
Abstract
Genomic prediction of the extreme forms of adult body height or stature is of practical relevance in several areas such as pediatric endocrinology and forensic investigations. Here, we examine 770 extremely tall cases and 9,591 normal height controls in a population-based Dutch European sample to evaluate the capability of known height-associated DNA variants in predicting tall stature. Among the 180 normal height-associated single nucleotide polymorphisms (SNPs) previously reported by the Genetic Investigation of ANthropocentric Traits (GIANT) genome-wide association study on normal stature, in our data 166 (92.2 %) showed directionally consistent effects and 75 (41.7 %) showed nominally significant association with tall stature, indicating that the 180 GIANT SNPs are informative for tall stature in our Dutch sample. A prediction analysis based on the weighted allele sums method demonstrated a substantially improved potential for predicting tall stature (AUC = 0.75; 95 % CI 0.72–0.79) compared to a previous attempt using 54 height-associated SNPs (AUC = 0.65). The achieved accuracy is approaching practical relevance such as in pediatrics and forensics. Furthermore, a reanalysis of all SNPs at the 180 GIANT loci in our data identified novel secondary association signals for extreme tall stature at TGFB2 (P = 1.8 × 10−13) and PCSK5 (P = 7.8 × 10−11) suggesting the existence of allelic heterogeneity and underlining the importance of fine analysis of already discovered loci. Extrapolating from our results suggests that the genomic prediction of at least the extreme forms of common complex traits in humans including common diseases are likely to be informative if large numbers of trait-associated common DNA variants are available.
Similar content being viewed by others


Introduction
Adult human body height or stature, due to its high heritability (>0.8) (Carmichael and McGue 1995; Macgregor et al. 2006; Phillips and Matheny 1990; Silventoinen et al. 2000), quantitative nature, easy accessibility, and accurate measurability, serves as a reference trait for studying the genetics of human complex traits (Aulchenko et al. 2009; Campbell et al. 2005; Hirschhorn et al. 2001). Until early 2008, however, only a few genes were known to be involved with normal height variation in general human populations (Perola et al. 2007; Silventoinen et al. 2003; Visscher et al. 2006). In 2008, three genome-wide association studies (GWAS) comprising tens of thousands of individuals highlighted 54 genetic loci displaying statistically significant association with body height (Gudbjartsson et al. 2008; Lettre et al. 2008; Weedon et al. 2008). Forty of these loci (74.1 %) had not been previously implicated in human body height, and many were from outside the biological pathways known or expected to regulate human growth. These studies on one hand convincingly confirmed the polygenic nature of adult height and on the other hand revealed several new genetic pathways involved in growth regulation, such as Hedgehog signaling and basic cell cycle regulation. In 2010, a meta-analysis of all available GWAS data from 46 studies comprising 180,000 individuals raised the number of known genetic loci associated with human height to at least 180 (Lango Allen et al. 2010). This work was done under the auspices of the Genetic Investigation of ANthropocentric Traits (GIANT) Consortium, referred to as the GIANT study below. In spite of the large number of genetic loci identified, the associated single nucleotide polymorphisms (SNPs) according to the authors only explained about 10 % of the sex- and age-adjusted height variance in the study populations (Lango Allen et al. 2010). The GIANT study pointed out that allelic heterogeneity is likely a frequent feature for human polygenic traits in general, and for adult body height in particular.
The ability to predict the extreme forms of adult body height based on the parental or the donor’s DNA has important practical implications in pediatric endocrinology (Unrath et al. 2012) as well as in forensic investigation (Kayser and de Knijff 2011), to name only two examples. Using 54 height-associated SNPs available in 2009, Aulchenko and colleagues (2009) constructed a genomic profile and showed that extreme forms of human body height were predictable with only limited accuracy. In the present study, we aim to update the predictive capacity for human tall stature available with the extended list of 180 height-associated SNPs from the GIANT study using a European study population from the Netherlands that was enriched by people of tall stature. We focus on tall stature here because it had been shown previously that the effects of height-associated common DNA variants are consistent with the predicted polygenic effects in extremely tall but less so in extremely short individuals (Chan et al. 2011).
Methods
The Dutch tall cohort (DT)
From the records of the Division of Pediatric Endocrinology at the Erasmus University Medical Center, Sophia Children’s Hospital, we identified former patients who attended this clinic for evaluation of tall stature. Eligible subjects were traced using municipal registries and invited by mail to participate in this study. We also identified several healthy tall individuals through advertisement in local specialized shops, sports centers, and institutions of higher education in Rotterdam. Subjects eligible for participation fulfilled the following inclusion criteria: (1) standard deviation score (SDS) above +1.88 SD according to Dutch standards (http://www.tno.nl/groei), which corresponds to the 3 % upper tail of the height distribution in Dutch adults [approximately >195 cm in men and >180 cm in women at age 30, after correcting for secular trend (Fredriks et al. 2000)]; and (2) Dutch European ancestry defined as being born to Dutch parents who themselves were born in the Netherlands. Three subjects with endocrine or metabolic disorders or primary or secondary growth disorders were excluded. Subjects fulfilling the inclusion criteria who had been treated with high-dose sex steroids to limit growth in adolescence were eligible to participate and were considered as being tall. Participants were invited to visit the outpatient clinic of the Erasmus Medical Center. Height was measured using a stadiometer (SECA 225; SECA, Hamburg, Germany). Genomic DNA was extracted from venous blood using standard methods. After phenotypic and genotypic quality controls, the DT cohort included 462 unrelated Dutch tall individuals. These tall subjects are independent of the GIANT study.
The Rotterdam Study (RS)
The RS is a population-based prospective study including a main cohort (RS-I) and two extensions (RS-II and RS-III) (Hofman et al. 1991, 2007, 2009). The height and genetic data regarding RS subjects have been described in the GIANT paper. In brief, all RS participants were examined in detail at baseline including a quantitative measurement of body height using a stadiometer. The Medical Ethics Committee of Erasmus University Medical Center approved the study protocol and all study participants provided written informed consent. After genomic and phenotypic quality controls, the current study included 5,748, 2,152, and 1,999 participants from RS-I, RS-II, and RS-III, respectively. The tall stature in RS was set as the sex- and age-adjusted residuals >1.88 standard deviations (193 cases in RS-I, 49 in RS-II, and 66 in RS-III).
