
Abstract
Plastid genomes (plastomes) are part of the integrated compartmentalised genetic system of photoautotrophic eukaryotes. They are highly redundant and generally dispersed in several regions (nucleoids) within organelles. DNA quantities and number of DNA-containing regions per plastid vary and are developmentally regulated in a way not yet understood. Reliable quantitative data describing these patterns are scarce. We present a protocol to isolate fractions of pure plastids with varying average sizes from leaflets (≤1 mm) and leaves of different developmental stages continuously up to maturity (25 cm) from Beta vulgaris L. (sugar beet) to determine DNA amounts per organelle. The approach is based on plastid purification from homogenates of moderately fixed tissue by differential and isopycnic gradient centrifugations and on application of two different DNA specific colorimetric reactions after removing potentially interfering compounds. The sensitive fluorochrome DAPI (4′,6-diamidino-2-phenylindole) was used to estimate numbers and emission intensity of nucleoids per plastid. The amounts determined ranged from 0.15 to 4.9 × 10−2 pg DNA for plastids of 1→8 μm average diameter, corresponding from approximately a dozen to 330 genome equivalents per organelle and on average four to seven copies per nucleoid. The ratio of plastid/nuclear DNA changed continuously during leaf development from as little as 0.4% to about 20% in fully developed leaves. On the other hand, mesophyll cells of mature leaves differing in ploidy (di-, tri- and tetraploid) appeared to maintain a relatively constant nuclear genome/plastome ratio, equivalent to about 1,700 copies per C-value.
Similar content being viewed by others


Introduction
One of the intriguing, though not yet understood, features of the compartmentalised eukaryotic genome is a high degree of reiteration of organelle genomes. Published data indicate that diploid leaf parenchyma cells of higher plants with 30–40 chloroplasts, for instance, may possess between several hundred and >10,000 copies of the approximately 150,000 bp plastid genomes (plastomes). There is evidence that nuclear genome/plastome ratios change during development, but also that at a given differentiation status plastome copy numbers are adjusted with cellular ploidy, keeping that ratio relatively constant [summarised in Butterfass (1979), Herrmann and Possingham (1980)]. Redundancy of organelle genomes appears to be a general phenomenon since all organisms studied possess multicopy organelle genomes in their cells.
Within plastids, DNA is organised in a prokaryotic fashion (Ris and Plaut 1962). Early semiquantitative light microscopic autoradiography of plastids released from thymidine labelled tissue, ultrastructural work including serial sectioning of DNA exposing, protease treated leaf tissue and later the use of the DNA-specific fluorochrome DAPI provided information about the arrangement of DNA within plastids and about variation of DNA amounts per organelle and nucleoid (Herrmann 1968; 1970; Herrmann and Kowallik 1970; Kowallik and Herrmann 1972; James and Jope 1978; Coleman 1979; Kuroiwa et al. 1981). These studies established that the DNA is usually found in a single or in several regions (nucleoids) within the organelle stroma, or organised peripherally in a ring-shaped configuration, e.g., below the outermost thylakoid lamellae (girdle lamellae) in some algal taxa, notably Chromophytes (Bisalputra and Bisalputra 1969; Gibbs et al. 1974). It was also shown that the number of nucleoids, DNA quantity and sizes of plastids are usually correlated (Herrmann 1970; Kowallik and Herrmann, 1972; James and Jope 1978; Kuroiwa et al. 1981). Tiny plastids, such as proplastids, and mitochondria often contain a single nucleoid or a few nucleoids only (Herrmann and Kowallik 1970; Kuroiwa et al. 1981; Fujie et al. 1994). Nucleoid number increases usually with organelle size to ≥20 in fully developed leaf chloroplasts (Kowallik and Herrmann 1972; Scott and Possingham 1980; Miyamura et al. 1986). Furthermore, nucleoids can have different quantities of DNA, i.e., they are generally polyploid, and the degree of their ploidy can vary, even within the same organelle. This has been inferred from varying numbers of silver grains per cluster in autoradiographs, from fibril length and volume differences (0.01–0.3 μm3) of DNA regions in situ (Kowallik and Herrmann 1972), and from differences in emission intensity of individual fluorescent DNA spots of an organelle (James and Jope 1978; Coleman 1978, 1979; Miyamura et al. 1986). Independent evidence for nucleoid ploidy came from the isolation of various kinds of complex multigenomic plastid DNA (ptDNA) structures (Herrmann and Possingham 1980; Oldenburg and Bendich 2003; Scharff and Koop 2007). For instance, fibril lengths of rapidly sedimenting associations of predominantly supercoiled plastid DNA attached to protein patches that can be prepared from controlled partial chloroplast lysates and are reminiscent to isolated bacterial nucleoids (e.g. Drlica et al. 1978) can amount up to a dozen plastome equivalents (Herrmann and Possingham 1980; for membrane association of ptDNA see Herrmann and Kowallik 1970, Kowallik and Haberkorn 1971).
While cytological and semiquantitative aspects of organelle DNA have been appropriately documented, quantitative data on ptDNA and its developmental changes are varied and often contentious, partly due to the fact that unbiased DNA determinations and even more so their changes with organelle development are technically not easy to achieve. This is particularly relevant for higher plants with their functionally different cell types, tissues and complex developmental patterns of organelles. The enormous morphological plasticity of the plastid which can differentiate into various structurally and metabolically specialised forms is accompanied by complex structural and quantitative changes of its DNA complement. Moreover, chloroplast numbers or volumes and ptDNA quantities per cell depend on nuclear genome dosage and change proportionally with ploidy levels (summarised in Butterfass 1979). This nucleo-cytoplasmic relationship, a concept introduced by Strasburger (1893), is under nucleogenetic control and contributes to the continuity of plastids. Approximately one third of the nuclear gene complement is involved in management of the genetic and functional integration of the energy-transducing organelles (Herrmann 1997), but as yet none of the factors determining plastids numbers, sizes and modifications has been identified (cf. McCourt et al. 1987).
To obtain a coherent quantitative ontogenetic picture of organelle genomes, determinations per cell would require knowledge of amounts of ptDNA per cell, nuclear ploidy, numbers of organelles per cell, organelle sizes and their variation within a cell, numbers of nucleoids per organelle, and ploidy levels of individual nucleoids, but these parameters were barely investigated in combination. Quantifications of ptDNA are relatively easy in cases in which DNA species differ sufficiently in base composition to separate in CsCl equilibrium or relaxation gradients, such as from Chlamydomonas, Euglena, Antirrhinum or Oenothera, for which amounts of organelle DNA per cell usually range from 5 to >20% for plastid DNA and from 0.5 to 4% for mitochondrial DNA. However, in most vascular plants studied physicochemical properties of plastid and nuclear DNA are similar. This and methodical insufficiencies render quantifications more difficult, and data may often reflect only approximate DNA contents (see “Discussion”). It is therefore not surprising that values of DNA copies per organelle vary substantially, ranging from total absence through a few (5) to >1,000 copies in plastids, that comparable variation as well as different developmental patterns were reported for ptDNA per cell, and that findings to that subject are frequently not consistent or controversial, even for the same organism and from the same lab (Bennett and Radcliff 1975; James and Jope 1978; Jope et al. 1978; Pascoe and Ingle 1978; Lamppa and Bendich 1979; Scott and Possingham 1980; 1983; Boffey and Leech 1982; Lawrence and Possingham 1986; Baumgartner et al. 1989; Oldenburg and Bendich 2004; Li et al. 2006; Shaver et al. 2006; Zoschke et al. 2007; Rowan et al. 2004; 2009; see also “Discussion”).
