protein 63 kDa (CEP63), co-segregating with developmental dyslexia in this pedigree. This mutation is novel, and predicted to be highly damaging for the function of the protein. 3D modelling suggested a distinct conformational change caused by the mutation. CEP63 is localised to the centrosome in eukaryotic cells and is required for maintaining normal centriole duplication and control of cell cycle progression. We found that a common polymorphism in the CEP63 gene had a significant association with brain white matter volume. The brain regions were partly overlapping with the previously reported region influenced by polymorphisms in the dyslexia susceptibility genes DYX1C1 and KIAA0319. We hypothesise that CEP63 is particularly important for brain development and might control the proliferation and migration of cells when those two events need to be highly coordinated.
Similar content being viewed by others

Introduction
Disabilities in cognitive processes such as dyslexia are a major problem worldwide, although estimates of their prevalence vary greatly (Katusic et al. 2001; Roeleveld et al. 1997). Developmental dyslexia (DD) is the most common learning problem in children, manifesting as deficiencies in reading and writing skills (Francks et al. 2002; Kere 2014; Shaywitz et al. 1990). These difficulties arise despite normal intelligence, presence of educational opportunities and independently of the social environment. Individuals affected by DD show impairments in multiple correlated cognitive processes, especially the relation between letters and sounds, but the difficulties can often be traced to phonological processes (Elliott and Grigorenko 2014; Tunmer and Greaney 2010) which typically remain stable over time (Svensson and Jacobson 2006). Secondary consequences may include problems in reading comprehension and reduced interest in reading. DD can occur in multiple generations in families and studies have shown a relative high heritability (Shaywitz 1998).
DD is considered a complex trait in which variants in several genes may influence brain development. Some of these genes may execute their effect by interacting with environmental factors (Schumacher et al. 2007). In such cases, the power of genetic assays to identify the causative genetic factors is relatively low. This means that genetic studies of DD must either be very extensive or strategic to maximise the findings of the molecular biological analyses. Previous research has implicated genetic factors contributing to increased risk (Kere 2014) of developing reading and writing deficiencies although some studies using unrelated individuals have not been replicated. The difficulty in finding strong genetic association between common polymorphisms and DD was recently illustrated by the findings of the EU-funded project NeuroDys. This consortium used individual genotyping of 19 single-nucleotide polymorphisms (SNPs) in DYX1C1, DCDC2, KIAA0319, and the MRPL19/C2ORF3 locus using case–control samples from multiple research groups from throughout Europe. Despite this ambitious and multi-center approach, no SNPs reached genome-wide significance (Becker et al. 2014) and no single SNPs were associated with DD in samples from more than one population. This may potentially be explained by differences in diagnosis between countries, genetic heterogeneity among the different populations or insufficient power due to the heterogeneity of the disease, or a combination of all those factors.
Family studies have proven to be valuable in identifying genetic factors associated with increased risk in common traits (Brunham and Hayden 2013), including dyslexia (Hannula-Jouppi et al. 2005; Taipale et al. 2003). In fact, many of the genes originally identified in families inheriting dyslexia have led to new insights in the function of the genes (Lamminmaki et al. 2012; Tammimies et al. 2013) and multiple studies have replicated the original findings (Scerri and Schulte-Korne 2010). In the current study, we describe our approach to identify a novel potential susceptibility gene for DD. This is based on exome sequencing of selected individuals from an extended family from Sweden with familial DD. Due to the diagnosis and family structure, we assumed an autosomal dominant inheritance of DD with high or complete penetrance. Our aim was to identify rare, high-risk disease variants that would, in this particular pedigree, strongly influence disease risk. Identification of such variants might give us new leads on genes and pathways that are important also for the more common forms of dyslexia. Here, we describe and characterise a novel two-base mutation in CEP63 resulting in an amino acid substitution co-segregating with DD suggesting CEP63 as a new candidate gene for DD. Correlation of genotype to phenotype using computational structure predictions can aid the identification of potential damaging or deleterious non-synonymous variants (Kumar and Purohit 2012). As protein structures for CEP proteins are typically not available, we utilised in silico tools for 3D modelling of the predicted wild-type and mutated CEP63 proteins. Finally, we discuss the potential functional implications of the mutation and suggest future prospects to evaluate the effect of the mutated CEP63 on cellular functions.
Materials and methods
Ethics statement
Ethical permission for genetic analysis and collection of test data in this study was granted by the Faculty of Medicine Research Ethics Committee at the University of Gothenburg (L-225-99) and the regional ethics committee (EPN) Stockholm (Dnr 2013/214-31/3). Informed consent was given by all participating individuals.
Participants
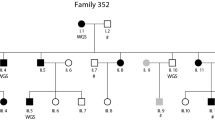
A three-generation family (n = 62) was recruited from a local reading and writing center in southern Sweden as described previously (Svensson et al. 2011). The proband was tested and diagnosed with severe reading and writing disabilities. Thirty-five percent of the family members (22/62 individuals) were diagnosed with dyslexia (Svensson et al. 2011), twelve of them are represented in Fig. 1a. A drawing of the full pedigree is available in Fig. S1.

