
Abstract
Inositol 1,4,5-trisphosphate receptor (IP3R) and ryanodine receptor (RyR) are homologous cation channels that mediate release of Ca2+ from the endoplasmic/sarcoplasmic reticulum (ER/SR) and thereby are involved in many physiological processes. In previous studies, we determined that when the D2594 residue, located at or near the gate of the IP3R type 1, was replaced by lysine (D2594K), a gain of function was obtained. This mutant phenotype was characterized by increased IP3 sensitivity. We hypothesized the IP3R1-D2594 determines the ligand sensitivity of the channel by electrostatically affecting the stability of the closed and open states. To test this possibility, the relationship between the D2594 site and IP3R1 regulation by IP3, cytosolic, and luminal Ca2+ was determined at the cellular, subcellular, and single-channel levels using fluorescence Ca2+ imaging and single-channel reconstitution. We found that in cells, D2594K mutation enhances the IP3 ligand sensitivity. Single-channel IP3R1 studies revealed that the conductance of IP3R1-WT and -D2594K channels is similar. However, IP3R1-D2594K channels exhibit higher IP3 sensitivity, with substantially greater efficacy. In addition, like its wild type (WT) counterpart, IP3R1-D2594K showed a bell-shape cytosolic Ca2+-dependency, but D2594K had greater activity at each tested cytosolic free Ca2+ concentration. The IP3R1-D2594K also had altered luminal Ca2+ sensitivity. Unlike IP3R1-WT, D2594K channel activity did not decrease at low luminal Ca2+ levels. Taken together, our functional studies indicate that the substitution of a negatively charged residue by a positive one at the channels’ pore cytosolic exit affects the channel’s gating behavior thereby explaining the enhanced ligand-channel’s sensitivity.
Similar content being viewed by others
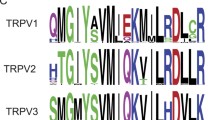


Introduction
The endoplasmic/sarcoplasmic reticulum (ER/SR) have specialized Ca2+ release channels that mediate intracellular Ca2+ signals, which regulate many cellular processes as diverse as fertilization, apoptosis, muscle contraction, transcription, secretion, learning, and memory [1, 2, 29]. These Ca2+ release channels are the inositol 1,4,5-trisphosphate receptors (IP3Rs) and ryanodine receptors (RyRs), each having 3 different isoforms. Although in most cells, a mix of the 3 isoforms is present, there are some tissues where only one type predominates. For example, the type 1 IP3R (IP3R1) is the predominant intracellular Ca2+ release channel in cerebellum [23, 33], while the type 2 RyR (RyR2) predominates in the heart [32, 34]. The IP3Rs and RyRs share a high degree of functional and structural homology [28, 30, 41, 42, 45] which can be exploited to advance our understanding of these channels.
Production of cytosolic IP3 occurs when extracellular ligands (e.g., hormones, neurotransmitters) bind to Gq-protein–coupled receptors in the surface membrane to initiate an intracellular signaling cascade [1, 23, 36]. IP3R channel activity is primarily regulated by the availability of IP3 and Ca2+ [5, 27, 37, 38]. Specifically, single-IP3R1 channels are activated when they bind IP3, and the extent of IP3R1 activation (i.e., opening duration/frequency) is a bell-shaped function of the cytosolic-free Ca2+ concentration [5, 7, 24, 37, 38]. The bell-shaped cytosolic Ca2+ dependency of the IP3R1 is explained by the presence of a cytosolic Ca2+ activation site and a putative lower affinity cytosolic Ca2+ inhibition site [20, 31, 43]. An intense research topic of single IP3R1 Ca2+ control has been the mechanism by which intra-ER Ca2+ modulates the channel activity [4, 18, 44, 47, 48]. Specifically, the controversial mechanisms that have been investigated are (a) intra-ER (luminal) Ca2+ interaction with a luminal regulatory site on the channel itself [8, 44, 48], or (b) on a closely associated regulatory protein [15, 47, 49], and/or (c) luminal Ca2+ flowing through open IP3Rs (lumen-to-cytosol) acting on the IP3R’s cytosolic Ca2+ activation and/or Ca2+ inhibitory sites [4, 17, 49, 50]. This latter process is commonly referred to as feedthrough (FT) Ca2+ regulation. Overall, the IP3R1 ligand regulatory scenario involves allosterically interacting inputs [24,25,26, 53]. For example, Mak et al. (1998) proposed that cytosolic IP3 allosterically “tunes” cytosolic IP3R Ca2+ inhibition. Likewise, cytosolic ATP is a complementary allosteric regulator of IP3R gating [26, 48].
Single RyR activity is also governed simultaneously by multiple synergistically acting ligands. For example, single RyR2 function is controlled by cytosolic ATP and Ca2+ (via its cytosolic Ca2+ activation and inhibition sites) and by luminal Ca2+ (via Ca2+ binding sites in the channel and accessory proteins) [10, 14, 22, 52]. In regard to the luminal [Ca2+] control, Chen et al., using RyR2 site-directed mutagenesis of negatively charged residues within the channel’s permeation pathway, identified the RyR2-E4872 residue as a critical site involved in RyR2 luminal Ca2+ regulation [9]. Furthermore, functional and structural analyses revealed that the negatively charged RyR2-E4872 residue in one subunit forms a salt-bridge with the positively charged RyR2-R4874 residue in the neighboring subunit in the closed state. This inter-subunit network of electrostatic interactions is believed to stabilize the closed state of RyR2. Thus, a model was put forward in which Ca2+ binding to E4872 disrupts the electrostatic interactions with neighboring amino acids favoring the transition from closed to open state [9, 35].