In this communication, we present a direct approach to determine average DNA amounts per plastid and their changes during development of sugar beet leaves, and relate the data obtained to structural aspects including those detailed previously (Herrmann 1970; Herrmann and Kowallik 1970; Kowallik and Herrmann 1972). The experimental protocol prevents fragmentation of organelles and chromatin contamination of purified organelle fractions, and should be applicable to all materials from which plastids can be readily isolated. The data presented differ from published studies on beet leaves but substantiate our conclusions from previous cytological work.
Materials and methods
Leaf material and treatment
As in our previous work, sugar beet (Beta vulgaris L. var. Kleinwanzlebener Marta) was chosen for study, because of the availability of genetically defined ploidy lines, of trisomic lines with inhibited plastid division, and because developmental organelle, DNA and nucleoid patterns resembled that described from many other plants (e.g., James and Jope 1978). Leaves of different developmental stages were collected from beet roots that were propagated in a greenhouse in wet sand after leaves had been removed following harvest. Before leaf excision, the plant material was kept in darkness for two days. Newly grown leaves or leaflets were grouped into seven distinct developmental classes (Fig. 1). Material for the first fraction which included vegetation tips and very first off-white to pale yellow leaflets up to 4 mm (Fig. 1f) was excised with scalpel and forceps. Fractions II–IV (Fig. 1c–e) consisted of yellow to moderately green leaflets of 0.4–0.9 cm (II), 1.0–2.2 cm (III), or 2.5–3.5 cm (IV), all still with curled margins, fractions V–VII of green leaves of 4.5–7 cm (V, Fig. 1b), 7–12 cm (VI, Fig. 1a) and leaves that were expanded to nearly mature size (15–25 cm), fully green with a glossy surface (VII). Only fully developed leaves were taken from trisomic line IV which contained large plastids that differed markedly in size from those of their eudiploid sister plants (Butterfass 1967, 1979; Herrmann 1969). Immediately after harvest, the material was cut into pieces of approximately 0.5 × 0.5 cm and fixed by vacuum infiltration in 5% buffered, weakly hypotonic formaldehyde for at least 20 h (0.1 M sucrose, 20 mM Tris–HCl, 2 mM EDTA, pH 7.2). The fixative was replaced by buffer without sugar. Fixation stabilised nuclei and organelles and, due to the loss of osmotic response, largely prevented their fragmentation as well as the breakdown of envelope membranes during mechanical treatments and tonicity changes in the course of subcellular fractionation. Starch grains that were observed occasionally remained in the organelle (Herrmann 1982). Fixation eliminated the serious problem of contamination of the organelle fractions by chromatin, but as chosen still allowed enzymatic removal of RNA, reduction of protein and enzymatic extraction of DNA (Herrmann and Kowallik 1970).

Sugar beet leaflet and leaf classes used for plastid isolation (a–f). Plastids were isolated and purified as described in the text. i Large chloroplasts from trisomic leaves. j Chloroplasts from 7 to 12 cm-leaves shown in a, k plastids from 2.5 to 3.5 cm-leaves (c), l from 0.4 to 2.2 cm-leaflets (d, e), and m from 0.2–0.9 cm-leaflet fractions (e, f). g Nuclear and protoplast fraction removed during plastid purification (arrowhead protoplast; arrows indicate well preserved nucleoli); h mitochondrial fraction with some plastids left (arrows). Subcellular fractions were viewed with a Zeiss phase contrast microscope and pictures taken at the same magnification. Bar 10 μm
Isolation and characterisation of plastids
Fixed and chilled leaf material was homogenised four times for 3 s with low speed in a threefold volume of sterilised ice-cold medium (0.2 M sorbitol, 25 mM HEPES, 2 mM MgCl2, 3 mM EDTA, 5 mM ß-mercaptoethanol, pH 7.4) in a Waring Blender. The homogenate was filtered through three layers of sterilised (80°C) nylon gauze (Schweizerische Nylongaze AG, Zürich) of defined pore sizes, 5 μm for plastids of fractions I and II (Fig. 1e, f), 10 μm for those of fractions III and IV (Fig. 1c, d), 20 μm for chloroplasts of fraction V–VII (Fig. 1a, b), and 50 or 100 μm for chloroplasts from trisomic material. The plastids were then pelleted by centrifugation for 2 min at 2,500–5,000×g, depending on organelle size, and washed in homogenisation buffer twice. They were further purified by isopycnic centrifugation in continuous sucrose gradients (35–72%, supersaturated) for 45 min at 25,000×g and 4°C in an SW40 rotor (Spinco-Beckman). A cushion of Fluorinert (density 1.92 g cm−3) was included to separate plastids of fraction I from nuclei. The initial differential centrifugation steps removed most mitochondria (Fig. 1h) and nuclei from small plastids, the gradient step protoplasts, protoplast fragments and nuclei (Fig. 1g). Plastid containing bands, usually in the lower third of the tubes, were collected and diluted fourfold with buffer lacking sucrose. Plastids were then quantitatively pelleted by centrifugation for 5 min at 5,000×g and 4°C, and washed once under the same conditions. All steps were performed semisterile and at low temperature. Fractions were continuously monitored by phase contrast microscopy during all stages of purification and subsequent treatments. The final pellets were resuspended in a tenfold volume of 0.4 × sodium saline citrate puffer (150 mM NaCl, 15 mM tri-sodium citrate, pH 7.2) and a drop of chloroform was added to each tube to prevent bacterial growth. Aliquots were taken from all preparations for electronmicroscopy and organelle counting in a Thoma hematocytometer on a phase contrast microscope (Fig. 1j), before each fraction was divided into four or six equal parts.
For size determinations, fixed plastids were immobilised on albumin-glycerol coated slides with a stream of cold air (Herrmann 1970). The chosen treatment did not notably change plastid sizes (cf. Menke 1966). Two diameters per organelle were measured in surface view; 1.5 and 2 μm were taken as thickness for small and large organelles, respectively (see Figs. 2 and 3 in Herrmann and Kowallik 1970). For each sample, average sizes were calculated from 20 to 40 randomly selected chloroplasts. Ultrastructural analyses were performed using standard procedures and a Zeiss electron microscope EM 912.