Exome sequencing in a family with DD shows affected individuals co-segregating a novel CEP63 mutation. a Family members with confirmed DD are denoted by black filled symbols. White symbols indicate individuals for which testing excluded DD diagnosis. Question marks indicate uncertain DD diagnosis. Generations are marked with roman numbers to the left of the image and individuals are counted from left to right. Affected individuals IV:1, IV:5, IV:7, IV:9, V:6, V:7 and V:8, as well as healthy individuals III:2, IV:6 and IV:11 were selected for exome sequencing. Family members for whom DNA was available with a Sanger sequencing-verified c.686_687insdelTT CEP63 mutation are shown with ±, representing heterozygous mutation, whereas +/+ represents wild-type CEP63 alleles. No testing for DD was available for individuals marked with n/a. b Sanger sequencing chromatograms for representative wild-type (+/+) and c.686_687delinsTT (±) family members. c Schematic representation of the CEP63 gene region. Exons are numbered and depicted as filled black boxes. The c.686_687delinsTT mutation predicts a p.R229L in exon 7
Criteria for developmental dyslexia and assessment procedure
Particular weight was given to phonological assessments. Likewise, a self-reported history of reading difficulties was important concerning the affected status. To be regarded as dyslexic, the participants had to fail at least two tests of phonological ability plus a positive history of reading problems. In addition, if there was no self-report of reading and writing disabilities, the participant had to fail at least three phonological tests and two manifest tests of literacy such as word recognition and orthographic choice. A test performance below 1 standard deviation (SD) compared to the norms on these tests was considered a fail, as it has been the case in several previous studies (Nopola-Hemmi et al. 2001; Schulte-Korne et al. 1998; van der Leij et al. 2001). The interview and the questionnaire were also used as a complement to either confirming or rejecting a diagnostic of dyslexia, especially in uncertain cases and for children below 13 years.
Fifty-six of the participating family members completed tests in the assessment battery. For 10- to 13-year olds, the tests were adapted to their age. Two 9-year-old children, who were interviewed and completed some reading tests, were regarded as dyslexic. For one child below the age of 9, only personal records from the parents were available. Children below the age of 7 were not included in the investigation.
In total, 10 reading and spelling tests (word recognition, oral word reading, letter chain, orthographic choice, phonological choice, pseudo-word reading, spoonerism, spelling, reading comprehension, listening comprehension) covering both phonological and orthographic decoding skills, self-reported history of reading ability, years in school and short-term memory were included. Furthermore, data from a non-verbal intelligence test (Raven et al. 1983) were available to exclude low extremes (below the 10th percentile) on this test even though no participants were excluded for that reason. The test battery in the present investigation was similar to that used in many other genetic studies concerning dyslexia (Fagerheim et al. 1999; Fisher et al. 1999; Grigorenko et al. 1997; Kaplan et al. 2002; Olson et al. 1989). The participants that failed on two to four tests were classified as uncertain cases if there were no previous reports of reading and writing disabilities.
DNA samples and ion proton exome sequencing
Blood samples were initially collected from the participants and DNA was prepared as previously described (Svensson et al. 2011). We re-measured available DNA concentration of available genomic DNA from family members using Qubit BR dsDNA assay (Life Technologies, USA). Individuals III:2, IV:1, IV:5, IV:6, IV:7, IV:9, IV:11, V:6, V7 and V:8 were selected for exome sequencing (Fig. 1a). All DNA samples used for exome sequencing were tested for DNA degradation using Genomic DNA ScreenTape analysis (Agilent Technologies, USA). Subsequently, 100 ng non-degraded genomic DNA per sample was used in AmpliSeq library preparations followed by Ion Proton (Life Technologies, USA) sequencing at the Science for Life Laboratory, Uppsala, Sweden, according to standard protocols. AmpliSeq target regions are available upon request. The Ion Proton sequencing was performed on P1 chips producing 200-bp sequence reads.
Variant calling, validation and analysis
Sequence reads were aligned to the hg19 genome assembly using a proprietary Ion Proton pipeline (Life Technologies, USA). Single-nucleotide variants (SNVs) and small insertions and deletions (indels) were called for each sample individually using the Torrent Suite Software (Life Technologies, USA). We aimed to identify highly penetrant variants contributing to a monogenic trait in the family members. First, we selected variants found in all affected family members after exome sequencing. Next, to enrich for novel and very rare variants, we filtered out any SNVs present in an in-house database (Ameur et al. 2014) of 700 exome data variants (Uppsala Genome Center, Uppsala, Sweden). SNV validation was carried out using Sanger capillary sequencing using standard protocols (KIGene, Karolinska Institutet, Solna, Sweden). Based on the inheritance pattern of DD in the family, the transmission of SNVs among affected individuals was analysed assuming a dominant inheritance pattern with high penetrance.
Analysis of CEP63 conservation and expression
The UniProtKB/Swiss-Prot database (www.uniprot.org/uniprot/) was queried for the amino acid sequences of CEP63 homologues in multiple species to understand the conservation and likely impact of the p.R229L amino acid change. The amino acid sequences for human CEP63 homologues in orangutan, crab-eating macaque, rat, mouse, chicken and zebrafish were aligned for comparison.
RNA expression of CEP63 was determined by CAGE in phase 1 and 2 of FANTOM5 data, adapted from the Zenbu promoterome browser at fantom.gsc.riken.jp/zenbu/. The human protein atlas (http://www.proteinatlas.org/) was used to assess tissue protein expression profiles.
Pathway analysis
We performed a search for CEP63 interaction partners using the Ingenuity Pathway Analysis (IPA) software (Qiagen, CA, USA). Network enrichment analysis was performed using FunCoup (Functional Coupling tool, http://funcoup.sbc.su.se/search/) for all interactions partners found in the IPA analysis (Schmitt et al. 2014).