The corresponding IP3R1 site to RyR2-E4872 residue is the IP3R1-D2594 at the transmembrane region 6 (TMR6). Figure 1a shows the location of this residue in a 3D structure of the rat IP3R1 channel in a non-conductive closed state (4.7 Å resolution; [12]). The IP3R results from the assembly of four subunits arranged around the central channel axis, where two major regions can be defined, the bulky cytosolic region connected via “stalk” densities to the transmembrane regions (TMRs; [12]). The boxes show a close-up view of the ion conduction pathway, along the membrane plane (bottom) and from the cytosol (top). In orange, the F2586 residues are shown (F2585 mouse) which have been suggested to serve as the pore gate. Away from the gate, in the ion conduction pathway, the D2595 (D2594 mouse; red) and R2597 (R2596 mouse; blue) residues are marked. Similarly, to RyR2-R4874 residue, IP3R1-R2596 have been suggested to participate in a network of TMR interactions that could play a role in transmitting signals to the gate, in part, through an interaction between neighboring TMR6 helices [13]. Figure 1b details the amino acid sequence alignment of the region containing the IP3R1-D2594 and the corresponding site RYR2-E4872 in different species. Although Bhanumathy et al. did not find a substantial change in the IP3R1 Ca2+ signaling when the D2594 residue (Fig. 1, residue in red) was replaced by alanine mutagenesis [6], we have recently shown that its substitution with a positive charge (IP3R1-D2594K) results in a gain-of-function phenotype, while its alanine substitution (IP3R1-D2594A) induces a reduction of its function. To gain mechanistic insights into the functional significance of IP3R1-D2594K, a mouse model was created. The ITPR1‐D2594K+/− mutant mice exhibited pathological ramifications that included male infertility, azoospermia, and acrosome loss [46]. These studies indicated that changing the electric nature of IP3R1-D2594 residue allosterically influences the IP3R1 channel’s activity. We speculated that changing the charge of this residue gives rise to electrostatic repulsion between side chains favoring the opening of the channel. Therefore, we hypothesized the IP3R1-D2594 residue determines the ligand sensitivity of the channel by electrostatically affecting the stability of the closed and open states of the channel. Consequently, the aim of the present work was to define for the first time the molecular mechanism by which the IP3R1-D2594K mutation induces a gain-of-function phenotype. To this end, we measured the influence of the D2594K mutation on the IP3 activation, cytosolic Ca2+ sensitivity, and luminal Ca2+ dependency of IP3R1 at the cellular, subcellular, and single-channel levels. Our results indicate that replacing this negative charge at the pore’s cytosolic exit with a positive charge (D2594K) dramatically potentiated the IP3R1 channel’s IP3, cytosolic, and luminal Ca2+ sensitivity. Our interpretation is that this residue influences the efficacy of cytosolic (IP3 and Ca2+) and luminal (Ca2+) ligands perhaps by electrostatically influencing the stability of the closed and / or open state of the channel, and thus the operation of the IP3R gating process.

Closed IP3R1 channel structure. a Illustration of the 3D structure of IP3R1 (PDB ID: 3JAV; PyMOL) viewed from the cytosolic side down through the channel and along the membrane plane. The residue of interest D2594 is marked in red. Marked in blue are the conserved positively charged residues and in orange are the residues that correspond to the suggested IP3R1 channel’s pore gate. b Amino acid sequence alignment for the region containing IP3R1-D2594 and the corresponding site RYR2-E4872. Green line identifies the IP3R1 transmembrane segment 6 (aa 2564–2596) accordingly to PDB: 3JAV. Sequence alignment was done using the following IP3R1 sequences: Mus musculus (UniProt ID: P11881), Rattus norvegicus (UniProt ID: P29994), Homo sapiens (UniProt ID: Q14643). RYR2 sequences used in alignment include Mus musculus (UniProt ID: E9Q401), Rattus norvegicus (UniProt ID: B0LPN4), and Homo sapiens (UniProt ID: Q92736). Alignment was performed using Clustal Omega (Sievers & Higgins, 2018)
Materials and methods
Generation and culture of stable inducible IP3R1 expressing cell lines
Briefly, as previously described [46], stable inducible WT- and D2594K-IP3R1 expressing HEK293 cell lines were generated using full-length ITPR1 cDNA that was sub-cloned into an inducible expression vector, pcDNA5/FRT/TO. The recombinant expression of rat IP3R1 was achieved by the Flp-In T-REx-293 system (Invitrogen). Cells were incubated at 37 °C under 5% CO2 with DMEM supplemented with 0.1 mM nonessential amino acids, 2 mM L‐glutamine, 100 units of penicillin/ml, 1% streptomycin/ml, and 5% fetal bovine serum. Cells were selected for Flp positive in 200 µg/ml hygromycin selection medium. Induction of IP3R1-WT or IP3R1-D2594K expression was initiated upon incubation with DMEM containing 1 μg/ml tetracycline.
High throughput recording of global Ca2+ transients
To evaluate the changes in cytosolic Ca2+ concentration, stable inducible HEK293 cells were subcultured in poly-d-lysine pretreated black wall/clear bottom 96-well plates and incubated at 37 °C under 5% CO2. Each 96-well plate was split to have IP3R1-WT and -D2594K seeded cells. For the corresponding IP3R1 expression, after 24 h, cells were induced with tetracycline (1 µg/ml). On the day of the experiment, the media was removed and replaced with Fluo-8 NW (ATT Bioquest) solution containing 10 × pluronic F127 Plus and KRH solution (2 mM Ca2+, 125 mM NaCl, 5 mM KCl, 6 mM glucose, 1.2 mM MgCl2, 25 mM HEPES, pH 7.4). After 60-min incubation at room temperature, Fluo-8 NW solution was removed, and each well was rinsed twice with KRH 0 Ca2+ solution. KRH 0 Ca2+ solution was added back to each well, and the 96-well plate was placed into FLIPR® Tetra High Throughput Cellular Screening System (Molecular Devices). Each well on the plate was simultaneously challenged with KRH 0 Ca2+ solution plus different concentrations of ATP (Sigma-Aldrich) to induce Ca2+ release via IP3R1s. LED excitation wavelength of 490 nm and a 520-nm bandpass emission filter were used to acquire images. The images were captured at every 2 or 4 s. Each determination was done by duplicate at 3 different days. The resulting Fluo-8 NW fluorescent signals were measured for each well using ScreenWorks software (Molecular Devices).