Biochemical approaches; DNA determinations
The aliquots of the organelle fractions mentioned above were transferred to 2 ml Eppendorf tubes. A defined amount of 14C-labelled DNA was added to selected samples to monitor DNA recovery during probe handling. Four of the six parts of each fraction were treated with preincubated (1 h) nuclease-free trypsin and Pronase P (1 mg/ml each) for 3 h at 40°C. A mixture of DNase-free RNase A (40 μg/ml) and RNase T1 (10 μg/ml) was then added and incubation continued for another 2 h at room temperature (with one extra enzyme addition). The treatment resulted in a significant reduction of stroma proteins and RNA (Herrmann and Kowallik 1970). The Eppendorf tubes were sealed with dialysis membrane, inverted, and the enzymatically treated samples dialysed against 0.4 × sodium saline citrate buffer (150 mM NaCl, 15 mM tri-sodium citrate, pH 7.2) for 2 h. Subsequently, they were extracted with ice-cold 75% ethanol, 100% ethanol, ethanol/ether (1:1) and ether at −5°C and centrifuged at low speed after each step, until the supernatants appeared pigment-free.
The surface dried final off-white or brownish probes were extracted twice with 5% perchloric acid (PCA) at 4°C in the same tube. They were then adjusted to 5 N PCA (or 5% trichloro acetic acid), heated for 15 min at 70 or 85°C (90°C with trichloro acetic acid), cooled down in an ice bath and clarified by centrifugation. The pellets were re-extracted once under the same conditions, the supernatants combined and, after measuring their volumes, used for colorimetric reactions with diphenylamine/acetaldehyde (30°C for 18 h) and indole/HCl (boiling water bath for 10 min). The first PCA hydrolysis released more than 95% of the reacting material; no colorimetric reaction was noted with the PCA pre-extract. Alternatively, in some instances the enzymatically treated and dialysed organelle suspensions were digested with a mixture of DNase I (electrophoretically purified, 30 μg/ml; Boehringer, Mannheim, Germany) and snake venom phosphodiesterase (20 μg/ml; Boehringer) at 37°C for 2 h and the supernatants obtained after centrifugation (15,000×g, 10 min) taken for colorimetry.
Dische’s diphenylamine (p.a., Merck no. 803010) and indole (p.a., Merck no. 104730) reagents as modified by Burton (1956) and Keck (1956), respectively, for higher specificity, higher sensitivity and low susceptibility to interfering compounds were used for colorimetric DNA determinations. Both assays operate at somewhat different concentration ranges, the indole reaction being more sensitive. Absorbances were taken at 600 μm (diphenylamine) and 490 μm (indole) in a Beckman DK2A dual path spectrophotometer using a microcell (100 μl capacity). The colorimetric reactions were calibrated with samples containing defined amounts of deoxyadenosine or hydrolysates from salmon sperm or calf thymus DNA (Boehringer). A buffer blank, initially also comparably treated solutions of the enzymes used or albumine blanks, all containing no DNA, served as references. Hyperchromicities (A100°C /A25°C ) of single stranded vs. double stranded DNA served as purity criterion for reference DNAs and were between 1.39 and 1.41. Recovery of radioactivity from radiolabelled DNA added to selected tubes was ≥88%, so corrections for loss of DNA were never large.
Cytology of leaf tissue; DAPI staining; high resolution light microscopy
Excised leaflets or leaf sectors were fixed in 4% glutaraldehyde in 50 mM phosphate buffer, pH 7.2, treated with Cellulase “Onozuka” R10 and Macerozyme R10 (both Yakult Honsha, Tokyo, Japan) for 2–4 h depending on tissue as described in Kuchuk et al. (1998), washed in buffer and stored at 4°C until use. Small tissue sectors were gently squeezed under a cover glass and handled in one of the following ways. For organelle counts and morphometry samples were viewed in a Leica DMRD/BE light microscope and evaluated with software “analysis” (version 2.1; SIS.Soft-Imaging Software GmbH, Münster, Germany). Ploidy levels were determined cytophotometrically with Feulgen stained samples embedded in Euparal (Roth, Karlsruhe, Germany) using a Leica MPV photometer attached to the camera port of the microscope and by organelle counting as described by Butterfass (1979). For fluorescence microscopy, squeezed preparations were frozen on dry ice, cover glasses removed, the preparations air dried and mounted in a drop of DAPI solution (2 μg DAPI/ml McIlvaine’s buffer, 2:1 mixed with glycerol [Merck, “for fluorescence microscopy”]). The preparations were sealed with rubber cement (Marabu) and examined in a Nikon Eclipse 80i or Zeiss Axioplan epifluorescence microscope using an immersion 100× UV objective. Images were taken with a Nikon CCD monochrome camera DS-2Mc-U1. In cases where a cell was oriented in several focal planes, five to seven picture frames were digitally captured, each at a different focal plane. The frames were stacked and combined using standard macro commands of CombineZM software (http://www.hadleyweb.pwp.blueyonder.co.uk).
Results
DNA amounts of organelles differing in average size; purity of plastid fractions
A developmental leaf series and mature leaves of trisomy line IV of sugar beet were used to prepare pure fractions of organelles differing in average sizes. Figure 1 illustrates six of the eight leaf classes chosen for study as well as representative samples of the purified organelle fractions or of fractions discarded during subcellular purification derived from them. Figure 1i shows the relatively large chloroplasts of trisomy IV cells due to impaired chloroplast division (Butterfass 1967, 1979 ; Herrmann 1969). Figure 1j presents an example of hematocyometer counting of chloroplasts from 7 to 12 cm-leaves (fraction VI) shown in Fig. 1a. The organelle fraction depicted in Fig. 1k corresponds to 2.5–3.5 cm-leaves (IV, Fig. 1c), that of Fig. 1l to 0.4–0.9 cm-leaflets (II/III, Fig. 1e or occasionally Fig. 1d), and that of Fig. 1m to plastids from vegetation tips/leaflets (I/II, Fig. 1f or 1e). Fig. 1g and h demonstrate fractions that were removed during purification, i.e., fixed protoplasts, protoplast fragments and unbroken nuclei, and the mitochondrial/microsomal fraction, respectively. Note that nuclei and nucleoli are well preserved (Fig. 1g). DAPI staining, employed to assess the levels of contaminating nuclear DNA, gave no fluorescing nuclear or chromatin signal in more than 20,000 plastids monitored in triplicate in those plastid preparations used finally (data not shown). Mitochondrial contamination was negligible as well and varied between one per 650 to 2,350 plastids in individual preparations as judged electronmicroscopically (data not shown; for ultrastructural sections of comparable organelle fractions see Fig. 7 in Schmitt and Herrmann 1977). The envelopes of most plastids, particularly those of smaller sizes, were preserved.