Structural modelling of CEP63
To investigate the probable effects of the p.R229L mutation in CEP63, we analysed the predicted 3D structure of the protein either with the Arginine (R) or the Leucine (L) at position 229 of the protein. We utilised the RaptorX (Kallberg et al. 2012) tool (from the University of Chicago, US, raptorx.uchicago.edu) for 3D structure predictions with default parameters. This method aims at optimising the modelling to the specific target being used, while integrating, into the modelling, a variety of biological signals. Multiple secondary structure templates are also used, if possible, to aid in the modelling of the specific target sequence.
Pdb files from RaptorX were visualised further in Jmol: an open-source Java viewer for chemical structures in 3D (http://www.jmol.org/).
Brain imaging
We utilised brain imaging in a set of 76 healthy controls to test for correlation of CEP63 genetic variants to brain structure, in particular white matter volume. These individuals have been studied previously (Darki et al. 2012, 2014) and were genotyped using whole-genome SNP arrays (Affymetrix Genome-wide SNP array 6.0). To select variants for the brain imaging analysis, we chose all genotyped SNPs within the CEP63 gene region and used Haploview version 4.2 (Barrett et al. 2005) and the tagger option with default setting (pair-wise marker correlation r 2 > 0.8). Minimum minor allele frequency was set to 20 %. Two SNPs, rs7619451 and rs9868985, together tagged (D′ = 1) the linkage disequilibrium (LD) region within CEP63.
Structural brain images were collected by three-dimensional T1-weighted magnetization prepared rapid gradient echo (MP-RAGE) sequence with TR = 2300 ms, TE = 2.92 ms, field of view of 256 × 256 mm2, 256 × 256 matrix size, 176 sagittal slices, and 1 mm3 isotropic voxel size. Scanning was repeated three times with the same parameters, each 2 years apart. To segment the brain into grey matter, white matter and CSF, Diffeomorphic Anatomical Registration Through Exponentiated Lie Algebra (DARTEL) was performed. The white matter segmented images were then smoothed with an 8-mm Gaussian kernel and were analysed using a flexible factorial design using the Statistical Parametric Mapping (SPM) software package. Each SNP was entered as main factor and the model was corrected for age, sex, handedness and total white matter volume. The gene interactions by age and sex were also considered in the model.
Results
Targeted exome sequencing and validation
High molecular weight genomic DNA from seven affected and three unaffected individuals was used in exome sequencing (Fig. 1a). The sequencing produced an average of 37 (29-39) million reads for all samples resulting in an average sequence coverage of 90.3 % (88.4–92.4 %) on target regions and a mean sequence depth of 73–130× (average 99×) over each base. Aligned sequencing reads were submitted to the European Nucleotide Archive (ENA) at EMBL-EBI and are available as study accession number PRJEB9294.
The variant calling predicted on average 50556 SNPs per sample (SD = 6524). We hypothesised that the dominant inheritance pattern of DD in the pedigree is due to a heterozygous DNA mutation segregating with DD in all affected individuals. An exception from the dominant inheritance hypothesis was made for individual IV:9 (Fig. 1a). No phenotypic data was available from his mother and DD has a frequency of 5–10 % in the general population, it was thus unclear from which parent the DNA variant would be transmitted (Fig. 1a). Initial variant calling and filtration were performed on the first sequencing run of eight exomes using DNA from individuals IV:1, IV:5, IV:6, IV:7, IV:9, V:6, V:7 and V:8. We found in total 283 variants, of which 142 are non-synonymous, present in each and every affected individual (including IV:9) while not found in the unaffected (Table S1). Thereafter, we applied a series of variant filtering steps to pinpoint the variant segregating with DD in the family. To enrich for rare or novel variants any of the 283 variants present in an in-house database of 700 exomes were removed. The remaining 16 variants all had a minor allele frequency below 1 % (1000Gphase3EUR and ESP6500), as summarised in Table S2. To further restrict the number of variants shared exclusively among affected family members, a second exome sequencing run including unaffected individuals III:2 and IV:11 was performed. We chose to proceed with the analysis of variants in genes with experimentally validated brain expression profiles. These analyses resulted in a total of eight variants located in the CEP63, DNAJC11, GET4, INTS5, PAQR9, RNF152, TNRC18 and ZNF507 genes, five of which are non-synonymous.
Due to the inheritance pattern of DD in the pedigree, we chose to validate the five non-synonymous variants in the CEP63, GET4, INTS5, RNF152 and ZNF507 genes using Sanger sequencing and all individuals from the family (Fig. 1a) for which DNA was available. The evidence for all polymorphisms was supported by at least 20 sequence reads carrying the alternative allele (Table S2). The novel c.T775G (p.259C>G) variant in GET4 was found to be monomorphic and thus represents an error in the exome sequencing or variant calling. A novel variant in INT5S (c.3026C>T, p.P1009L) was predicted to be present in individual IV:9 (Table S2). However, upon Sanger sequencing, the mother (III:4, uncertain affection status) was shown not to carry the variant and as the father is unaffected, the transmission of the variant from a common ancestor was not confirmed. Furthermore, unaffected individuals III:2 and IV:11 carried the predicted variant in ZNF507 (c.634C>G p.P212A), leading us to exclude this polymorphism from further analysis (Table S2). The predicted variant in RNF152 (c.359G>A, p.R120H) was also found in unaffected individual IV:6 and was thus also excluded from further analysis (Table S2).