Linescan confocal microscopy imaging of Ca2+ puffs
Ca2+ puffs were recorded with linescan confocal microscopy in tetracycline-induced HEK293 cells expressing either IP3R1-WT or IP3R1-D2594K. Cells were plated on glass bottom (0.13–0.17 mm thick) plates and induced for 48 h. On the day of the experiment, cells were incubated for 30 min in normal KRH external solution containing 2 mM Ca2+ for ER loading, followed by 45-min incubation in the dark and at room temperature in 1 ml KRH solution containing the fluorophore Cal-520 AM (AAT Bioquest). Cal-520 AM 45 µM stock solution was prepared by dissolving 50 µg of Cal-520 AM in 20% pluronic acid in DMSO (Molecular Probes) in KRH solution. The incubation period was ended by rinsing the cells with dye-free normal external solution at room temperature for 15 min. To record Ca2+ puffs, Cal-520 was excited with the 488 nm line of an argon laser, and the emitted fluorescence was collected through a 515-nm long pass emission filter. Cal-520 images were recorded in bidirectional linescan mode with 256 × 512 pixels per frame at 4 Hz and x-time series images at 10 Hz. For consistency, all linescan confocal microscope settings were kept the same for all experiments (laser power: 5%, photomultiplier gain: 140, pin size: 0.9, pixel size: 0.63). All experiments started with perfusion with external KRH 0 Ca2+ solution for 1 min followed by 2-min perfusion of 20 nM ATP. Fluorescence images were analyzed with Elements v4.4 (Nikon) and ImageJ v1.52 (NIH) software. Briefly, Ca2+ puffs were interactively detected as areas of high fluorescence compared with the standard deviation (threshold arbitrarily set between 5 and 6.5 standard deviations) of the background fluorescence. Puff duration was measured as the dwell time at the 50% of the peak amplitude (full-duration-half-maximum; FDHM). Puffs frequency was expressed in Hz as number of Ca2+ puffs per cell, while their kinetic attributes were estimated by measuring time to peak (time from baseline to maximal), rise time (time from 20 to 80% maximal amplitude), and decaying time (from 80 to 20% maximal amplitude; Lock et al., 2015). All puff parameters were measured, pooled, and averaged for either IP3R1-WT or IP3R1-D2594K.
Isolation of microsomal vesicles
Crude microsomes were obtained from control (WT), ITPR1‐D2594K+/− and -D2594A+/− mutant knock-in (KI) mice models developed previously [46]. Mouse cerebellar microsomes were prepared as described previously [51] with some modifications. Briefly, cerebella were homogenized in a glass-Teflon Potter homogenizer in solution A (10 mM Tris-maleate, pH 6.8, 0.5 mM DTT, and protease inhibitor cocktail; Roche cOmplete EDTA free; Sigma-Aldrich). The homogenate was centrifuged for 10 min at 4000 × gmax. The supernatant was re-centrifuged for 5 min at 15,000 × gmax, and the pellet was resuspended in solution B (10 mM Tris-Maleate, 100 mM KCl, pH 6.8, and 0.5 mM DTT). Then, the supernatant was centrifuged for 90 min at 165,000 × gmax, and the resulting pellet was resuspended in an appropriate volume of solution B. All pellets were frozen in liquid nitrogen and stored at − 80 °C.
Single-channel recordings
Reconstitution of IP3R1 channels into artificial planar lipid bilayers was performed as previously described [39]. Briefly, planar lipid bilayers were formed on ~ 100-µm-diameter hole in Teflon septa, separating two compartments: cis and trans. Bilayers were formed using a 5:3:2 mixture of phosphatidylethanolamine, phosphatidylserine, and phosphatidylcholine (Avanti Polar Lipids) dissolved in decane to a final concentration of 50 mg/ml. Microsomes were added to the solution in cis that was held at ground potential. IP3-gated channel activity was measured at room temperature in the presence of 2 mM ATP-Tris and various concentrations of IP3 using Cs+ as current carrier. The trans side of the bilayer mimics the luminal (intra-ER) compartment, where the voltage was applied. Solutions (unless otherwise specified) contained symmetrical 250 mM CsCH3SO3, 20 mM HEPES pH 7.4, and 1 mM EGTA (70 nM or 140 nM free Ca2+). Channel sidedness was determined by IP3 sensitivity. Stock solutions of IP3 and Ca2+ (Sigma-Aldrich) were made to obtain the desired final concentrations. The cytosolic and luminal free Ca2+ concentrations were calculated by MaxChelator [3]. Calculations of total Ca2+ needed to obtain a specified free Ca2+ concentration were based on the presence of 1 mM EGTA for the determination of cytosolic Ca2+ and IP3 dependency. For the luminal Ca2+ dependency, 0.5 mM EGTA, 0.5 mM BAPTA was used in the cis solution while trans solution contained 0.5 mM EGTA, 0.5 mM BAPTA, and 0.5 mM dibromo-BAPTA. Unitary current was processed by a patch-clamp amplifier (Axopatch 200B, Axon Instruments) that was connected, via Ag/AgCl electrodes, to the cis and trans solutions through agar bridges. Data were filtered at 2 kHz and digitized at 20 kHz for computer analysis using pClamp 10 (Axon Instruments). Opening events were detected from the filtered records by the half-amplitude threshold crossing technique [11]. Events with durations < 300 µs were not included in the analysis. Channel open probability was measured from the idealized records longer than 3 min. The number of channels in each experiment was estimated from the maximal number of stacked opening events observed in the bilayer and corroborated in the total amplitude histogram of the raw data by the presence of multiple peaks [16]. IP3R1 channel currents depicted as positive (upward deflections of the current) reflect cation flux from the trans (luminal) to the cis (cytosolic) compartment. Likewise, the downward deflections of the current reflect the cation flux in opposite direction. Single-channel gating kinetics analysis was done by constructing dwell-time histograms for the open and closed times and fitted with the corresponding probability density functions. Mean open and closed times were calculated as the sum of rate constants exiting each gating state [11].