The DNA amounts and genome copies per plastid and their changes with average organelle sizes obtained from the respective organelle fractions are presented in Table 1 and Fig. 2. The results were calculated on a mass basis of 1.5184 × 10−16 g per chromosome. Data obtained from aliquoted samples coincided usually within ±5%; no significant difference was noted between DNA hydrolysates obtained by acidic or enzymatic treatments nor between the two colorimetric reactions in appropriate absorbance ranges (0.08–1.2). Although there was some variation, especially with organelle fractions from smaller leaves, the values of DNA per plastid were remarkably consistent between individual experiments. Figure 2 illustrates that there is an accumulation of DNA per organelle with increasing size throughout all developmental stages studied. Plastids of <1–2 μm diameter contained in the order of 1.5–5 × 10−3 pg of DNA equivalent to 10–30 genome copies, plastids of 5 μm diameter in the order of 18 × 10−3 pg, corresponding to 110–120 genome equivalents. The relationship between organelle size and DNA amount also holds for giant chloroplasts from trisomic material (Fig. 1i). In five independent determinations, mesophyll chloroplasts of trisomy type IV plants with average diameters between 6.6–8.7 μm and organelle volumes between 68 and 119 μm3 contained between 216 and 324 plastome copies or 3.3–4.9 × 10−2 pg DNA (data not shown).

DNA quantities per plastid during organelle development, determined by colorimetry of DNA hydrolysates from purified organelle fractions and plotted as a function of organelle volumes (see “Materials and Methods”). The data are listed in Table 1
Two sets of experiments were conducted to relate DNA amounts per plastid to DNA regions (nucleoids) and to estimate amounts of ptDNA per cell including their variation with both development and ploidy.
Estimates of nucleoids per plastid
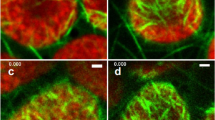
The DNA-specific fluorescent dye DAPI was employed to visualise DNA within plastids. This fluorochrome is considered to be sensitive enough to detect DNA of a single plastid genome copy (James and Jope 1978). Fixed, enzymatically softened and squeezed tissue explants were utilised because they allow the preparation of well spread cells in which plastids can be accurately counted (Figs. 3, 4). Fluorescing spots, varying in size and shape from as large as 3 μm in length to the limits of resolution, were visible in virtually all organelles; the patterns were lost after pre-treatment with DNase, but remained unaffected by RNase or Proteinase K (data not shown). Figures 3 and 4 depict representative developmental series of cells and corresponding DAPI fluorograms, respectively. The number of fluorescing spots per plastid increases with organelle size (Fig. 4, Table 1) and illustrates that the emission intensity of individual spots within an organelle can vary significantly (Fig. 4i, j and k). Plastids of 1–2 μm in diameter contained between 1 and 5 fluorescing regions (Fig. 4a), those of 4–5 μm between 17 and >30 (Fig. 4f, i), and those of 6.5–8.7 μm from trisomic material in the order of 42–65 (data not shown). Comparing these figures with the corresponding number of plastome equivalents per organelle (see above and Table 1) establishes that each fluorescing DNA region contains on average 4–7 copies (see “Discussion”). Note that there was generally no significant intensity difference between DNA regions in organelles of different sizes or from cells differing in ploidy (Fig. 4a–f vs. g, h or i). However, a tendency to lower emission per spot was observed for small plastids. Conversely, a trend to higher emission was noted for organelles of expanding leaves or of organelles of comparable sizes containing lower numbers of DNA regions.

Representative cells from central lamina areas of leaf material shown in Fig. 1a–f that was used for morphometry and organelle isolation. a–e A developmental series of diploid, f–j triploid, and k–o tetraploid sugar beet. a, f, k reflect cells from 0.2 to 0.9 cm-leaflet fractions I/II (Fig. 1f, e), b, g, l 0.5–2.2 cm-leaflet fractions II/III (Fig. 1d, e), c, h, m 1.0–2.2 cm-leaf fraction III (Fig. 1d), d, i, n 2.5–6 cm-leaf fractions IV–V (Fig. 1c, b), and e, j, o 15–25 cm-leaf fraction VII. Note size variation of individual chloroplasts in o (arrow). Bar 10 μm

Representative DAPI stained squashed cells of from Beta leaflets or leaves of different developmental stages as shown in Fig. 1a–f. a–f Cells from diploid material of 0.2–0.9 cm-leaf fractions I/II (a; see Fig. 1e), 1.0–2.2 cm leaf fraction III (b, c; Fig. 1d), 2.5–3.5 cm-leaf fraction IV (d, Fig. 1c), 4.5–7 cm-leaf fraction V (e, Fig. 1b) and 15–25 cm mature leaves of fraction VII (f). g, h Cells from triploid material, g corresponding to c/d, and h to d/e. i Tetraploid cell of leaf fraction VII; j and k illustrate mature plastids at larger magnification to visualise individual nucleoids. Note differences in emission intensity between individual spots of an organelle. Arrows in l, j and k DNA spots fluorescing close to the detection limit (see text). Bar in f 10 μm (a–i), in j 5 μm (j and k)
Estimates of cellular nuclear genome/plastome ratios
In order to obtain an idea about magnitude and changes of plastid/nuclear DNA ratios per cell, it was necessary to determine plastid numbers for cells differing in developmental stage and ploidy. Plastids were monitored from cells of leaflets and leaves (Fig. 1a–f) as well as from mesophyll cells of di-, tri and tetraploid sugar beet lines with comparable nuclear background. Five plants per leaf class were chosen for study and the plastids of at least 25 cells per sample were counted. Cells of different developmental and ploidy stages differ markedly in their chloroplast complement (Fig. 3). As usual, more or less bimodal patterns were obtained, reflecting cells in G1 and G2 (data not shown; Butterfass 1979), and again, the amount of DNA present per plastid as estimated visually from fluorescence emission and nucleoid numbers was related to organelle size (Table 1). Juvenile diploid cells from 0.2 to 0.8 cm-leaflets of fractions I and II in which plastids could be accurately counted contained between 6 and 9 tiny plastids (Figs. 3a, b, 4a, b). In spongy parenchyma cells from mature leaves (15–25 cm, fraction VII) on average 29.2 ± 6.7 chloroplasts were present per cell; in tri- and tetraploid cells the numbers ranged from 41.6 ± 7.6 and 50.3 ± 9.1, respectively. These figures correspond largely to those reported in the literature (Mochizuki and Sueoka 1955; Butterfass 1959, 1979). Organelle sizes were comparable among mature spongy mesophyll cells of different ploidy levels. Diploid cells usually contained relatively uniformly sized organelles, although in some cases variation could be observed (e.g., Fig. 3o). In summary, there was an about 4-fold increase in plastid number, 5-fold increase in organelle diameter, and a >30-fold increase in plastid genome equivalents per cell from diploid 0.2–0.5 cm-leaflets of fraction I to mature 15–25 cm-leaves of the same ploidy level (fraction VII; Table 1). In contrast, virtually no change in plastome-to-genome ratio was noted in mature spongy mesophyll cells differing in ploidy; averages estimated for plastid genomes per cell were 3390 (2×; average of expts. 32–37 in Table 1), 5,220 (3×) and 6,480 (4×) which corresponds to 20.5, 21.1 and 20.0% of the cellular DNA, respectively.