Microarray genotyping and quality control (QC)
Details regarding genotyping, SNP imputation and QC of RS individuals can be found elsewhere. DT individuals were de novo genotyped using Illumina Human610Quad array (with 620,901 markers, both SNPs and CNV probes) according to standard procedures. The standard QC of the samples included the analysis of genotyped called sex, call rate, heterozygosity, homozygosity, assessment of cryptic family relationships and assessment of population admixture/stratification. Multidimensional scaling (MDS) analysis was conducted using identity-by-state (IBS) pair-wise distances with reference to individuals of the HapMap Phase II panel (http://hapmap.ncbi.nlm.nih.gov/). Potential population stratification was initially controlled by exclusion of individuals of non-Northwestern European origin defined as those deviating more than 4 standard deviations from the mean of the HapMap CEU panel. Genotypes with minor allele frequency (MAF) > 1 %, SNP call rate > 98 % and Hardy–Weinberg equilibrium (HWE) P > 1 × 10−4 were then imputed to 2,543,887 SNPs using MACH (Li et al. 2009) with reference to the phased autosomal chromosomes of the HapMap CEU Phase II panel (release 22, build 36). Pair-wise IBS matrix between individuals was recalculated using a subset of pruned SNPs (N = 50,000) that are in approximate linkage equilibrium. No close relatives were identified. We merged DT with RS using Genome Studio software v2010 and compared the SNP call rate by cohort status and excluded 13,330 SNPs with a significant difference (P < 0.01). This QC step is very important to reduce the potential false positives due to differential missingness between cohorts.
Association analysis at 180 height-associated regions
The tall stature was considered as a binary trait (tall or non-tall). Since tall stature was defined using sex- and age-adjusted residuals, sex and age were no longer considered in subsequent analyses. We selected the 180 SNPs described in the Supplementary Table 1 of the GIANT paper (Lango Allen et al. 2010). Association was tested using logistic regression where allelic Odds ratios (ORs) were derived. We consider P values <0.05 as statistically significant. Next, we extended the 180 GIANT SNPs to 180 regions defined as ±200 kBp from each side of the GIANT SNPs and conducted association analysis as well as conditional association analysis (conditional on the effect of the GIANT SNP in each region) searching for secondary signals, where the minimal P values were Bonferroni corrected for the number of SNPs in each region. Haplotypes were inferred based on genotypes using the expectation–maximization algorithm implemented in R library haplo.stats. We tested whether the observed ORs are consistent with the expected ones, based on the previously estimated effect sizes from the GIANT study using the method proposed by Chan et al. In brief, we calculated the expected OR and its expected 95 % confidence interval for each SNP by estimating the odds of the effect allele versus the non-effect allele in the tall cases and controls assuming a standard normal distribution for the standardized height based on the formula provided by Chan et al., except that we used 1.88 for integration (representing 3 % tallest) instead of using ±2.326 (representing 1 % two-sided tails).
Prediction analysis
There are several competing methods available for predicting tall stature from DNA variants. In this study, we selected the weighted allele sums (WAS) (Aulchenko et al. 2009) method to allow direct comparison with the previous study of Aulchenko, also because its performance was not significantly lower than the other methods tested in our dataset (see Supplementary materials and Supplementary Fig. 1a, b). The WAS is a weighted prediction of height for each individual based on either logistic regression betas from the training set of the current study (80 % of samples) or the previously estimated betas of linear regressions from the GIANT study. The model performance was cross-validated in the remaining 20 % samples (number of cross-validations = 1,000), where the prediction accuracies were derived. We derived the area under the receiver operating characteristic (ROC) curves, or AUC (Janssens et al. 2004), as the accuracy estimate. AUC is the integral of ROC curves, which ranges from 0.5 representing total lack of prediction to 1.0 representing completely accurate prediction.
Prediction analyses were conducted using four different lists of SNPs as predictors: (1) 180 SNPs randomly selected over the microarray, (2) 54 SNPs previously used by Aulchenko et al. (2009), (3) 180 GIANT SNPs (Lango Allen et al. 2010), (4) 180 GIANT plus two additional SNPs from current study showing significant secondary association with tall stature in a reanalysis of the 180 regions. All prediction analyses were conducted using R scripting and R libraries “e1071”, “glmnet”, “nnet”, “rpart” and “randomForest” accepting default tuning parameters, and the AUC values were derived using R library “verification”, all freely available from the CRAN website (http://cran.r-project.org/).
GWAS
We conducted a case–control designed GWAS for tall stature in 10,361 Dutch Europeans (see Supplementary materials, Supplementary Fig. 2).
Results
A binary tall stature was set as the sex- and age-adjusted height residuals >1.88 standard deviations (770 tall cases and 9,591 non-tall controls, Fig. 1). More than half (60 %) of the tall cases were from the Dutch tall cohort (N = 462), which was a completely independent cohort from the GIANT study population, whereas the remaining tall individuals as well as the normal height controls came from the Rotterdam study previously included in the GIANT meta-analysis (Lango Allen et al. 2010). Among the 180 height-associated SNPs reported in the GIANT study and tested here, 166 (92.2 %) had the same height-increasing alleles in our case–control samples as reported in the GIANT study; 75 (41.7 %) were nominally significantly associated with tall stature (P < 0.05); and none of the significant associations were in the opposite allele directions (Supplementary Table 1). The probability of observing 75 or more significant P values out of 180 tests is extremely small (P = 7.1 × 10−50) under the null binomial distribution. We tested whether the observed ORs are consistent with the expected ORs based on the previously estimated effect sizes from the GIANT study. Similar to that observed by Chan et al., the number of SNPs with observed OR greater than expected OR was no different from expectation under the model of equal effect sizes in tall cases and controls (97/180 SNPs, P = 0.11, Supplementary Table 1). A multivariate logistic regression including all 180 SNPs showed highly consistent results with the analysis of individual SNPs, where 70 (38.9 %) were nominally significant and all of these 70 were directionally consistent with the GIANT study (Supplementary Table 1). These results indicate that the 180 GIANT SNPs are highly informative for tall stature in our dataset, which motivated us to carry out a series of prediction analyses of tall stature utilizing these DNA variants.