Statistics
All studies were designed so that only one parameter was changed at a time. To test for differences between groups, we used Student’s t test (two‐tailed with Welch correction), and the two-way ANOVA with Šídák’s post hoc correction using GraphPad Prism software. Data is reported as mean ± SEM. Differences were considered statistically significant at p < 0.05.
Results
Effects of IP3R1-D2594K on intracellular Ca2+ dynamics
To evaluate the functional impact of IP3R1 mutations on the cellular Ca2+ signaling, the IP3R1-WT and IP3R1-D2594K were expressed in HEK 293 cells. IP3R-mediated response was induced through the production of endogenous IP3 by stimulating Gq-protein-coupled purinergic receptors in the surface membrane with ATP. Intracellular Ca2+ signals were recorded with automated fluorescence plate reader in Fluo-8 NW-loaded cells. Figure 2a compares averaged fluorescence Ca2+ signals evoked by ATP with no added Ca2+, in cells expressing IP3R1-WT or IP3R1-D2594K. The evoked Ca2+ transients were larger in cells harboring the mutated IP3R channel compared to the WT. Figure 2b shows pooled measurements of intracellular Ca2+ transient amplitude. Maximal change in fluorescence expressed as ΔF/F0 is plotted as a function of the extracellular ATP stimulus (WT filled, mutant open circles). It can be observed that cells expressing IP3R1-D2594K produced a significantly larger signal at each [ATP] tested. The reproducibility of this response was examined by performing agonist dose–response curves by duplicate in three separate experiments. ATP stimuli greater than 1 µM evoked a significantly larger Ca2+ release transients when the mutant IP3R1 was expressed.

Concentration-dependent Ca2+ release induced by ATP. a Traces show typical intracellular Ca2+ transients in HEK 293 cells expressing IP3R1-WT (left) or IP3R1-D2594K (right) evoked by the indicated ATP concentrations. b Maximal Ca.2+ transient amplitude plotted as a function of [ATP] from cells expressing IP3R1-WT (filled circles) and IP3R1-D2594K (open circles). Data were fitted by a Hill equation with an EC50 of 10.9 ± 0.75 and 3.3 ± 0.7 µM for WT and mutant IP3R1, respectively (n = 6 independent plates for each [ATP]). Data points are mean ± SEM. Significance was determined using unpair t test *p value 0.0002 and two-way ANOVA with Šídák test p < 0.0001
Effects of IP3R1-D2594K on Ca2+ puffs
To determine if local Ca2+ release events are altered by the mutated IP3R1, Ca2+ puffs were imaged in intact HEK cells expressing IP3R1-WT or IP3R1-D2594K. IP3R-mediated Ca2+ puffs are analogous to RyR-mediated Ca2+ sparks in striated muscle. Compared to the global Ca2+ transients, the attributes of Ca2+ puffs reflect closely the molecular IP3R functional properties, such as unitary conductance and single-channel gating (opening/closing) kinetics. Ca2+ puffs were promoted by addition of 20 nM of extracellular ATP. Our dose–response titration experiments ranging from 10 to 100 nM indicated that 20 nM ATP consistently promoted Ca2+ puffs and rarely induced global intracellular Ca2+ transients. Note these local Ca2+ signals are not the result of extracellular Ca2+ entry because the extracellular solution contained 0 mM Ca2+. Ca2+ puffs were measured during 2-min periods that were preceded and followed by control periods (no extracellular ATP present). Figure 3a shows example line scan images of Ca2+ puffs recorded in cells expressing IP3R1-WT or IP3R1-D2594K channels. Confocal images obtained under these conditions indicate that HEK cells expressing IP3R1-D2594K channels exhibit substantially higher Ca2+ puff activity elicited by ATP than those expressing IP3R1-WT. Subcellular areas with high Ca2+ puff activity were identified and selected as regions of interests (ROIs). In these selected regions, x-time images were generated with 10-µm kymograph lines to define Ca2+ puffs’ properties, which are better visualized by their fluorescence intensity profile plotted under each x-time image. From this type of fluorescent traces, Ca2+ puff kinetic attributes (i.e., rise time, time to peak, decay time, and half duration) were measured. Frequency of occurrence was directly quantified from selected ROIs in the x–y linescan images. Figure 3b reveals that IP3R1-D2594K-expressing cells showed more than 2.5-fold higher Ca2+ puffs frequency (in Hz, 0.1 ± 0.002 vs 0.04 ± 0.004 puffs/cell), while the mutant puffs’ kinetics exhibited significantly longer decay (2.28 ± 0.09 vs 0.35 ± 0.01 s), longer half duration (2.39 ± 0.09 vs 0.68 ± 0.02 s), longer time to peak (1.43 ± 0.04 vs 0.57 ± 0.02 s), and longer rise time (0.76 ± 0.03 vs 0.35 ± 0.01 s) than their counterpart from WT. To evaluate the contribution of HEK293 cells endogenous IP3Rs to Ca2+ release events, IP3R1-D2594K and IP3R1-WT cells were not induced with tetracycline, and linescan confocal images were recorded during application of external ATP. Under these conditions, infrequent or no intracellular Ca2+ signals were recorded in either cell type, indicating that the activity of endogenous IP3R was not detectable. Overall, these cellular results (Figs. 2 and 3) indicate the IP3R1-D2594K exhibits a gain of function phenotype.