Discussion
Direct measurements of DNA amounts per plastid used in this study rely both on purified plastid fractions from weakly fixed leaf tissue and on quantitative recovery of DNA from these fractions. Fixation can overcome the major source of inaccuracy by nuclear (and mitochondrial) contamination. The method circumvents potential limitations of conventional quantitative approaches used in that field, such as by class III chloroplasts (Larsson et al. 1971; electron microscopy in Schmitt and Herrmann 1977) in isolated intact, DNase treated, chloroplasts, by cross-reaction between plastid and (contaminating) nuclear DNA in reassociation kinetics presumably due to DNA promiscuity (Herrmann et al. 1974; Ayliffe et al. 1998; Huang et al. 2005), or by DNA compaction differences, organelle orientation, emission in non-linear ranges at high local DNA concentrations, focal plane differences, possible fluorescence quenching, or gradual fading of the DNA-DAPI complex in quantitative DAPI fluorescence microspectrometry (e.g., Coleman 1978; James and Jope 1978). We showed that the purified fixed organelle suspensions were practically free of contaminating nuclear DNA. This and the high reproducibility of data from split samples independent of procedural detail in sample treatment, DNA hydrolysis, and different colorimetric assays indicate that our DNA determinations are reliable. The biochemical data obtained refine previous estimates of DNA quantities per organelle, per individual DNA regions and per cell. The data bear on four aspects of general interest to the genetic system of plastids and eukaryotic cell:
-
(1)
DNA amounts and organelle sizes
The amounts of DNA per organelle determined, from approximately 2 fg or a dozen genome equivalents in 1 μm-plastids of cells of post-primordial leaflets to >100 copies in mesophyll chloroplasts of about 5 μm in diameter of mature leaf tissue, reflect an approximately ninefold increase of plastome copies during a 50-fold or more increase of plastid volume in the material analysed (Table 1, Fig. 2). The increase was continuous and includes giant chloroplasts from trisomic mesophyll material with 200–300 copies in up to 65 DNA regions. Some variation was determined, mostly at earlier developmental stages, but there was no pronounced discontinuous increment during the leaf development studied, notably no transitory or unusual increase or reduction in DNA amount per organelle deviating from the general nucleoid pattern and relationship between organelle size and DNA quantity occasionally reported by others (e.g., Tymms et al. 1983). Later, the DNA amounts per organelle remained constant (Fig. 2), including in chloroplasts from “stationary phase”-leaves with matt surfaces at early stages of ageing. Cytological and biochemical data are consistent (Figs. 2, 3, 4).
The DNA amounts determined are relatively high. Usually, data range between 15 and 40 plastome copies per plastid from developing tissue and between 5 and 50 in fully developed organelles, but in various instances values substantially exceeded 100 copies, i.e., 250, 320 up to 1,000 copies (for references see Introduction). Disregarding variation, an increase in plastome copies and comparable nucleoid patterns were reported from various vascular plants and probably reflect a general feature of developing leaves. Different patterns were described for later stages however, constant amounts or a decrease or even complete loss of ptDNA during etioplast/chloroplast transition and/or as leaves expand, mature or senesce. Although there is no reason a priori why both patterns should not occur, in several instances data are contentious, usually for methodical reasons (e.g., Li et al. 2006, Rowan et al. 2009). Both constant as well as declining ptDNA amounts in (nearly) mature and/or senescing material were recently reported for Arabidopsis and tobacco grown under comparable conditions (Li et al. 2006; Zoschke et al. 2007; Rowan et al. 2009). Our findings add to this controversy. They deviate from previously reported data from beet obtained by reassociation kinetics showing a reduction from 104 to 29 copies in plastids of 1.8–4.9 μm in leaves of 2–30 cm and from >150 to <60 copies per plastid for leaves of 2–10 mm in its relative spinach (Scott and Possingham 1980; Tymms et al. 1983). Plausible explanations for these contradictory results will be discussed below in point 3.
-
(2)
Plastids are generally polyenergidically organized, plastid nucleoids generally polyploid
Inspite of an intense nuclear staining with DAPI, the relatively faint fluorescent DNA regions in chloroplasts could be visualised with changing focal planes, and counted (Fig. 4). As DNA amounts, numbers of fluorescing spots increased with increasing organelle sizes. The DAPI patterns resembled those obtained from previous autoradiographic work (Herrmann 1970), are commensurate with ultrastructural work (Herrmann and Kowallik 1970; Kowallik and Herrmann 1972) and with reports from other vascular plants (e.g., James and Jope 1978; Kuroiwa et al. 1981; Miyamura et al. 1986). The intensity of spots remained more or less constant during development. Plastids with a single, strongly fluorescing central DNA spot or with peripheral DNA rings, which are usually not uniformly stained and appear as closely-spaced “beads on a string”, reported from developing plastids or etioplast-like organelles of wheat and oat (Hashimoto 1985; Miyamura et al. 1986, 1990) were occasionally observed in Beta (e.g., Fig. 4b, g and h), but not as general morphotypes. This also applies to some other temporal DNA arrangements described which appear to be often prescribed by structural features.
Genome equivalents per plastid calculated from average amounts of DNA per organelle exceeded without exception the average number of DNA regions for the corresponding organelle size class, on average four to sevenfold (Table 1). This substantiates that plastids are generally polyenergidic (for nomenclature see Herrmann and Possingham 1980), in analogy to multinucleate or comparably organised prokaryotic cells, and that individual nucleoids are generally polyploid, on average tetra- to heptaploid. Furthermore, ultrastructural section series across entire plastids (e.g., Fig. 1 in Kowallik and Herrmann 1972) and DAPI microspectrometry on individual DNA regions (Fig. 4, Coleman 1978; 1979; James and Jope 1978; Kuroiwa et al. 1981; Miyamura et al. 1986) suggested that DNA amounts per individual region can vary at least by a factor of 10 indicating that the degree of ploidy of individual nucleoids can differ accordingly, even within the same organelle. The difference in fluorescent intensity after DAPI staining between strongly fluorescing nucleoids and those exhibiting only faintest visible individual spots corroborates this for Beta plastids (Fig. 4i, j and k). We have found no evidence that sugar beet proplastids, which frequently contain only a single nucleoid (Figs. 3a, f, k, 4a), are haploid, as reported from Brassica juncea (Kuroiwa et al. 1981). The topological problem, of how multiple plastid genome equivalents of 45 μm unit length (Herrmann and Possingham 1980; Bendich 2004; Scharff and Koop 2007) or >1,000 μm bacterial chromosomes are folded, replicate, and separate into DNA regions of 0.01–0.3 μm3 in organelles of 1–5 μm in diameter or in 1 μm-bacterial cells, remains an unresolved, highly challenging scientific topic.