A single non-synonymous variant was confirmed by Sanger sequencing to segregate with DD in the family using all available DNA samples. This mutation is a novel complex polymorphism involving two adjacent bases at position chr3:134264558-9 (build 37), resulting in a p.R229L c.686_687delinsTT (NM_001042383) variant in exon 7 (Fig. 1a–c) of the centrosomal protein 63 kDa (CEP63). The G/T variant predicting the non-synonymous amino acid substitution is very rare with a minor (T) allele frequency of 0.01 % in the European population (ExAC, http://exac.broadinstitute.org/). We found that that the chr3:134264558 position is highly conserved [Gerp (Cooper et al. 2005) score 5.53] and the G/T variant is predicted to be probably damaging by PolyPhen-2 (score 1) (Adzhubei et al. 2010) and by SIFT (score 0) (Kumar et al. 2009), see Table S2. Due to the amino acid substitution and potential functional significance of the c.686_687delinsTT in CEP63, we undertook further analysis of the CEP63 protein and the novel mutation.
Differential expression profiles of CEP63 transcript variants
We investigated publicly available data on the RNA expression of CEP63 to get an understanding of how dysregulation of CEP63 might contribute to risk of DD. The FANTOM5 promoterome data browser reveals that CEP63 is mainly transcribed from a cluster of transcription start sites (TSSes) at the first exon of the full-length CEP63 transcript. The expression of transcripts originating from this TSS cluster is best described as ubiquitous (Fig. S2). Another set of transcripts is transcribed from a region within the gene, resulting in transcripts starting from exons 4–9. These transcripts show a very different expression pattern, being mainly expressed in brain, particularly in the medial temporal gyrus and the substantia nigra (Fig. S3).
Conservation and structural modelling of CEP63
The c.686_687delinsTT mutation in CEP63 results in an in-frame amino acid substitution (p.R229L). The position is highly conserved in vertebrates (Fig. 2). A sequence of regularly spaced hydrophobic residues in the proximity of pR229L seems to be conserved in all the species we studied, and changing the charged side chain of arginine to the hydrophobic side chain of a leucine could very plausibly interfere with this pattern.

ClustalW alignment of the sequence of amino acids 200-255 of human CEP63 isoform 1 (Q96MT8-1) as well as the homologous sequence for orangutan (PONAB, Pongo abelii), crab-eating macaque (MACFA, Macaca fascicularis), rat (RAT, Rattus norvegicus), mouse (MOUSE, Mus musculus), chicken (CHICK, Gallus gallus) and zebrafish (DANRE, Danio rerio). Hydrophobic residues are marked in purple. The p.R229L mutation found to be co-segregating with DD in the current study is in bold and marked with an arrow
To predict the structural defects in the tertiary structure of the mutated protein, we attempted to model the mutation in 3D. Our hypothesis was that the 3D structure of the protein might change slightly (conformation change, domain change, access of binding sites, etc.). In the prediction from this modelling, we could show that the mutated residue abolishes a hinge region between two coiled-coil regions (Fig. 3) thus disrupting the three-dimensional structure of CEP63. Pdb files of the results are available upon request.

Predicted tertiary structure of wild-type (wt) CEP63 carrying an arginine at site 229 of the protein and mutated (mut) CEP63 with a leucine at the same amino acid position. In the wild-type protein (Arg229), amino acid 229 is part of a hinge segment between two coiled-coil domains, (a, b). In the mutated protein (c) (Leu229), this hinge segment is predicted to be lost. The prediction was made with the RaptorX tool (University of Chicago, US, http://www.raptorx.uchicago.edu) using the default parameters
Pathway analysis and interaction partners of CEP63
We used the Ingenuity Pathway Analysis (IPA) software for analyses of protein–protein interactions involving CEP63. Here, we present evidence of direct, experimentally validated, protein–protein interactions (i.e. no intermediate proteins) of CEP63 with 13 proteins: CEP152, DISC1, DST, DTNB, EXOC4, MACF1, NCOR2, NDEL1, PPP1R13B, PPP4R1, SMARCE1, SYNE1 and TBC1D15 (Table S3; Fig. S4). Despite extracting these interactions and applying a functional coupling analysis, using FunCoup, we did not reveal any direct interconnection among any of the 13 binding targets of CEP63. Based on these results, we speculate that each of the interacting partners might interact at separate cellular locations and/or time-point, with CEP63.
Genetic effects on white matter volume
Developmental dyslexia is likely a neurological disease, and given the high brain expression of certain CEP63 transcripts in human (Figs. S2, S3), we aimed at investigating the impact of genetic variations within CEP63 on brain structure, more specifically on white matter volume. Here, we used available genotypes from a whole-genome SNP array and MRI data to correlate common variants with white matter volume in a set of 76 healthy controls (Darki et al. 2012, 2014). Interestingly, we showed that the AA/AC genotypes of rs7619451 are significantly associated with higher white matter volume in the right hemisphere overlapping with right superior longitudinal fasciculus and posterior part of corpus callosum (peak coordinate: 28, −55, 29; p = 0.0076, corrected at the cluster level with p < 0.01). The cluster of significant effect on white matter volume for rs7619451 in the right temporo-parietal region partly overlaps with a region previously found significant for rs3743204 and rs6935076 from the dyslexia candidate genes DYX1C1and KIAA0319, respectively (Fig. S5) (Darki et al. 2012). We extracted the mean white matter volume from the significant region in the right hemisphere as well as its homologous area in the left hemisphere. Rs7619451 was significantly associated with white matter volume in both left (p = 0.006) and right (p = 0.003) regions of interest.