Elementary Ca2+ release events induced by ATP. a Ca2+ puffs recorded from HEK293 cells expressing IP3R1-WT or IP3R1-D2594K during perfusion of 0 Ca2+ KRH external solution containing 20 nM ATP. Fluorescence kymograph images and traces show Ca2+ puffs time course and frequency. b Pool data comparison of Ca2+ puffs amplitude, kinetic attributes, and frequency of occurrence evaluated from averaged traces recorded from HEK293 cells expressing either IP3R1-D2594K (open circles; n = 64 cells) or IP3R1-WT (filled circles; n = 60 cells). Data points are mean ± SEM, unpair t test with Welch’s correction had a p < 0.0001
Effects of IP3R1-D2594K on unitary currents
Since these studies were conducted in a recombinant heterologous system, it was possible that the augmented responses observed with the IP3R1-D2594K mutant could have resulted from an increased receptor expression. To address this possibility, IP3R1-WT and -D2594K single-channel activity was evaluated from our WT and KI mice. The corresponding IP3R1 microsomal fractions from cerebellum were reconstituted in planar lipid bilayers, and single-channel activity was studied under control conditions. As shown in Fig. 4a, the current amplitude as a function of voltage for the mutant channel was very similar to that observed in the WT. Using Cs+ as charge carrier, the unitary conductance of 248 ± 5 pS was not significantly different between the IP3R1-WT and the IP3R1-D2594K channels (Fig. 4b). Like IP3R1-WT, the IP3R1-D2594K Cs+ conductance was reduced when Ca2+ flux was favored by increased luminal [Ca2+] (see Fig. 8a and Online Resource 1), being consistent with the anomalous mole fraction effect previously described for IP3R1 [4].

Ion conductance of single IP3R1-WT and IP3R1-D2594K channels. Symmetrical solutions contained (in mM): 250 CsCH3SO3, 1 EGTA, 0.00014 free Ca2+, 10 HEPES, and pH 7.4. Solution in the cytosolic side of the channel also contained (in mM) 0.010 IP3 and 2 ATP. a Sample single WT and mutant (D2594K) IP3R1 recordings obtained at the indicated membrane potentials. Arrows labelled “c” mark the closed non-conducting state. b Pooled data (mean ± SEM; n = 3–8) of unitary current from WT (filled circles) and D2594K (open circles) IP3R1 plotted as a function of membrane potential and fitted by linear regression with a conductance of 248 ± 5 pS
Effects of IP3R1-D2594K on IP3 sensitivity
To gain insight about the mechanism for the enhanced response to ATP by cells expressing IP3R1-D2594K, its ligand sensitivity was determined at the single-channel level. Figure 5 compares the cytosolic IP3 sensitivity of the WT and mutant IP3R1 channels. Representative single IP3R1 recordings with 50 nM and 5 µM IP3 present are shown (Fig. 5a). The frequency of WT and mutant channel openings is higher at 5 µM IP3 (compared to 50 nM). However, the mutant IP3R1 is far more active (compared to WT) at 5 µM IP3. Figure 5b plots pooled open probability normalized by the number of channels present in the bilayer (expressed as nPo) of WT and mutant channels as a function of cytosolic [IP3]. These data were well fitted by a Hill equation with significantly different EC50 values (660 nM for WT and 406 nM for mutant). In addition, IP3 activated the mutant IP3R to a significantly higher Po level. The maximal Po reached at 10 µM IP3 was 3.6-fold higher for the IP3R1-D2594K compared to the WT channel. Thus, the IP3R1-D2594K mutation increased the sensitivity and efficacy of IP3 activation.