Combined with organelle counts per cell (Table 1, Figs. 3 and 4), the per-organelle-data presented provide information about average quantities of ptDNA per cell as well as about nuclear genome/plastome ratios and their changes.
-
(3)
Plastome copy numbers per cell during leaf development
Available data consistently show that meristematic, generative or embryonic cells contain not only much smaller (1 μm), but also fewer organelles with less DNA in a single or a few nucleoids (summarised in Butterfass 1979; see also Introduction for references). Organelle counts from (diploid) sugar beet range between 5 and 18 proplastids or leucoplasts in meristematic cells; root tip and guard cell mother cells contain approximately half-a-dozen tiny plastids (Butterfass 1979). Since nucleoid sizes and fluorescence intensities of small plastids in Beta are comparable to those of leaf chloroplasts (Fig. 4), the synergistic increase of both organelle size and number seen during development suggested that genome/plastome ratios at a given ploidy level are not constant during ontogenesis (Herrmann and Possingham 1980). The data demonstrate a remarkably low fraction of ptDNA, in the order of 0.5% or less, in total cellular DNA of diploid 0.5–0.9 cm-leaflets of fractions I and II (Fig. 1e, f). Concomitant with DNA amounts in plastids (Fig. 2, Table 1), this value increased approximately 40-fold, to >20%, more or less continuously during development up to the 4.5–12 cm leaf fraction VI (or V) of the same ploidy (Fig. 1b, c). Data range from about 100 plastome copies in diploid near-embryonic cells to 3,500 in mature diploid cells (Table 1). The latter figure compares to 11.4% ptDNA or between 1,100 and 1,900 chromosome copies per Beta cell (with undefined ploidy) reported by Tymms et al. (1983). For comparable tissue from spinach, values range from 3,300–5,700 (Scott and Possingham 1980; Scott and Possingham 1983) to as high as 12,500 copies per cell (Lawrence and Possingham 1986), a variance which the authors attribute to the different approaches (reassociation kinetics, microphotometry) employed. The latter value would correspond to an atypically high fraction (47%) of ptDNA per cell and nearly equal the nuclear 2C value of spinach (Bennett et al. 1982; equivalent to approximately 13,550 plastome copies). The most plausible explanation for the quite diverse observations on the Chenopodiacean species lies in methodical inconsistencies, outlined in the introductory paragraph of this discussion chapter, possibly also in growth or ploidy differences of the material (soil vs. liquid culture, ploidy levels were usually not provided) as the unusually high average chloroplast numbers per cell (65) suggests (for cellular organelle numbers see below).
As for DNA amounts of plastids, available data generally describe an increase of ptDNA amounts per cell early in leaf development, and basically two patterns, constant amounts or decline or loss, later including senescence (for references see Introduction). In Beta, organelle numbers and ptDNA amounts per cell remained more or less constant (Fig. 2, Table 1). As for plastids, no evidence was found that plastome copy numbers per cell declined during or early after leaf maturation, i.e., in mesophyll cells of early ageing “stationary-phase” leaves with matt surfaces. Senescent leaves were not investigated in our study, however. Both patterns, constant amounts as well as gradual decrease or loss following an increase in amount in juvenile material, were described from wheat, though from seedlings of different age (3 vs. 7 days; Boffey and Leech 1982; Miyamura et al. 1990). It is conceivable that various ways exist to regulate ptDNA copy numbers per cell and nuclear genome/plastome ratios in a species-specific manner, but in some instances, as for Arabidopsis, maize, tobacco and now beet, observations are controversial, predominantly due to methodical reasons (see above and Li et al. 2006; Zoschke et al. 2007; Rowan et al. 2009).
-
(4)
Ploidy levels and plastome copy number per cell
Tissues usually reflect ploidy patterns, since soma cells frequently endopolyploidise. Nuclear and cell sizes, plastid numbers and/or volumes per cell vary with nuclear ploidy. Chloroplast counts from cells of comparable differentiation status, which from sugar beet leaf parenchyma cells are available from seven ploidy levels (haploid to octoploid; Mochizuki and Sueoka 1955; Butterfass 1959, 1979), showed that organelle number and nuclear genome dosage are linearly related, independent of whether ploidy differences occur in the same individual (endopolyploidy) or in lineages of identical genotype differing in ploidy (meristematic polyploidy; Butterfass 1979). Chloroplast counts from fully developed mesophyll cells of di-, tri- or tetraploid material used here are commensurate with published data. Numbers changed with a factor of approximately 1.7 per doubling of nuclear chromosome number. Since organelles in Beta cells of different ploidy had comparable sizes and nucleoid patterns (Figs. 3 and 4), the increase in chloroplast number must necessarily be accompanied by corresponding changes of plastome copies per cell. The fraction of ptDNA in total cellular DNA found was remarkably constant for this cell type, independent of ploidy level in the order of 20%, which corresponds to about 1,700 chromosome copies per C value (1.25 pg or 1.22 × 109 bp; Bennett 1972). This indicates that nuclear genome/plastome ratios at a given differentiation stage may be more precisely balanced than mere changes of plastid numbers would suggest. It should be borne in mind however that chloroplast sizes and numbers depend on a balance of growth and division. Also, DNA synthesis, nucleoid division, organelle and cell division are separable events. Based on the organelle size/number relationship, a distinct genome/plastome ratio upon a nuclear ploidy change can in principle be achieved in two ways, either by changing organelle numbers and keeping organelle sizes “constant”, or by maintaining organelle numbers per cell, but changing organelle size or volume. Both cases exist (Butterfass 1979), but reality may often lie somewhere between these two extremes. Indeed, plastids of different sizes in a given cell (Fig. 3o), in which smaller organelles often reside in pairs, or differences in emission intensity of nucleoids indicate that not all plastids and nucleoids divide or not all plastid chromosomes of a nucleoid replicate simultaneously. This complicates DNA quantifications. Nevertheless, the ploidy data reflect a second mechanism, presumably of nuclear control, that must operate for plastids in higher plants, besides the one that alters nuclear genome/plastome ratios one that keeps the subgenome ratio relatively constant (Herrmann and Possingham 1980). Along with a third mechanism, the control of differences in multiplication rates between plastids of related species (summarised in Chiu et al. 1988), the outlined findings reflect highly complex, phylogenetically relevant regulatory networks operating to integrate organelles into cells.