The distribution of sex- and age-adjusted height residuals (z-score) in the four studied Dutch subpopulations. Dutch Tall cohort (DT) = 462, Rotterdam Study 1 cohort (RS-I) = 5,748; Rotterdam Study 2 cohort (RS-II) = 2,152; Rotterdam Study 3 cohort (RS-III) = 1,999; total N = 10,361. The vertical blue dashed line indicates the cut-off value (1.88 SD) for cases
A WAS prediction analysis using 180 SNPs repeatedly and randomly selected from the whole SNP microarray dataset gave an average AUC of 0.52, slightly higher than the null value of 0.50 (Table 1). This finding is not surprising given the highly polygenic nature of the trait, i.e., one may expect some of the randomly selected SNPs being associated with height simply by chance. Conducting the WAS prediction analysis using the 54 SNPs previously applied for tall stature prediction by Aulchenko et al. (2009) gave a mean cross-validated AUC of 0.67 (95 % CI 0.63–0.70, Table 1) in our study population for predicting the 3 % tallest, which was consistent with the one reported by Aulchenko et al. (AUC = 0.65 for predicting the 5 % tallest) in the RS-I population, which is partly (25 % of all cases and 58 % of all controls) overlapping with the samples used here. Repeating the prediction analysis using the 180 GIANT SNPs, however, resulted in a substantial improvement in prediction accuracy with a mean cross-validated AUC of 0.75 (95 % CI 0.72–0.79, Table 1) in our sample set. The WAS score calculated using the 180 GIANT SNPs also showed a significant increase from non-tall (mean −1.68) to tall individuals (mean −0.31, P t test < 1 × 10−300, Fig. 2). In terms of the percentage of variance explained (the sex- and age-adjusted residuals), the 180 GIANT SNPs (cross-validated mean R 2 = 12.14 %, 95 % CI 10.35–14.07) could explain more than twofold increased variance of sex- and age-adjusted body height than that explained by the 54 SNP previously used by Aulchenko et al. (cross-validated mean R 2 = 5.40 %, 95 % CI 3.75–6.97, Table 1).

Distribution of weighted allele sums (WAS) in 770 tall and 9,591 non-tall Dutch Europeans. a Histogram; b density plot. The WAS score was calculated for each individual using the 180 GIANT SNPs as regression beta × the number of minor alleles (not necessary the height-increasing allele)
The above prediction accuracies were estimated using the WAS score weighted by the logistic regression coefficients in our Dutch sample. Weighting the WAS score directly using the linear regression betas from the GIANT paper also resulted high comparable accuracy estimates (AUC = 0.74 and R 2 = 12.03 %), indicating that our prediction model is likely extendable to other European populations. Further restricting the prediction analysis only in RS sample also resulted comparable accuracy estimates (AUC = 0.75, R 2 = 12.16 %), indicating that the accuracy is unlikely over-estimated because the RS sample was part of the GIANT study. This is expected because the Rotterdam sample represents only a small fraction of the GIANT study, and also because the majority of the tall cases in the current study were ascertained independent of the Rotterdam study and were not used in the GIANT study. Several genes/loci were highlighted to contain SNPs contributing the prediction accuracy with ∆AUC > 0.01, such as ADAMTS10, PCSK5, LTBP2, EFEMP1, PTCH1/FANCC, CABLES1, NPR3 and ADAMTSL3, representing the best tall stature DNA predictors in our study. Yang et al. (2010) has proposed that reanalysis of the already discovered regions may reveal new variants further explaining phenotypic variance. In this study, the prediction accuracy could be slightly (AUC = 0.76, 95 % CI 0.73–0.80, Table 1) but significantly (P < 0.01, t test based on 1,000 cross-validations) improved by including two newly identified SNPs (rs12048049 in TGFB2 and rs10869665 in PCSK5) showing significant secondary association with tall stature (see below).
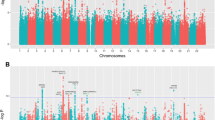
We reanalyzed all SNPs flanking the 180 GIANT SNPs (±200 kBp, total 36,442 SNPs), which did not reveal additional SNPs with genome-wide significance except two signals at LTBP2 and ADAMTS10 (Fig. 3a). The observed P values at these 180 regions far deviated from the expected ones under the null hypothesis (Fig. 3b). This can be largely explained by linkage disequilibrium (LD) around known signals, supporting the involvement of these regions in tall stature. A conditional analysis (conditioned on the GIANT SNPs in each region), however, revealed two different SNPs in two different regions being highly significantly associated with tall stature: rs12048049 in TGFB2 (P = 1.8 × 10−13) and rs10869665 in PCSK5 (P = 7.8 × 10−11, Fig. 3c). For both regions, individual SNPs were associated with tall stature at only nominal significance (0.001 < P < 0.05). We, therefore, further conducted a diplotype analysis for the associated SNPs and identified several large ORs by contrasting different diplotype groups. For example, the AC/GC diplotype group of the two SNPs rs6684205 (with alleles AG, from GIANT) and rs12048049 (CG, the secondary signal) at TGFB2 had a significantly increased fraction of tall stature compared to the reference AC/AC group (OR = 5.20, 95 % CI 3.24–8.33, P = 8.01 × 10−12, Table 2). Likewise, the GC/AT group for rs11144688GA and rs10869665CT at the PCSK5 locus had a significantly decreased fraction of tall stature than the reference GC/GC group (OR = 0.38, 95 % CI 0.27–0.55, P = 1.92 × 10−7, Table 2). These secondary association signals were new to the GIANT findings, which may be explained by different study designs. The conditional analysis in the GIANT study included 225 SNPs as covariates and was conducted in each stage-I cohort then meta-analyzed, whereas our analysis was iteratively conditioned on each GIANT SNP using individual-level data. The observed P values from the conditional analysis also substantially deviated far from the null (Fig. 3d). The remaining LD at these loci alone cannot fully explain this massive deviation including the highly significant secondary signals at TGFB2 and PCSK5, which strongly suggest that allelic heterogeneity plays an important role not only for normal height variation but also for tall stature in humans.