Cytosolic IP3 sensitivity of single IP3R1-WT and IP3R1-D2594K channels. a Cytosolic and luminal solutions contained symmetrical 250 mM CsCH3SO3, 1 mM EGTA, 140 nM free Ca2+, pH 7.4, with 2 mM ATP and the indicated IP3 concentration in the cytosolic side. Sample single IP3R1-WT and IP3R1-D2594K recordings made at + 30 mV are shown with 50 nM or 5 µM cytosolic IP3 present. Arrows labelled “c” mark the closed non-conducting state. Upward deflections represent channel openings. b Open probability expressed as nPo of IP3R1-WT and IP3R1-D2594K channels plotted as a function of log [IP3]. Pooled data (mean ± SEM; n = 3–9) were fitted by a Hill equation with an EC50 of 660 amf 406 nM and a Hill coefficient of 1.8 and 2 for IP3R1-WT (filled circles) and IP3R1-D2594K (open circles), respectively. Statistical significance between WT and mutant data points was evaluated with an unpaired t test with p < 0.05 and two-way ANOVA with Šídák test p < 0.0001
Effects of IP3R1-D2594K on cytosolic Ca2+ sensitivity
In a previous study, we found that when the IP3R1-D2594 residue was replaced by alanine, the concentration-dependent response to IP3 was reduced, while replacing it by a lysine or arginine, the response was substantially increased [46]. One possible mechanism to explain these effects is that the D2594 residue determines the channel sensitivity to cytosolic Ca2+. To evaluate this possibility, the experiments shown in Fig. 6 compare the cytosolic Ca2+ sensitivity of single IP3R1-WT with that of D2594 mutated IP3R1 channels. Representative single IP3R1 channel activity obtained in 70 nM, 300 nM, and 1 µM cytosolic Ca2+ is shown (Fig. 6a). At 70 nM cytosolic Ca2+, openings of all three IP3R1 channels are brief and relatively rare, but for IP3R1-WT and IP3R1-D2594K, they become longer and more frequent when the cytosolic Ca2+ is increased to 300 nM. Further increasing the cytosolic Ca2+ to 1 µM reduced the activity of these three types of IP3R1 channels. Still, the activity of the IP3R1-D2594K channel at all tested cytosolic Ca2+ levels was considerably greater than the WT channel. Although the IP3R1-D2594A activity also exhibited a bell-shaped dependency, the opening events remained brief and scarce at the three cytosolic Ca2+ concentrations. Figure 6b plots nPo of IP3R1-WT, IP3R1-D2594K, and IP3R1-D2594A channels as a function of cytosolic Ca2+. These experiments were conducted in the presence of 10 μM cytosolic IP3 and 70 nM luminal Ca2+. The cytosolic Ca2+ dependency of all three channels exhibited a bell-shaped relationship, but the IP3R1-D2594K activity was considerably larger at all tested [Ca2+]. These data were well fitted by a biphasic Hill equation with the following EC50 and IC50 values: 78 and 562 nM for IP3R1-WT, 101 and 1380 nM for IP3R1-D2594K, and 89 and 399 nM for IP3R1-D2594A, respectively. Across the range of all Ca2+ levels tested, cytosolic Ca2+ activated the IP3R-D2594K to a significantly larger Po level. The maximal Po reached near 300 nM Ca2+ was roughly threefold higher for the IP3R1-D2594K than for the other two channel types. This shows that the D2594K mutation also increases the cytosolic Ca2+ activation efficacy without substantially altering the channel’s cytosolic Ca2+ sensitivity given the similarity of EC50 values.

Cytosolic Ca2+ dependency of IP3R1-WT, IP3R1-D2594K, and IP3R1-D2594A. The cytosolic and luminal solutions respectively contained (in mM): 500/50 CsCH3SO3, 1/1 EGTA, various/0.00007 [Ca2+]free, 2/0 ATP, 0.01/0 IP3 at pH 7.4. a Single-channel activity from IP3R1-WT (left), IP3R1-D2594K (center), and IP3R1-D2594A (right), recorded at 0 mV at the indicated cytosolic [Ca2+]. Downward deflections indicate channel openings. b Open probability as a function of Log10 cytosolic [Ca.2+] for IP3R1- WT (filled circles), IP3R1-D2594A (gray circles), and IP3R1-D2594K (open circles). Pooled data shown as mean ± SEM (n = 9–12) were fitted with a biphasic Hill equation. Significance was determined using ANOVA with Šídák test with p values 0.006 (WT vs. D2594K) and 0.962 (WT vs. D2594A)
Effects of IP3R1-D2594K on luminal Ca2+ sensitivity
The mechanism of luminal Ca2+ regulation has been the focus of intense research [4, 8, 18, 24, 44, 48,49,50]; therefore, the effect of IP3R1-D2594K on this property was investigated. Figure 7a shows sample single IP3R1-WT and IP3R1-D2594K channel recordings with 10 µM luminal Ca2+, 10 µM cytosolic IP3, and 70 nM free cytosolic Ca2+. The mutant IP3R1 is significantly more active than the WT channel. A possible mechanism of luminal Ca2+ regulation would involve luminal Ca2+ ions feeding through (FT) an open IP3R1, as they move from lumen of the ER to cytosol, and binding to the channel’s own cytosolic Ca2+ activation and inhibition sites. Logically, the degree of Ca2+ regulation by FT will depend on the amplitude of Ca2+ flux through the open channel, which varies with both the trans-membrane Ca2+ gradient and the ER membrane potential. Figure 7b left compares the activity of IP3R1-WT (filled triangles) with IP3R1-D2594K (open triangles) normalized to the initial nPo, as a function of luminal [Ca2+] at − 40 mV of membrane potential, which favored cytosol-to-lumen Ca2+ flux. Under these conditions, the luminal Ca2+ dependency was essentially eliminated, as indicated by the straight lines fitted to the data points. In contrast, Fig. 7b right shows that at + 40 mV, which favored the lumen-to-cytosol Ca2+ flux, the luminal Ca2+ dependency of both types of channels becomes bell-shaped, resembling the cytosolic Ca2+ dependency. This bell-shaped behavior indicates the predominance of activation over inhibition at low luminal [Ca2+], and the predominant inhibitory effect as luminal [Ca2+] is increased. However, the activity of IP3R1-D2594K (open circles) as a function of luminal [Ca2+] was more than threefold greater than that of the WT channel (filled circles) and peaked at about one order of magnitude lower luminal [Ca2+] (10 vs 100 μM, respectively). These dramatic effects observed across most of luminal Ca2+ levels tested were unlikely due to the mutant conducting a larger flux because as shown in Fig. 4, the mutation does not alter IP3R1 unitary conductance.