Moreover, the association of rs7619451 with reading comprehension scores from those 76 individuals was assessed using a mixed linear model considering three longitudinal measures of reading ability, each collected two years apart. The reading score was entered as dependent variable, and the SNP genotype was set as a factor. Age, sex, handedness and the SNP interaction with age were considered as covariates. The main effect of rs7619451 [F (1, 88.98) = 5.938, p = 0.017] as well as its interaction by age [F (1,91.62) = 5.518, p = 0.021] was found significant for reading comprehension.
Discussion
A previous whole-genome linkage analysis using SNP genotyping (10 k Affymetrix array) of the family members under study-yielded inconclusive results (Svensson et al. 2011). Here, we used phenotypic data and validated variants detected after exome sequencing of genomic DNA from key family members to identify the risk variant co-segregating with DD in this family. We could identify a mutation segregating with DD in CEP63, a gene not implicated in DD previously. The mutation found in the family is a two base-pair variant predicting a p.R229L substitution in CEP63 at a position between two of the six coiled-coil domains. CEP63 is, together with at least 30 other proteins of the CEP family, localised to the centrosome (Kumar et al. 2013), an organelle that functions as the major microtubule-organising center in eukaryotic cells and is essential for proper cell cycle progression. Several alternatively spliced variants of CEP63 with a variable number of protein coding exons have been found. However, their biological validity has not yet been determined experimentally. Exon 7, which contains the mutation described here, is shared among a majority of the annotated CEP63 transcripts. CEP63 is likely needed in all or most cells of the human body as seen from its ubiquitous expression. However, our observations of the TSSs of the various transcripts indicate that there may also be a tissue-specific set of transcripts that are uniquely important for the brain. If these transcripts are affected by the p.R299L variant, this may help explain why carriers of the mutation show developmental dyslexia but no other noticeable phenotype. Presently, we can only speculate about the exact mechanism for this effect.
Several studies report on a link between DD candidate genes and centrosome and cilia functions. DYX1C1 was identified as a DD candidate gene more than a decade ago (Taipale et al. 2003) and has since then been replicated as a DD gene by genetic association studies. Interestingly, the murine homologue Dyx1c1 is upregulated in ciliogenesis and exogenous DYX1C1 is localised to the centrosome (Hoh et al. 2012). Furthermore, we and others have linked reduced DYX1C1 levels to ciliary defects using a mouse model (Tarkar et al. 2013) and a zebrafish model system (Chandrasekar et al. 2013). Recent reports have described mutations in the dyslexia candidate gene DCDC2 in individuals with renal-hepatic ciliopathy or recessive deafness (Grati et al. 2015; Schueler et al. 2015). We speculate that amino acid substitutions in centrosomal proteins and proteins involved in ciliogenesis can increase risk of developmental dyslexia. The amino acid position and the severity of the alterations in chemical properties of the mutated protein might govern the observed/resulting phenotypic effect.
As CEP63 is expressed in brain and involved in mechanisms of cell division, we hypothesised that individuals heterozygous for CEP63 mutations may present mild defects in neuronal cell migration and proliferation leading to the cognitive problems associated with DD. Defects in neuronal cell proliferation and migration can manifest as altered density of white matter in the brain. As demonstrated earlier, SNPs in DD candidate genes affect white matter volume in a cohort of normally developing children and young adults from Sweden (Darki et al. 2012). In this study, we saw that there was a significant correlation between genotypes of rs7619451, located in CEP63, and white matter volume in the right hemisphere of healthy individuals. The cluster of significant effect further overlapped with a region previously found to be significant for SNPs within the DD candidate genes DYX1C1 and KIAA0319 (Darki et al. 2012). While this does not directly prove the p.R229L mutation in CEP63 as the cause of DD in the family under study, it supports our hypothesis that subtle genetic variation in CEP63 might contribute to normal variation in the mechanisms underlying DD. It also highlights further the emerging role of centrosome function in neuronal growth and maintenance.
Our Ingenuity Pathway Analysis shows that CEP63 directly interacts with multiple target proteins in the cell and these interactions are experimentally validated. The interacting targets could be subject to further analyses as it is tempting to speculate that the CEP63 mutation may alter the protein structure and result in reduced or loss of protein complex formation with one or several CEP63 binding proteins. Interestingly, the CEP63 p.R229L mutation is located within the CEP152 binding region (Firat-Karalar et al. 2014) and the mutation may possibly alter the protein–protein interaction between CEP63 and CEP152. This must, however, be verified experimentally.