Regional Manhattan plot for SNPs associated with tall stature at the 180 GIANT loci (total 36,442 SNPs) in 770 tall and 9,591 non-tall Dutch Europeans. a Initial results; b QQ plot of the initial results; c conditioned on the GIANT SNPs in each locus; d QQ plot of the conditional analysis
Finally, we compared our data with two recent candidate gene studies on tall stature. The growth hormone (GH)/insulin-like growth factor-1 (IGF-1) axis is a key regulator of somatic growth in humans. We observed significant genetic association with tall stature for IGF2BP2 (rs720390, P = 4.1 × 10−2), IGF2BP3 (rs12534093, P = 1.1 × 10−3), IGF1R (rs2871865, P = 4.7 × 10−4) and GH1 (rs2665838, P = 1.5 × 10−2) but not for GHSR (rs572169, P = 0.37, Supplementary Table 1). These findings are highly consistent with a recent candidate gene study focusing on the GH axis in relation to extreme tall stature (Hendriks et al. 2011a). Recently, a candidate gene study of HMGA2 reported an effect of rs1042725 in extremely tall stature (OR for C allele = 1.53, 95 % CI 1.02–2.28; P = 0.03) (Hendriks et al. 2011b). In our study of an increased sample size, this SNP also showed significant but smaller effect on tall stature (OR for C allele = 1.15, 95 % CI 1.03–1.27; P = 0.01), whereas another HMGA2 SNP rs1351394 reported by the GIANT study showed a slightly larger effect (OR for T allele = 1.17, 95 % CI 1.05–1.29; P = 3.45 × 10−3, Supplementary Table 1).
Discussion
With the present study we demonstrated that a large fraction of the 180 SNPs previously associated with normal height in Europeans is also associated with tall stature in Europeans. Notably, the 180 SNPs were previously identified in samples from 46 studies, comprising 183,727 individuals with recent European ancestry from various regions of Europe, whereas our study was carried out in a European population from a single country (i.e., the Netherlands). The vast majority of these 180 previously normal height-associated SNPs in our study had odds ratios for tall stature that were directionally consistent with those for normal height in the GIANT study. These results confirm the findings of Chan et al. (2011) in that height-associated common DNA variants show predicted polygenic effects in tall individuals, i.e., the observed ORs are consistent with the expected ORs from the previously estimated effect sizes from the GIANT study. However, this does not contradict the hypothesis that rare variants may still explain a substantial portion of the trait heritability.
Our data demonstrate that common SNPs allow the genetic prediction of tall stature with an accuracy that considerably exceeds a previous attempt. Aulchenko and colleagues (2009) previously established a genomic profile based on 54 height-associated SNPs available in 2009, which showed a relatively low predictive accuracy (AUC = 0.65 for predicting a person falling into 5 % tallest of their study and AUC = 0.67 for predicting the tallest 3 % in the current study). These authors also estimated that in order to achieve an AUC of 0.80, one needs to explain at least three times the amount of phenotypic variance that is explained by the 54 SNPs they used. On the other hand, they showed that the traditional approach of predicting height using mid-parental height, the so-called Victorian Galton’s method, achieved an AUC of 0.84 when predicting the tallest 5 %. The authors suspected that the Victorian Galton’s method will long stay unsurpassed in terms of predictive accuracy. Unfortunately, this approach is useless in those practical applications of height prediction where parental height is usually not available, such as in forensic investigations. Here, we empirically demonstrate that the capacity of the extended list of 180 SNPs in predicting tall stature summed up to a reasonably high accuracy with AUC of 0.75, which far surpassed that of Aulchenko et al. (AUC of 0.65 or 0.67) and gets closer to albeit not yet surpassing the non-genetic Galton’s method. However, the genomic prediction accuracy for tall stature obtained here is likely to be further improvable by the outcomes of future work on the genetic basis of human heights. Considering the increasingly smaller effect sizes of the remaining common variants to be discovered by GWAS, it is rather unlikely that the genomic prediction accuracy can be improved substantially by undiscovered common variants at novel loci even if extremely large studies are being conducted (ongoing extension of GIANT study). A more promising approach to further improve the genomic predictability of tall stature may be by considering genetic interactions and compound alleles of all variants within the already discovered loci as will become feasible with next generation exome or whole genome sequencing data.
The ability to accurately predict the extreme forms of adult human body height using DNA variants is of practical relevance in several applications. In pediatric endocrinology for instance, the early knowledge about abnormal statures is important for making clinical decisions on growth advancing and height-limiting treatments (Hendriks et al. 2010, 2011c, 2012). Current practices in predicting the expected adult body height of a child relies on bone age (Thodberg et al. 2009) and mid-parental height (Su et al. 2011). Although the accuracy of the Galton’s mid-parental height method was estimated as high as AUC of 0.84, its applicability may be hampered by assortative mating and poor parent–offspring correlation. These methods may at least be improvable by including the child’s or parental DNA variants as additional predictors.
Another application of DNA-based prediction of tall human stature is in Forensic DNA Phenotyping (FDP), a young field of forensic genetics aiming to provide investigative leads in suspect-less criminal cases via providing externally visible characteristics of the sample donor directly from DNA obtained from the crime scene (Kayser and de Knijff 2011; Kayser and Schneider 2009). Successful examples of FDP include DNA prediction of certain eye and hair color categories, which can be predicted with a much higher accuracy from a much smaller number of SNPs (Branicki et al. 2011; Liu et al. 2009a). For these traits, DNA test system have already been developed and validated for practical forensic case work applications (Walsh et al. 2011a, b, 2012a, b) and are started to be used in routine forensic practice.