Luminal Ca2+ sensitivity of single IP3R1-WT and IP3R1-D2594K channels. The cytosolic and luminal solutions respectively contained (in mM) 250/250 CsCH3SO3, 0.5/0.5 EGTA, 0.5/0 BAPTA, 0.00007/various [Ca2+]free, 0.01/0 IP3, 2/0 ATP and pH 7.4. a Sample single IP3R1-WT and IP3R1-D2594K recordings made at + 30 mV with 10 µM luminal free Ca2+ present. Arrow labelled “c” mark the closed non-conducting state. Upward deflections represent channel openings. b Left, IP3R1-WT (filled triangles) and -D2594K (open triangles) nPo plotted as a function of luminal [Ca2+] at negative applied voltages. Data are mean ± SEM (n = 3–9) and were fitted by linear regression. b Right, nPo of IP3R1-WT (filled circles) and -D2594K (open circles) plotted as a function of luminal [Ca.2+] at positive applied voltages. Data are mean ± SEM (n = 4–9) fit by a biphasic Hill equation. Statistical significance at positive potentials by unpaired t test with Welch’s correction had a p value 0.0015
Discussion
IP3R and RyR share many structural and functional similarities. In RyR2, the negatively charged residue E4872 electrostatically interacts with the positively charged residue R4874 in the neighboring subunit to stabilize the closed state. When open, E4872 moves away from R4874, but closer to the negatively charged residue E4878. These residues (E4872 and E4878) are thought to be involved in electrostatic interaction with luminal Ca2+ [19, 35]. The corresponding E4872 residue in IP3R1 is D2594, but its role in IP3R function is unknown. We have previously found that when the D2594 residue’s charge was neutralized, the IP3 sensitivity of IP3R1 was reduced, but the IP3 sensitivity increased if it was replaced by a positive residue like lysine or arginine [46]. In this work, we found that in intact cells, this substitution increased the efficacy of activation by IP3 (shifted the EC50), while at the subcellular level, it increased the frequency, amplitude, and duration of Ca2+ puffs. These actions could be explained by differences in cell densities, expression levels, and/or altered receptor regulation. To address these possibilities, we defined how the D2594K alters IP3R1 function at the single-channel level. We showed (Fig. 5b) the mutation increased Po and shifted the IP3 EC50, indicative of an increase in the channel’s IP3 sensitivity. However, this could also be explained in part by changes in either IP3R1 cytosolic and/or luminal [Ca2+] regulation. We found that IP3R1-D2594K, like its WT counterpart, exhibited a bell-shaped cytosolic-free [Ca2+] dependency, but the mutant exhibited higher Po even when the lumen-to-cytosol Ca2+ flux was minimized by reducing the Ca2+ driving force with 70 nM luminal [Ca2+]. As seen in Fig. 6, the channel activity at both 70 and 300 nM cytosolic [Ca2+] was considerably higher in the mutant channel than in the WT with low luminal [Ca2+]. Likewise, WT and D2594K channels were both sensitive to luminal [Ca2+], but the mutant’s Po was higher as well. Although potential effects of D2594K on ionic permeability/selectivity were not ruled out, our Ca2+/Cs+ selectivity experiments suggest this possibility is unlikely (see Fig. 4 and Online resource 1). Taken together, it appears that this mutation near the channel’s gate simultaneously alters the IP3 and Ca2+ (cytosolic and luminal) sensitivities of the channel to produce its clear gain-of -function phenotype. This is consistent with the notion that the D2594 residue stabilizes the IP3R1 closed state, as it has been proposed by Ogawa’s group for the corresponding residue in RyR2, where specific interactions within the pore stabilize and prevent hyperactivity [21]. Furthermore, this possibility has already been suggested for the IP3R as point mutations in the channel permeation pathway alter intra/inter-domain interactions and have secondary regulatory actions [7, 40].
Specifically, we speculate that IP3R1 D2594 residue electrostatically interacts with positively charged R2596 in the neighboring subunit. This forms a network of salt bridges that stabilizes the IP3R1 closed state. This makes the R2594 residue key for IP3R1 stabilization and prevention of potentially deleterious hyperactivity.
As mentioned, the IP3R1-D2594 residue is homologous to RyR2-E4872 which (in RyR2) influences luminal Ca2+ regulation [9]. Replacing RyR2-E4872 for alanine abolished luminal Ca2+ dependency, but it was restored by introducing a glutamate residue nearby (G4871E). Thus, we sought to better detail the potential influence of IP3R1-D2594 on luminal Ca2+ regulation. Figure 8 shows the actions of luminal [Ca2+] and membrane potential on the duration of IP3R1-WT opening and closing events. Single IP3R1-WT channel activity obtained at three different luminal [Ca2+] at + 40 mV (Fig. 8a) illustrates the clear luminal [Ca2+] dependency of IP3R1 Po. Note the unitary Cs+ current is small at 5 mM luminal (as Ca2+ competes with Cs+ for occupancy of the pore). The duration of IP3R1 opening and closing events varied with luminal [Ca2+]. We analyzed mean open time (MOT) and mean closed time (MCT) separately because lumen-to-cytosol Ca2+ flux carried by an open channel may act on cytosolic Ca2+ regulatory sites and extend mean open time (MOT; [50]). Intuitively, lumen-to-cytosol Ca2+ flux should not influence closed channel function. In Fig. 8b, MOT was normalized (to its initial value) and plotted as a function of the electrochemical Ca2+ driving force (EDF, measured as the difference between the membrane potential and the Ca2+ Nernst potential; Em − ECa). Interestingly, single IP3R1 MOT displayed a bell-shaped dependency on EDF. As indicated by the vertical arrows, MOT decreased as the ER [Ca2+] is reduced. Arrows represent the expected EDFs at room temperature, for an ER that is full of Ca2+ (solid arrow), a depleted ER (dash arrow), and an extremely depleted ER (square dot arrow). Note that the activation phase lies within the physiological range of electrochemical driving forces for Ca2+. The MOT behavior is reminiscent of the IP3R1’s bell-shaped cytosolic Ca2+ sensitivity (see Fig. 6) and strongly suggests luminal Ca2+ feeds through an open IP3R1 and acts on its cytosolic Ca2+ activation and inactivation sites. Interestingly, the mean closed time (MCT) also exhibited a profound dependency on EDF as illustrated in Fig. 8c. In fact, the experimental data points were well described by the bell-shaped curve fitted to the MOT (double-Hill equation scaled and inverted; Fig. 8c; dashed curve). Although our results cannot definitively rule out the presence of a luminal Ca2+ binding site (in the channel itself and/or in an accessory protein), our analysis indicates that the IP3R1 dependence on luminal [Ca2+] is strongly influenced by the Ca2+ ions passing through the IP3R1. Unfortunately, the same type of kinetic analysis could not be performed on IP3R-D2594K channels because multiple (not single) mutant channels were typically present. However, based on our cytosolic and luminal [Ca2+] studies, we believe mutant channel dwell times would likely exhibit similar dependencies on EDF.