The CEP63 and DEUP1 genes were produced from a single gene locus by gene duplication leading to proteins with related but specialised functions. The CEP63 paralog DEUP1 promotes mother centriole-independent centriole amplification in multi-ciliated differentiated cells (Zhao et al. 2013). Unlike DEUP1, CEP63 is required for normal mother centriole-dependent centriole duplication by recruitment of CEP152 and regulates mitotic entry through binding and recruitment of CDK1 to the centrosome (Brown et al. 2013; Loffler et al. 2011). Studies in U2OS cells revealed that CEP63 is localised at the proximal end of the paternal centriole where it forms a ring structure in complex with CEP57 and CEP152 (Lukinavicius et al. 2013). Interestingly, homozygosity for a nonsense mutation in CEP63 was reported in three cousins, from a consanguineous Pakistani family, presenting with microcephaly and short stature (Seckel syndrome-6, OMIM 614728) (Sir et al. 2011). Microscopy imaging using fluorescent antibodies in primary human white blood cells showed partly disordered or incomplete CEP152 ring structures in CEP63+/− individuals (carriers), or complete loss of CEP152 rings in CEP63−/− cells (Sir et al. 2011). Recently, it was shown that mice deficient in Cep63 display a Seckel syndrome phenotype with reduced adult weight accompanied by mitotic errors leading to p53-dependent neuronal progenitor cell death (Marjanovic et al. 2015).
Also, it remains to be seen if any of the previously reported DD candidate gene products form protein complexes with centrosomal proteins in general and CEP63 in particular. Future in vitro assays using exogenous mutated CEP63 could be initiated to investigate relevant cellular processes, such as centriole duplication and neuronal differentiation. These experiments could reveal the effect of gene mutations on centrosome function and, potentially link mutations in CEP63 to the phenotype of developmental dyslexia.
To conclude, we describe a novel CEP63 mutation in an extended three-generation family from Sweden and suggest CEP63 as a new candidate gene for DD. Further functional analysis of the mutated CEP63 protein may reveal aberrant cellular functions and implicate the centrosome as an important cellular structure in the biology underlying developmental dyslexia.
References
Adzhubei IA et al (2010) A method and server for predicting damaging missense mutations. Nat Methods 7:248–249. doi:10.1038/nmeth0410-248
Ameur A, Bunikis I, Enroth S, Gyllensten U (2014) CanvasDB: a local database infrastructure for analysis of targeted- and whole genome re-sequencing projects. Database (Oxford) 2014. doi:10.1093/database/bau098
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21:263–265. doi:10.1093/bioinformatics/bth457
Becker J et al (2014) Genetic analysis of dyslexia candidate genes in the European cross-linguistic NeuroDys cohort. Eur J Hum Genet 22:675–680. doi:10.1038/ejhg.2013.199
Brown NJ, Marjanovic M, Luders J, Stracker TH, Costanzo V (2013) Cep63 and cep152 cooperate to ensure centriole duplication. PLoS One 8:e69986. doi:10.1371/journal.pone.0069986
Brunham LR, Hayden MR (2013) Hunting human disease genes: lessons from the past, challenges for the future. Hum Genet 132:603–617. doi:10.1007/s00439-013-1286-3
Chandrasekar G, Vesterlund L, Hultenby K, Tapia-Paez I, Kere J (2013) The zebrafish orthologue of the dyslexia candidate gene DYX1C1 is essential for cilia growth and function. PLoS One 8:e63123. doi:10.1371/journal.pone.0063123
Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S, Sidow A (2005) Distribution and intensity of constraint in mammalian genomic sequence. Genome Res 15:901–913. doi:10.1101/gr.3577405
Darki F, Peyrard-Janvid M, Matsson H, Kere J, Klingberg T (2012) Three dyslexia susceptibility genes, DYX1C1, DCDC2, and KIAA0319, affect temporo-parietal white matter structure. Biol Psychiatry 72:671–676. doi:10.1016/j.biopsych.2012.05.008
Darki F, Peyrard-Janvid M, Matsson H, Kere J, Klingberg T (2014) DCDC2 polymorphism is associated with left temporoparietal gray and white matter structures during development. J Neurosci 34:14455–14462. doi:10.1523/JNEUROSCI.1216-14.2014
Elliott JG, Grigorenko EL (2014) The dyslexia debate. Cambridge Studies in Cognitive and Perceptual Development (14), Cambridge University Press, New York
Fagerheim T, Raeymaekers P, Tonnessen FE, Pedersen M, Tranebjaerg L, Lubs HA (1999) A new gene (DYX3) for dyslexia is located on chromosome 2. J Med Genet 36:664–669
Firat-Karalar EN, Rauniyar N, Yates JR 3rd, Stearns T (2014) Proximity interactions among centrosome components identify regulators of centriole duplication. Curr Biol 24:664–670. doi:10.1016/j.cub.2014.01.067
Fisher SE et al (1999) A quantitative-trait locus on chromosome 6p influences different aspects of developmental dyslexia. Am J Hum Genet 64:146–156. doi:10.1086/302190
Francks C, MacPhie IL, Monaco AP (2002) The genetic basis of dyslexia. Lancet Neurol 1:483–490
Grati M et al (2015) A missense mutation in DCDC2 causes human recessive deafness DFNB66, likely by interfering with sensory hair cell and supporting cell cilia length regulation. Hum Mol Genet 24:2482–2491. doi:10.1093/hmg/ddv009