Our GWAS in a Dutch European sample set enriched for tall individuals highlighted two loci being significantly associated with tall stature (LTBP2 and ADAMTS10), both overlapped with those loci previously reported with genome-wide significant association with normal stature in the GIANT study. Although the effective sample size (N ~ 25,600 from power calculation) is increased by the extreme design, this number is still far from comparable to the GIANT study, which used 134,000 individuals in the initial discover phase. Therefore, this sample is expected to have low power in finding new height loci unless specifically involved in extreme tall stature or much more involved in extremely tall than in normal stature. Our results suggest that such loci unlikely exist.
When analyzing the genomic regions that include the 180 GIANT SNPs, we found SNPs at two loci (LTBP2 and ADAMTS10) showing association with tall stature at the genome-wide significance level. Mutations in LTBP2 and ADAMTS10 are long known to cause autosomal recessive Weill-Marchesani syndrome (Dagoneau et al. 2004; Haji-Seyed-Javadi et al. 2012). We also found SNPs at two genes (TGFB2 and PCSK5) showing highly significant secondary association signals. This strongly suggests that allelic heterogeneity likely exists at least in these associated regions. Including these two newly identified SNPs in prediction model improved the prediction accuracy slightly but significantly, underling the importance of regional fine analysis of the already discovered loci/regions for finding new variants, as also proposed by Yang et al. (2010). TGFB2 encodes the transforming growth factor beta (TGF-β), a protein that controls proliferation, cellular differentiation, and other functions in most cells. The role of TGF-β signaling in growth and related disorders has been systematically reviewed (Le Goff and Cormier-Daire 2012). TGF-β is also known to prevent the impaired chondrocyte proliferation induced by unloading in growth plates of young rats (Zerath et al. 1997). PCSK5 encodes the proprotein convertase subtilisin/kexin type 5, the expression of which has been linked to developmental dynamics in mice (Liu et al. 2009b; Szumska et al. 2008) and zebra fish (Chitramuthu et al. 2010). Several loci did not show statistically significant association likely due to low MAF (<5 %, and thus low power) but showed consistent allele directions, such as PPARD/FANCE, CYP19A1 and ACAN, in which the possibility of existing rare functional mutations cannot be excluded. For example, exome sequencing recently identified amino acid-altering variants in ACAN influencing human height (Kim et al. 2012). In addition, our data supports the recent significant findings for the GH-related genes (Hendriks et al. 2011a) and HMGA2 (Hendriks et al. 2011b) on extreme tall stature, although their genetic effects appeared to be smaller in our sample. Overall, these data show that the genetics of tall stature is similar to that of normal height in humans, i.e., heritability is for a large part explained by many common variants with small effects and allelic heterogeneity is a frequent feature.
In summary, we showed that a large number of DNA variants previously implicated in normal height variation in Europeans are also involved in determining tall stature of Europeans. The obtained genomic prediction accuracy is considerably improved compared to a previous attempt. There is, however, still room for further improvement, which may partly rely on modeling the genetic interactions and allelic heterogeneities within those height-associated loci/regions. Nevertheless, the achieved accuracy is already relevant to fields of future practical applications of genomic height prediction such as pediatrics and forensics in that they clearly demonstrate the potential of DNA for predicting height, at least it is extremely tall form. Finally, extrapolating from our results suggests that the genomic prediction of at least the extreme forms of common complex traits in humans including common diseases are likely to be informative if large numbers of trait-associated common DNA variants are available.
References
Aulchenko YS, Struchalin MV, Belonogova NM, Axenovich TI, Weedon MN, Hofman A, Uitterlinden AG, Kayser M, Oostra BA, van Duijn CM, Janssens AC, Borodin PM (2009) Predicting human height by Victorian and genomic methods. Eur J Hum Genet 17:1070–1075. doi:10.1038/ejhg.2009.5
Branicki W, Liu F, van Duijn K, Draus-Barini J, Pospiech E, Walsh S, Kupiec T, Wojas-Pelc A, Kayser M (2011) Model-based prediction of human hair color using DNA variants. Hum Genet 129:443–454. doi:10.1007/s00439-010-0939-8
Campbell CD, Ogburn EL, Lunetta KL, Lyon HN, Freedman ML, Groop LC, Altshuler D, Ardlie KG, Hirschhorn JN (2005) Demonstrating stratification in a European American population. Nat Genet 37:868–872
Carmichael CM, McGue M (1995) A cross-sectional examination of height, weight, and body mass index in adult twins. J Gerontol A Biol Sci Med Sci 50:B237–B244
Chan Y, Holmen OL, Dauber A, Vatten L, Havulinna AS, Skorpen F, Kvaloy K, Silander K, Nguyen TT, Willer C, Boehnke M, Perola M, Palotie A, Salomaa V, Hveem K, Frayling TM, Hirschhorn JN, Weedon MN (2011) Common variants show predicted polygenic effects on height in the tails of the distribution, except in extremely short individuals. PLoS Genet 7:e1002439. doi:10.1371/journal.pgen.1002439