The enormous morphological variation in photosynthetic and non-photosynthetic organelle modifications, nucleoid patterns, DNA content per organelle and cell can be reconciled with numerous phenotypic observations reported in the literature, but is poorly understood. Explanations will perhaps contribute to our understanding of the functional implications of organelle structure. Genetically, the selective advantage for redundancy of organelle genomes, and its changes with development and nuclear genome dosage remain enigmatic. It was suggested that plastome reiteration may balance a lack of genetic recombination in the organelle (Muller’s ratchet; Martin and Herrmann 1998) or be related in some way to transcription to provide additional templates at distinct developmental stages (e.g., Bennett and Radcliff 1975; Scott and Possingham 1983). Findings that transcriptional activity of plastid genes and chromosomes does not primarily depend on copy number (Eberhard et al. 2002; Legen et al. 2002; Zoschke et al. 2007) and that highly redundant mitochondrial genomes of vascular plants recombine efficiently, as does the reiterated Chlamydomonas plastome, suggest however that a satisfactory explanation is likely to be more complex. Whatever the cause, it is evident that quantitative studies on organelle genome patterns and on regulatory networks ensuring the genetic and functional integration of the primary energy-transducing organelles into the overall plant genome are indispensable for understanding the eukaryotic cell and will be instrumental aspects for future research in this field.
Abbreviations
- PtDNA:
-
Plastid or chloroplast DNA
- DAPI:
-
4′,6-Diamidino-2-phenylindole
References
Ayliffe MA, Scott NS, Timmis JN (1998) Analysis of plastid DNA-like sequences within nuclear genomes of higher plants. Mol Biol Evol 15:738–745
Baumgartner BJ, Rapp JC, Mullet JE (1989) Plastid transcription activity and DNA copy number increase early in barley chloroplast development. Plant Physiol 89:1011–1018
Bendich AJ (2004) Circular chloroplast chromosomes: the grand illusion. Plant Cell 16:1661–1666
Bennett J, Radcliff C (1975) Plastid DNA replication and plastid division in the garden pea. FEBS Lett 56:222–225
Bennett MD (1972) Nuclear DNA content and minimum generation time in herbaceous plants. Proc Roy Soc Lond Ser B 181:109–135
Bennett MD, Smith JB, Heslop-Harrison JS (1982) Nuclear DNA amounts in angiosperms. Proc Roy Soc Lond Ser B 216:179–199
Bisalputra T, Bisalputra AA (1969) The ultrastrucuture of chloroplast of a brown alga Sphacelaria sp. I. Plastid DNA configuration–the chloroplast genophore. J Ultrastruct Res 29:151–170
Boffey SA, Leech RM (1982) Chloroplast DNA levels and the control of chloroplast division in light-grown wheat leaves. Plant Physiol 69:1387–1391
Burton K (1956) A study of the conditions and mechanism of the diphenylamine reaction for the colorimetric estimation of deoxyribonucleic acid. Biochem J 62:315–323
Butterfass T (1959) Ploidie und Chloroplastenzahlen. Ber dtsch bot Ges 72:440–451
Butterfass T (1967) Endopolyploidie und Chloroplastenzahlen in verschiedenartigen Zellen trisomer Zuckerrüben. Planta 76:75–84
Butterfass T (1979) Patterns of chloroplast reproduction. A developmental approach to protoplasmic plant anatomy. In: Cell biology monographs, vol. 6. Springer, Wien, p 205
Chiu W-L, Stubbe W, Sears BB (1988) Plastid inheritance in Oenothera: organelle genome modifies the extent of biparental plastid transmission. Curr Genet 13:181–189
Coleman AW (1978) Visualisation of chloroplast DNA with two fluorochromes. Exptl Cell Res 114:95–100
Coleman AW (1979) Use of the fluorochrome 4′6-diamidino-2-phenylindole in genetic and developmental studies of chloroplast DNA. J Cell Biol 82:299–305
Doležel J, Bartoš J, Voglmayr H, Greilhuber J (2003) Nuclear DNA content and genome size of trout and human. Cytometry 51A:127–128
Drlica K, Burgi E, Worcel A (1978) Association of the folded chromosome with the cell envelope of Escherichia coli: Nature of the membrane-associated DNA. J Bacteriol 134:1108–1116
Eberhard S, Drapier D, Wollman FA (2002) Searching limiting steps in the expression of chloroplast-encoded proteins: relations between gene copy number, transcription, transcript abundance and translation rate in the chloroplast of Chlamydomonas reinhardtii. Plant J 31:149–160
Fujie M, Kuroiwa H, Kawano S, Mutoh S, Kuroiwa T (1994) Behaviour of oganelles and their nucleoids in the shoot apical meristem during leaf development in Arabidopsis thaliana L. Planta 194:395–405
Gibbs SP, Cheng D, Slankis T (1974) The chloroplast nucleoid in Ochromonas danica. I. Three-dimensional morphology in light- and dark-grown cells. J Cell Sci 16:557–577
Hashimoto H (1985) Changes in distribution of nucleoids in developing and dividing chloroplasts and etioplasts of Avena sativa. Protoplasma 127:119–127
Herrmann RG (1968) Chloroplastengröße und inkorporierte 3H-Thymidinmenge. Autoradiographische Studien zur Frage: Gibt es genetisch mehrwertige Chloroplasten? Ber dtsch Bot Ges 81:332
Herrmann RG (1969) Are chloroplasts polyploid? Exptl Cell Res 55:414–416
Herrmann RG (1970) Multiple amounts of DNA related to the size of chloroplasts I. An autoradiographic study. Planta 90:80–96
Herrmann RG (1982) The preparation of circular DNA from plastids. In: Edelman M, Hallick R, Chua H (eds) Methods of chloroplast molecular biology. Elsevier/North Holland, Amsterdam, pp 254–279
Herrmann RG (1997) Eukaryotism, towards a new interpretation. In: Schenk HEA, Herrmann RG, Jeon KW, Müller NE, Schwemmler W (eds) Eukaryotism and symbiosis. Springer, Heidelberg, New York, pp 73–118
Herrmann RG, Kowallik K (1970) Multiple amounts of DNA related to the size of chloroplasts II. Comparison of electron-microscopic and autoradiographic data. Protoplasma 66:357–368
Herrmann RG, Possingham JV (1980) Plastid DNA—the plastome. In: Reinert J (ed) Results and problems in cell differentiation: chloroplasts, vol 10. Springer, Berlin, pp 45–96
Herrmann RG, Kowallik KV, Bohnert HJ (1974) Structural and functional aspects of the plastome. I. The organization of the plastome. Int Symp Plant Cell Diff Lisbon, Portugal Act Biol 14:91–110