Grigorenko EL, Wood FB, Meyer MS, Hart LA, Speed WC, Shuster A, Pauls DL (1997) Susceptibility loci for distinct components of developmental dyslexia on chromosomes 6 and 15. Am J Hum Genet 60:27–39
Hannula-Jouppi K, Kaminen-Ahola N, Taipale M, Eklund R, Nopola-Hemmi J, Kaariainen H, Kere J (2005) The axon guidance receptor gene ROBO1 is a candidate gene for developmental dyslexia. PLoS Genet 1:e50. doi:10.1371/journal.pgen.0010050
Hoh RA, Stowe TR, Turk E, Stearns T (2012) Transcriptional program of ciliated epithelial cells reveals new cilium and centrosome components and links to human disease. PLoS One 7:e52166. doi:10.1371/journal.pone.0052166
Kallberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, Xu J (2012) Template-based protein structure modeling using the RaptorX web server. Nat Protoc 7:1511–1522. doi:10.1038/nprot.2012.085
Kaplan DE et al (2002) Evidence for linkage and association with reading disability on 6p21.3–22. Am J Hum Genet 70:1287–1298. doi:10.1086/340449
Katusic SK, Colligan RC, Barbaresi WJ, Schaid DJ, Jacobsen SJ (2001) Incidence of reading disability in a population-based birth cohort, 1976–1982, Rochester. Minn Mayo Clin Proc 76:1081–1092. doi:10.4065/76.11.1081
Kere J (2014) The molecular genetics and neurobiology of developmental dyslexia as model of a complex phenotype. Biochem Biophys Res Commun 452:236–243. doi:10.1016/j.bbrc.2014.07.102
Kumar A, Purohit R (2012) Computational investigation of pathogenic nsSNPs in CEP63 protein. Gene 503:75–82. doi:10.1016/j.gene.2012.04.032
Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4:1073–1081. doi:10.1038/nprot.2009.86
Kumar A, Rajendran V, Sethumadhavan R, Purohit R (2013) CEP proteins: the knights of centrosome dynasty. Protoplasma 250:965–983. doi:10.1007/s00709-013-0488-9
Lamminmaki S, Massinen S, Nopola-Hemmi J, Kere J, Hari R (2012) Human ROBO1 regulates interaural interaction in auditory pathways. J Neurosci 32:966–971. doi:10.1523/JNEUROSCI.4007-11.2012
Loffler H et al (2011) Cep63 recruits Cdk1 to the centrosome: implications for regulation of mitotic entry, centrosome amplification, and genome maintenance. Cancer Res 71:2129–2139. doi:10.1158/0008-5472.CAN-10-2684
Lukinavicius G et al (2013) Selective chemical crosslinking reveals a Cep57-Cep63-Cep152 centrosomal complex. Curr Biol 23:265–270. doi:10.1016/j.cub.2012.12.030
Marjanovic M et al (2015) CEP63 deficiency promotes p53-dependent microcephaly and reveals a role for the centrosome in meiotic recombination. Nat Commun 6:7676. doi:10.1038/ncomms8676
Nopola-Hemmi J et al (2001) A dominant gene for developmental dyslexia on chromosome 3. J Med Genet 38:658–664
Olson R, Wise B, Conners F, Rack J, Fulker D (1989) Specific deficits in component reading and language skills: genetic and environmental influences. J Learn Disabil 22:339–348
Raven JC, Court JH, Raven J (1983) Manual for Raven’s progressive matrices and vocabulary scales (Sect. 7). London: HK Lewis
Roeleveld N, Zielhuis GA, Gabreels F (1997) The prevalence of mental retardation: a critical review of recent literature. Dev Med Child Neurol 39:125–132
Scerri TS, Schulte-Korne G (2010) Genetics of developmental dyslexia. Eur Child Adolesc Psychiatry 19:179–197. doi:10.1007/s00787-009-0081-0
Schmitt T, Ogris C, Sonnhammer EL (2014) FunCoup 3.0: database of genome-wide functional coupling networks. Nucleic Acids Res 42:D380–D388. doi:10.1093/nar/gkt984
Schueler M et al (2015) DCDC2 mutations cause a renal-hepatic ciliopathy by disrupting Wnt signaling. Am J Hum Genet 96:81–92. doi:10.1016/j.ajhg.2014.12.002
Schulte-Korne G et al (1998) Evidence for linkage of spelling disability to chromosome 15. Am J Hum Genet 63:279–282. doi:10.1086/301919
Schumacher J, Hoffmann P, Schmal C, Schulte-Korne G, Nothen MM (2007) Genetics of dyslexia: the evolving landscape. J Med Genet 44:289–297. doi:10.1136/jmg.2006.046516
Shaywitz SE (1998) Dyslexia. N Engl J Med 338:307–312. doi:10.1056/NEJM199801293380507
Shaywitz SE, Shaywitz BA, Fletcher JM, Escobar MD (1990) Prevalence of reading disability in boys and girls. Results of the Connecticut Longitudinal Study. JAMA 264:998–1002
Sir JH et al (2011) A primary microcephaly protein complex forms a ring around parental centrioles. Nat Genet 43:1147–1153. doi:10.1038/ng.971
Svensson I, Jacobson C (2006) How persistent are phonological difficulties? A longitudinal study of reading retarded children. Dyslexia 12:3–20
Svensson I, Nilsson S, Wahlstrom J, Jernas M, Carlsson LM, Hjelmquist E (2011) Familial dyslexia in a large Swedish family: a whole genome linkage scan. Behav Genet 41:43–49. doi:10.1007/s10519-010-9395-4
Taipale M et al (2003) A candidate gene for developmental dyslexia encodes a nuclear tetratricopeptide repeat domain protein dynamically regulated in brain. Proc Natl Acad Sci USA 100:11553–11558. doi:10.1073/pnas.1833911100
Tammimies K et al (2013) Molecular networks of DYX1C1 gene show connection to neuronal migration genes and cytoskeletal proteins. Biol Psychiatry 73:583–590. doi:10.1016/j.biopsych.2012.08.012
Tarkar A et al (2013) DYX1C1 is required for axonemal dynein assembly and ciliary motility. Nat Genet 45:995–1003. doi:10.1038/ng.2707