Chitramuthu BP, Baranowski DC, Cadieux B, Rousselet E, Seidah NG, Bennett HP (2010) Molecular cloning and embryonic expression of zebrafish PCSK5 co-orthologues: functional assessment during lateral line development. Dev Dyn 239:2933–2946. doi:10.1002/dvdy.22426
Dagoneau N, Benoist-Lasselin C, Huber C, Faivre L, Megarbane A, Alswaid A, Dollfus H, Alembik Y, Munnich A, Legeai-Mallet L, Cormier-Daire V (2004) ADAMTS10 mutations in autosomal recessive Weill-Marchesani syndrome. Am J Hum Genet 75:801–806. doi:10.1086/425231
Fredriks AM, van Buuren S, Burgmeijer RJ, Meulmeester JF, Beuker RJ, Brugman E, Roede MJ, Verloove-Vanhorick SP, Wit JM (2000) Continuing positive secular growth change in The Netherlands 1955–1997. Pediatr Res 47:316–323
Gudbjartsson DF, Walters GB, Thorleifsson G, Stefansson H, Halldorsson BV, Zusmanovich P, Sulem P, Thorlacius S, Gylfason A, Steinberg S, Helgadottir A, Ingason A, Steinthorsdottir V, Olafsdottir EJ, Olafsdottir GH, Jonsson T, Borch-Johnsen K, Hansen T, Andersen G, Jorgensen T, Pedersen O, Aben KK, Witjes JA, Swinkels DW, den Heijer M, Franke B, Verbeek AL, Becker DM, Yanek LR, Becker LC, Tryggvadottir L, Rafnar T, Gulcher J, Kiemeney LA, Kong A, Thorsteinsdottir U, Stefansson K (2008) Many sequence variants affecting diversity of adult human height. Nat Genet 40:609–615. doi:10.1038/ng.122
Haji-Seyed-Javadi R, Jelodari-Mamaghani S, Paylakhi SH, Yazdani S, Nilforushan N, Fan JB, Klotzle B, Mahmoudi MJ, Ebrahimian MJ, Chelich N, Taghiabadi E, Kamyab K, Boileau C, Paisan-Ruiz C, Ronaghi M, Elahi E (2012) LTBP2 mutations cause Weill-Marchesani and Weill-Marchesani-like syndrome and affect disruptions in the extracellular matrix. Hum Mutat 33:1182–1187. doi:10.1002/humu.22105
Hendriks AE, Boellaard WP, van Casteren NJ, Romijn JC, de Jong FH, Boot AM, Drop SL (2010) Fatherhood in tall men treated with high-dose sex steroids during adolescence. J Clin Endocrinol Metab 95:5233–5240. doi:10.1210/jc.2010-0435
Hendriks AE, Brown MR, Boot AM, Oostra BA, de Jong FH, Drop SL, Parks JS (2011a) Common polymorphisms in the GH/IGF-1 axis contribute to growth in extremely tall subjects. Growth Horm IGF Res 21:318–324. doi:10.1016/j.ghir.2011.08.001
Hendriks AE, Brown MR, Boot AM, Oostra BA, Drop SL, Parks JS (2011b) Genetic variation in candidate genes like the HMGA2 gene in the extremely tall. Horm Res Paediatr 76:307–313. doi:10.1159/000330764
Hendriks AE, Laven JS, Valkenburg O, Fong SL, Fauser BC, de Ridder MA, de Jong FH, Visser JA, van Ginneken AM, Boot AM, Drop SL (2011c) Fertility and ovarian function in high-dose estrogen-treated tall women. J Clin Endocrinol Metab 96:1098–1105. doi:10.1210/jc.2010-2244
Hendriks AE, Drop SL, Laven JS, Boot AM (2012) Fertility of tall girls treated with high-dose estrogen, a dose-response relationship. J Clin Endocrinol Metab 97:3107–3114. doi:10.1210/jc.2012-1078
Hirschhorn JN, Lindgren CM, Daly MJ, Kirby A, Schaffner SF, Burtt NP, Altshuler D, Parker A, Rioux JD, Platko J, Gaudet D, Hudson TJ, Groop LC, Lander ES (2001) Genomewide linkage analysis of stature in multiple populations reveals several regions with evidence of linkage to adult height. Am J Hum Genet 69:106–116. doi:10.1086/321287
Hofman A, Grobbee DE, de Jong PT, van den Ouweland FA (1991) Determinants of disease and disability in the elderly: the Rotterdam Elderly Study. Eur J Epidemiol 7:403–422
Hofman A, Breteler MM, van Duijn CM, Krestin GP, Pols HA, Stricker BH, Tiemeier H, Uitterlinden AG, Vingerling JR, Witteman JC (2007) The Rotterdam Study: objectives and design update. Eur J Epidemiol 22:819–829. doi:10.1007/s10654-007-9199-x
Hofman A, Breteler MM, van Duijn CM, Janssen HL, Krestin GP, Kuipers EJ, Stricker BH, Tiemeier H, Uitterlinden AG, Vingerling JR, Witteman JC (2009) The Rotterdam Study: 2010 objectives and design update. Eur J Epidemiol 24:553–572. doi:10.1007/s10654-009-9386-z
Janssens AC, Pardo MC, Steyerberg EW, van Duijn CM (2004) Revisiting the clinical validity of multiplex genetic testing in complex diseases. Am J Hum Genet 74:585–588. doi:10.1086/382052 (author reply 588–589)
Kayser M, de Knijff P (2011) Improving human forensics through advances in genetics, genomics and molecular biology. Nat Rev Genet 12:179–192. doi:10.1038/nrg2952
Kayser M, Schneider PM (2009) DNA-based prediction of human externally visible characteristics in forensics: motivations, scientific challenges, and ethical considerations. Forensic Sci Int Genet 3:154–161. doi:10.1016/j.fsigen.2009.01.012
Kim JJ, Park YM, Baik KH, Choi HY, Yang GS, Koh I, Hwang JA, Lee J, Lee YS, Rhee H, Kwon TS, Han BG, Heath KE, Inoue H, Yoo HW, Park K, Lee JK (2012) Exome sequencing and subsequent association studies identify five amino acid-altering variants influencing human height. Hum Genet 131:471–478. doi:10.1007/s00439-011-1096-4
Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, Willer CJ, Jackson AU, Vedantam S, Raychaudhuri S, Ferreira T, Wood AR, Weyant RJ, Segre AV, Speliotes EK, Wheeler E, Soranzo N, Park JH, Yang J, Gudbjartsson D, Heard-Costa NL, Randall JC, Qi L, Vernon Smith A, Magi R, Pastinen T, Liang L, Heid IM, Luan J, Thorleifsson G, Winkler TW, Goddard ME, Sin Lo K, Palmer C, Workalemahu T, Aulchenko YS, Johansson A, Carola Zillikens M, Feitosa MF, Esko T, Johnson T, Ketkar S, Kraft P, Mangino M, Prokopenko I, Absher D, Albrecht E, Ernst F, Glazer NL, Hayward C, Hottenga JJ, Jacobs KB, Knowles JW, Kutalik Z, Monda KL, Polasek O, Preuss M, Rayner NW, Robertson NR, Steinthorsdottir V, Tyrer JP, Voight BF, Wiklund F, Xu J, Hua Zhao J, Nyholt DR, Pellikka N, Perola M, Perry JR, Surakka I, Tammesoo ML, Altmaier EL, Amin N, Aspelund T, Bhangale T, Boucher G, Chasman DI, Chen C, Coin L, Cooper MN, Dixon AL, Gibson Q, Grundberg E, Hao K, Juhani Junttila M, Kaplan LM, Kettunen J, Konig IR, Kwan T, Lawrence RW, Levinson DF, Lorentzon M, McKnight B, Morris AP, Muller M, Suh Ngwa J, Purcell S, Rafelt S, Salem RM, Salvi E et al (2010) Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 467:832–838. doi:10.1038/nature09410
Le Goff C, Cormier-Daire V (2012) From tall to short: the role of TGFbeta signaling in growth and its disorders. Am J Med Genet C Semin Med Genet 160C:145–153. doi:10.1002/ajmg.c.31337
Lettre G, Jackson AU, Gieger C, Schumacher FR, Berndt SI, Sanna S, Eyheramendy S, Voight BF, Butler JL, Guiducci C, Illig T, Hackett R, Heid IM, Jacobs KB, Lyssenko V, Uda M, Boehnke M, Chanock SJ, Groop LC, Hu FB, Isomaa B, Kraft P, Peltonen L, Salomaa V, Schlessinger D, Hunter DJ, Hayes RB, Abecasis GR, Wichmann HE, Mohlke KL, Hirschhorn JN (2008) Identification of ten loci associated with height highlights new biological pathways in human growth. Nat Genet 40:584–591. doi:10.1038/ng.125
Li Y, Willer C, Sanna S, Abecasis G (2009) Genotype imputation. Annu Rev Genomics Hum Genet 10:387–406
Liu F, van Duijn K, Vingerling JR, Hofman A, Uitterlinden AG, Janssens AC, Kayser M (2009a) Eye color and the prediction of complex phenotypes from genotypes. Curr Biol 19:R192–R193. doi:10.1016/j.cub.2009.01.027
Liu S, Tang W, Fang J, Ren J, Li H, Xiao Z, Quarles LD (2009b) Novel regulators of Fgf23 expression and mineralization in Hyp bone. Mol Endocrinol 23:1505–1518. doi:10.1210/me.2009-0085
Macgregor S, Cornes BK, Martin NG, Visscher PM (2006) Bias, precision and heritability of self-reported and clinically measured height in Australian twins. Hum Genet 120:571–580. doi:10.1007/s00439-006-0240-z
Perola M, Sammalisto S, Hiekkalinna T, Martin NG, Visscher PM, Montgomery GW, Benyamin B, Harris JR, Boomsma D, Willemsen G, Hottenga JJ, Christensen K, Kyvik KO, Sorensen TI, Pedersen NL, Magnusson PK, Spector TD, Widen E, Silventoinen K, Kaprio J, Palotie A, Peltonen L (2007) Combined genome scans for body stature in 6,602 European twins: evidence for common Caucasian loci. PLoS Genet 3:e97. doi:10.1371/journal.pgen.0030097
Phillips K, Matheny AP Jr (1990) Quantitative genetic analysis of longitudinal trends in height: preliminary results from the Louisville Twin Study. Acta Genet Med Gemellol (Roma) 39:143–163
Silventoinen K, Kaprio J, Lahelma E, Koskenvuo M (2000) Relative effect of genetic and environmental factors on body height: differences across birth cohorts among Finnish men and women. Am J Public Health 90:627–630
Silventoinen K, Sammalisto S, Perola M, Boomsma DI, Cornes BK, Davis C, Dunkel L, De Lange M, Harris JR, Hjelmborg JV, Luciano M, Martin NG, Mortensen J, Nistico L, Pedersen NL, Skytthe A, Spector TD, Stazi MA, Willemsen G, Kaprio J (2003) Heritability of adult body height: a comparative study of twin cohorts in eight countries. Twin Res 6:399–408
Su PH, Wang SL, Chen JY (2011) Gender differences of final height contributed by parents’ height among healthy individuals. Pediatr Neonatol 52:183–189. doi:10.1016/j.pedneo.2011.05.003
Szumska D, Pieles G, Essalmani R, Bilski M, Mesnard D, Kaur K, Franklyn A, El Omari K, Jefferis J, Bentham J, Taylor JM, Schneider JE, Arnold SJ, Johnson P, Tymowska-Lalanne Z, Stammers D, Clarke K, Neubauer S, Morris A, Brown SD, Shaw-Smith C, Cama A, Capra V, Ragoussis J, Constam D, Seidah NG, Prat A, Bhattacharya S (2008) VACTERL/caudal regression/Currarino syndrome-like malformations in mice with mutation in the proprotein convertase Pcsk5. Genes Dev 22:1465–1477. doi:10.1101/gad.479408
Thodberg HH, Jenni OG, Caflisch J, Ranke MB, Martin DD (2009) Prediction of adult height based on automated determination of bone age. J Clin Endocrinol Metab 94:4868–4874. doi:10.1210/jc.2009-1429
Unrath M, Thodberg HH, Schweizer R, Ranke MB, Binder G, Martin DD (2012) Automation of bone age reading and a new prediction model improve adult height prediction in children with short stature. Horm Res Paediatr 78:312–319. doi:10.1159/000345875
Visscher PM, Medland SE, Ferreira MA, Morley KI, Zhu G, Cornes BK, Montgomery GW, Martin NG (2006) Assumption-free estimation of heritability from genome-wide identity-by-descent sharing between full siblings. PLoS Genet 2:e41
Walsh S, Lindenbergh A, Zuniga SB, Sijen T, de Knijff P, Kayser M, Ballantyne KN (2011a) Developmental validation of the IrisPlex system: determination of blue and brown iris colour for forensic intelligence. Forensic Sci Int Genet 5:464–471. doi:10.1016/j.fsigen.2010.09.008