Huang CY, Grünheit N, Achmadinejad N, Timmis JN, Martin W (2005) Mutational decay and age of chloroplast and mitochondrial genomes transferred recently to angiosperm nuclear chromosomes. Plant Physiol 138:1723–1733
James TW, Jope C (1978) Visualisation of fluorescence of chloroplast DNA in higher plants by means of the DNA-specific probe 4′6-diamidino-2-phenylindole. J Cell Biol 79:623–630
Jope CA, Kirai A, Wildman SG (1978) Evidence that the amount of chloroplast DNA exceeds that of nuclear DNA in mature leaves. J Cell Biol 79:631–636
Keck K (1956) An ultramicro technique for the determination of deoxypentose nucleic acid. Arch Biochem Biophys 63:446–451
Kishima Y, Mikami T, Harada T, Shinozaki K, Sugiura M, Kinoshita T (1986) Restriction fragment map of sugar beet (Beta vulgaris L.) chloroplast DNA. Plant Mol Biol 7:201–205
Kowallik KV, Haberkorn G (1971) The DNA-structures of the chloroplast of Prorocentrum micans (Dinophyceae). Arch Mikrobiol 80:252–261
Kowallik KV, Herrmann RG (1972) Variable amounts of DNA related to the size of chloroplasts. IV. Three-dimensional arrangement of DNA in fully differentiated chloroplasts of Beta vulgaris. J Cell Sci 11:357–377
Kuchuk N, Herrmann RG, Koop H-U (1998) Plant regeneration from leaf protoplasts of evening primrose (Oenothera hookeri). Plant Cell Rep 17:601–604
Kuroiwa T, Suzuki T, Ogawa K, Kawano S (1981) The chloroplast nucleus: distribution, number, size, and shape, and a model for multiplication of the chloroplast genome during chloroplast development. Plant Cell Physiol 22:381–396
Lamppa GK, Bendich AJ (1979) Changes in chloroplast DNA levels during development of pea (Pisum satrivum). Plant Physiol 64:126–139
Larsson C, Collin C, Albertsson P-Å (1971) Characterization of three classes of chloroplasts obtained by counter-current distribution. Biochim Biophys Acta 245:425–438
Lawrence ME, Possingham JV (1986) Microspectrofluorimetric measurement of chloroplast DNA in dividing and expanding leaf cells of Spinacea oleracea. Plant Physiol 81:708–710
Legen J, Kemp S, Krause K, Profanter B, Herrmann RG, Maier RM (2002) Comparative analysis of plastid transcription profiles of entire plastid chromosomes from tobacco attributed to wild-type and PEP-deficient transcription machineries. Plant J 31:171–188
Li W, Ruf S, Bock R (2006) Constancy of organellar genome copy numbers during leaf development and senescence in higher plants. Mol Gen Genom 275:185–192
Martin W, Herrmann RG (1998) Gene transfer from organelles to the nucleus: how much, what happens, and why? Plant Physiol 118:9–17
McCourt P, Kunst L, Browse J, Somerville CR (1987) The effects of reduced amounts of lipid unsaturation on chloroplast ultrastructure and photosynthesis in a mutant of Arabidopsis. Plant Physiol 84:353–360
Menke W (1966) Structure of the chloroplasts. In: Goodwin TW (ed) Biochemistry of chloroplasts, vol I. Academic, London, pp 3–18
Miyamura S, Nagata T, Kuroiwa T (1986) Quantitative fluorescence microscopy on dynamic changes of plastid nucleoids during wheat development. Protoplasma 133:66–72
Miyamura S, Kuroiwa T, Nagata T (1990) Multiplication and differentiation of plastid nucleoids during development of chloroplasts and etioplasts from proplastids in Triticum aestivum. Plant Cell Physiol 31:597–602
Mochizuki A, Sueoka N (1955) Genetic studies on the number of plastids in stomata. I Cytologia (Tokyo) 20:358–366
Oldenburg DJ, Bendich AJ (2003) Most chloroplast DNA of maize seedlings in linear molecules with defined ends and branched forms. J Mol Biol 335:953–970
Oldenburg DJ, Bendich AJ (2004) Changes in the structure of DNA molecules and the amount of DNA per plastid during chloroplast development in maize. J Mol Biol 344:1311–1330
Pascoe MJ, Ingle J (1978) Distinction between nuclear satellite DNAs and chloroplast DNA in higher plants. Plant Physiol 62:975–977
Ris H, Plaut W (1962) Ultrastructure of DNA-containing areas in the chloroplast of Chlamydomonas. J Cell Biol 13:383–391
Rowan BA, Oldenburg DJ, Bendich AJ (2004) The demise of chloroplast DNA in Arabidopsis. Curr Genet 46:176–181
Rowan BA, Oldenburg DJ, Bendich AJ (2009) A multiple-method approach reveals a declining amount of chloroplast DANN during development in Arabidposis. BMC Plant Biol 9:1–17
Scharff LB, Koop H-U (2007) Targeted inactivation of the tobacco plastome origins of replication A and B. Plant J 50:782–794
Schmitt JM, Herrmann RG (1977) Fractionation of cell organelles in silica sol gradients. In: Prescott DM (ed) Methods in cell biology, vol 15. Academic, London, pp 177–200
Scott NS, Possingham JV (1980) Chloroplast DNA in expanding spinach leaves. J Exptl Bot 31:1081–1092
Scott NS, Possingham JV (1983) Changes in chloroplast DNA levels during growth of spinach leaves. J Exptl Bot 34:1756–1767
Shaver JM, Oldenburg DJ, Bendich AJ (2006) Changres in chloroplast DNA during development in tobacco, Medicago tunculata, pea, and maize. Planta 224:72–82
Strasburger E (1893) Über die Wirkungssphäre der Kerne und die Zellgröße. Histol Beitr V:95–124
Tymms MJ, Scott NS, Possingham JV (1983) DNA content of Beta vulgaris during leaf cell expansion. Plant Physiol 71:785–788
Zoschke R, Liere K, Börner T (2007) From seedling to mature plant: Arabidopsis plastidial genome copy number, RNA accumulation and transcription are differentially regulated during leaf development. Plant J 50:710–722
Acknowledgments
We are grateful to Drs. Thomas Boerner, Klaus Kowallik and Peter Westhoff for comments, to Dr. Elizabeth Schroeder-Reiter for language editing, to Dr. Günter Strittmatter and Mr. Dietrich Hauer, KWS, Einbeck/Hannover for providing di-, tri- and tetraploid sugar beet lines, to Prof. Christian Jung, University Kiel, for providing seeds for trisomic lines IV and VIII, and to Dr. Eva Facher, Munich, for skilled advice in micromorphometry. This work was supported by the Deutsche Forschungsgemeinschaft (grant He 693 to R.G.H.).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by R. Hagemann.
This paper is dedicated to Prof. Dr. T. Butterfass.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Rauwolf, U., Golczyk, H., Greiner, S. et al. Variable amounts of DNA related to the size of chloroplasts III. Biochemical determinations of DNA amounts per organelle. Mol Genet Genomics 283, 35–47 (2010). https://doi.org/10.1007/s00438-009-0491-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-009-0491-1