Tunmer WG, Greaney K (2010) Defining dyslexia. J Learn Disabil 43:229–243
van der Leij A, de Jong PF, Rijswijk-Prins H (2001) Characteristics of dyslexia in a Dutch family. Dyslexia 7:105–124. doi:10.1002/dys.197
Zhao H et al (2013) The Cep63 paralogue Deup1 enables massive de novo centriole biogenesis for vertebrate multiciliogenesis. Nat Cell Biol 15:1434–1444. doi:10.1038/ncb2880
Acknowledgments
This study was supported by Grants from the Karolinska Institutet, the Magnus Bergvall Foundation, the Swedish Research Council, Swedish Brain Foundation (Hjärnfonden), the Knut and Alice Wallenberg Foundation and the Bank of Sweden Tercentenary Foundation (Riksbankens Jubileumsfond). The authors acknowledge support from the National Genomics Infrastructure (NGI)/Uppsala Genome Center and UPPMAX for providing assistance in massive parallel sequencing and computational infrastructure. Work performed at NGI/Uppsala Genome Center has been funded by RFI/VR and Science for Life Laboratory, Sweden. The authors also would like to acknowledge Professor Lennart Nilsson, Karolinska Institutet, for advice on structural modelling. Also, support from BEA and KIGene, Karolinska Institutet, for DNA quality controls and Sanger sequencing services.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Nucleotide sequence data reported are available in the DDBJ/EMBL/GenBank databases under the accession number PRJEB9294.
Electronic supplementary material
Below is the link to the electronic supplementary material.
439_2015_1602_MOESM1_ESM.pdf
Supplementary material 1: Fig. S1 Pedigree of a six-generation family segregating developmental dyslexia. Family members with confirmed DD are denoted by black filled symbols. White symbols indicate individuals for which testing excluded DD diagnosis. Question marks indicate uncertain DD diagnosis. (PDF 695 kb)
439_2015_1602_MOESM2_ESM.tiff
Supplementary material 2: Fig. S2 FANTOM5 expression of ubiquitous transcription start sites (TSSes). The top panel shows the genomic context within CEP63 (gene region marked in green) and the known transcripts below. The middle panel shows the levels of expression starting at each region. The green bars show the level of expression of CEP63. The cluster of TSSes at the start of CEP63 is highlighted (grey), and the bottom panel shows where the transcripts from this specific TSS cluster are expressed (green bars, sorted by expression levels). The highest expression is in neutrophils (234.97 rle normalised tags/library), but expression is seen in most tested tissues (for clarity, only some of the tissues are shown). (TIFF 1608 kb)
439_2015_1602_MOESM3_ESM.tiff
Supplementary material 3: Fig. S3 FANTOM5 expression of neuronal TSS. The top panel shows the genomic context within which CEP63 (gene region marked in green) lies and the known transcripts below. The middle panel shows the levels of expression starting at each region. The green bars show expression of CEP63. The cluster of TSSes inside the CEP63 gene is highlighted (grey), and the bottom panel shows where the transcripts from this TSS cluster are expressed (green bars, sorted by expression levels). The highest expression is seen in the medial temporal gyrus (220.52 rle normalised tags/library) and only a few tissues show any expression > 10 rle normalised tags/library (an arbitrary cut-off for determining expression in a tissue). (TIFF 3256 kb)
439_2015_1602_MOESM4_ESM.pdf
Supplementary material 4: Fig. S4 Thirteen direct protein–protein binding partners for CEP63. All molecules were experimentally validated according to analysis criteria in the IPA software. The shapes of the individual proteins in the figure denote the different protein families that the binding partners belong to (e.g. protein phosphatases PPP4R1 and PPP1R13B). (PDF 864 kb)
439_2015_1602_MOESM5_ESM.tif
Supplementary material 5: Fig. S5 Clusters (in yellow) from MRI data showing significant correlation between genotypes in rs7619451 and white matter volume in human brain. The AA/AC genotypes were significantly associated with larger white matter volume. The regions in the right hemisphere overlap with right superior longitudinal fasciculus and posterior part of corpus callosum (peak coordinate: 28, -55, 29; p = 0.0076, corrected at the cluster level with p < 0.01). (TIFF 1734 kb)
439_2015_1602_MOESM6_ESM.xlsx
Supplementary material 6: Table S1 Table of predicted variants shared among affected individuals using exome sequencing. (XLSX 35 kb)
439_2015_1602_MOESM7_ESM.xlsx
Supplementary material 7: Table S2 Table of shared predicted non-synonymous and synonymous variants after in-house database filtering. (XLSX 13 kb)
439_2015_1602_MOESM8_ESM.pdf
Supplementary material 8: Table S3 List of experimentally validated direct interacting protein partners to CEP63 based on Ingenuity analysis. (PDF 101 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Einarsdottir, E., Svensson, I., Darki, F. et al. Mutation in CEP63 co-segregating with developmental dyslexia in a Swedish family. Hum Genet 134, 1239–1248 (2015). https://doi.org/10.1007/s00439-015-1602-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-015-1602-1