Walsh S, Liu F, Ballantyne KN, van Oven M, Lao O, Kayser M (2011b) IrisPlex: a sensitive DNA tool for accurate prediction of blue and brown eye colour in the absence of ancestry information. Forensic Sci Int Genet 5:170–180. doi:10.1016/j.fsigen.2010.02.004
Walsh S, Liu F, Wollstein A, Kovatsi L, Ralf A, Kosiniak-Kamysz A, Branicki W, Kayser M (2012a) The HIrisPlex system for simultaneous prediction of hair and eye colour from DNA. Forensic Sci Int Genet. doi:10.1016/j.fsigen.2012.07.005
Walsh S, Wollstein A, Liu F, Chakravarthy U, Rahu M, Seland JH, Soubrane G, Tomazzoli L, Topouzis F, Vingerling JR, Vioque J, Fletcher AE, Ballantyne KN, Kayser M (2012b) DNA-based eye colour prediction across Europe with the IrisPlex system. Forensic Sci Int Genet 6:330–340. doi:10.1016/j.fsigen.2011.07.009
Weedon MN, Lango H, Lindgren CM, Wallace C, Evans DM, Mangino M, Freathy RM, Perry JR, Stevens S, Hall AS, Samani NJ, Shields B, Prokopenko I, Farrall M, Dominiczak A, Johnson T, Bergmann S, Beckmann JS, Vollenweider P, Waterworth DM, Mooser V, Palmer CN, Morris AD, Ouwehand WH, Zhao JH, Li S, Loos RJ, Barroso I, Deloukas P, Sandhu MS, Wheeler E, Soranzo N, Inouye M, Wareham NJ, Caulfield M, Munroe PB, Hattersley AT, McCarthy MI, Frayling TM (2008) Genome-wide association analysis identifies 20 loci that influence adult height. Nat Genet 40:575–583. doi:10.1038/ng.121
Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, Madden PA, Heath AC, Martin NG, Montgomery GW, Goddard ME, Visscher PM (2010) Common SNPs explain a large proportion of the heritability for human height. Nat Genet 42:565–569. doi:10.1038/ng.608
Zerath E, Holy X, Mouillon JM, Farbos B, Machwate M, Andre C, Renault S, Marie PJ (1997) TGF-beta2 prevents the impaired chondrocyte proliferation induced by unloading in growth plates of young rats. Life Sci 61:2397–2406
Acknowledgments
The authors are grateful to the study participants, the staff from the Rotterdam Study and the participating general practitioners and pharmacists. We thank Pascal Arp, Mila Jhamai, Marijn Verkerk, Lizbeth Herrera and Marjolein Peters for their help in generating the GWAS database, as well as Karol Estrada and Maksim V. Struchalin for their support in generating and analyzing imputed genomic data. This study was supported in part by funding from the Netherlands Forensic Institute (NFI), by a grant from the Netherlands Genomics Initiative (NGI)/Netherlands Organization for Scientific Research (NWO) within the framework of the Forensic Genomics Consortium Netherlands (FGCN), the Swedish Research Council (K2010-54X-15073-07-3), and by Ferring Pharmaceuticals, Eli Lilly and Company, and Ace Pharmaceuticals. The generation and management of GWAS genotype data for the Rotterdam Study are supported by the Netherlands Organisation of Scientific Research NWO Investments (nr. 175.010.2005.011, 911-03-012). This study is funded by the Research Institute for Diseases in the Elderly (014-93-015; RIDE2), the Netherlands Genomics Initiative/Netherlands Organisation for Scientific Research (NWO) project nr. 050-060-810. The Rotterdam Study is funded by the Erasmus MC University Medical Center Rotterdam, the Erasmus University Rotterdam, the Netherlands Organization for the Health Research and Development (ZonMw), the Research Institute for Diseases in the Elderly (RIDE), the Ministry of Education, Culture and Science of the Netherlands, the Ministry for Health, Welfare and Sports of the Netherlands, the European Commission (DG XII), the Municipality of Rotterdam and the Netherlands Genomics Initiative (NGI)/Netherlands Organization for Scientific Research (NWO) within the framework of the Netherlands Consortium on Healthy Ageing (NCHA). None of the funding agencies had influenced the design, execution or results of this study.
Conflict of interest
SLS Drop has received research grants from Ace, Ferring and Eli Lilly. All other authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
439_2013_1394_MOESM3_ESM.tif
Supplementary Fig. 1a Association result of the case–control designed GWAS for tall stature in 770 tall and 9,591 non-tall Dutch Europeans. A, Manhattan plot. (TIFF 1321 kb)
439_2013_1394_MOESM5_ESM.tif
Supplementary Fig. 2. Performance of six classifiers in predicting tall stature from DNA variants. Box plots of AUC values were generated for weighted allele sums (WAS); least absolute shrinkage and selection operator (LASSO); support vector machine (SVM), neuron networks (NN); classification and regression trees (CART); and random forest (RF). For each method, 1,000 AUC values were derived from cross-validations using randomly split training–testing samples (80 % - 20 %). Summary statistics in the box plots include median (black line), 25-75 % quartiles (grey box), minimum and maximum (dashed interval), and outliers (dots). A. using randomly selected 180 SNPs over the genome. B. using the 180 SNPs from the GIANT paper (Lango Allen et al. 2010). Note that this analysis included 20 % of controls and all cases because the computational burden of SVM scales with sample size. (TIFF 714 kb)
Rights and permissions
About this article
Cite this article
Liu, F., Hendriks, A.E.J., Ralf, A. et al. Common DNA variants predict tall stature in Europeans. Hum Genet 133, 587–597 (2014). https://doi.org/10.1007/s00439-013-1394-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-013-1394-0