Luminal Ca2+ dependency of single IP3R1-WT channels gating kinetics. The cytosolic and luminal solutions contained symmetrical 250 mM CsCH3SO3, 1 mM EGTA, 140 nM [Ca2+]free, pH 7.4. a Unitary Cs+ currents at + 40 mV with 2 mM ATP, 2 µM IP3 at the indicated 140 nM (upper), 60 µM (middle), or 5 mM (lower) free luminal Ca2+. Label “c” marks the closed non-conducting state. Upward deflections represent channel openings. b Normalized mean open time of IP3R1-WT channel plotted as a function of the electrochemical Ca2+ driving force (Em − ECa) in mV, where Em is the transmembrane potential applied to the bilayer (i.e., − 40, − 20, + 20, or + 40 mV) with the cis side held at ground and ECa is the Nernst potential of Ca2+ in mV, calculated at 22 ºC by the formula ECa = (RT/zCa2+F) ln([Ca2+]Cis/[Ca2+]Trans), where R is the universal gas constant, 8314 mJ/(K mol); F is Faraday’s constant, 96,485 C/mol; T is temperature, 295.15 K; and zCa2+ is the valence of Ca2+ ions, + 2. With these formulas, positive electrochemical driving forces indicate trans-to-cis Ca2+ fluxes. Data points were measured at − 40, − 20 mV (open circles), + 20, and + 40 mV (black circles). Data fitted with a double Hill equation (MOT = 1 + Range∙(1 + (Va50/EDF)^Ha) − 1 (1 + (EDF/Vi50)^Hi) − 1) with Va50 = 111.5 mV, Ha = 7.8, Vi50 = 170.7 mV, Hi = 3.7. Arrows represent the electrochemical driving force for Ca2+ at Em = 0 mV (the physiological transmembrane potential at the ER membrane) at full (1 mM), depleted (100 µM) or an extremely depleted (10 µM) ER, with 140 nM Ca2+ at the cytosol. c Normalized closed time of IP3R1-WT channel plotted as a function of the electrochemical Ca2+ driving force. Data points were measured at + 20 (dashed circles) and + 40 mV (gray circles). For comparison, the curve fitted to MOT (flipped and scaled) is included as a dashed line
In summary, a ligand gated channel’s gate interacts (likely allosterically) with ligand binding sites. Our data show the intra-pore near-the-gate IP3R1-D2594 residue critically contributes to ligand interactions (likely allosterically) of multiple IP3R1 binding sites. Thus, the IP3R1-D2594 residue is located in an ideal position to influence gating kinetics by establishing specific interactions within the pore to stabilize and prevent hyperactivity.
Data availability
The dataset generated during the present study are available from the corresponding author on reasonable request.
References
Berridge MJ (1993) Inositol trisphosphate and calcium signalling. Nature 361:315–325. https://doi.org/10.1038/361315a0
Berridge MJ (2016) The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol Rev 96:1261–1296. https://doi.org/10.1152/physrev.00006.2016
Bers DM, Patton CW, Nuccitelli R (1994) A practical guide to the preparation of Ca2+ buffers. Methods Cell Biol 40:3–29. https://doi.org/10.1016/s0091-679x(08)61108-5
Bezprozvanny I, Ehrlich BE (1994) Inositol (1,4,5)-trisphosphate (InsP3)-gated Ca channels from cerebellum: conduction properties for divalent cations and regulation by intraluminal calcium. J Gen Physiol 104:821–856. https://doi.org/10.1085/jgp.104.5.821
Bezprozvanny llya, Watras J, Ehrlich BE, (1991) Bell-shaped calcium-response curves of lns(l,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 351:751–754. https://doi.org/10.1038/351751a0
Bhanumathy C, da Fonseca PCA, Morris EP, Joseph SK (2012) Identification of functionally critical residues in the channel domain of inositol trisphosphate receptors. J Biol Chem 287:43674–43684. https://doi.org/10.1074/jbc.M112.415786
Boehning D, Mak D-OD, Foskett JK, Joseph SK (2001) Molecular determinants of ion permeation and selectivity in inositol 1,4,5-trisphosphate receptor Ca2+ channels. J Biol Chem 276:13509–13512. https://doi.org/10.1074/jbc.C100094200
Caroppo R, Colella M, Colasuonno A, DeLuisi A, Debellis L, Curci S, Hofer AM (2003) A reassessment of the effects of luminal [Ca2+] on inositol 1,4,5-trisphosphate-induced Ca2+ release from internal stores. J Biol Chem 278:39503–39508. https://doi.org/10.1074/jbc.M305823200
Chen W, Wang R, Chen B, Zhong X, Kong H, Bai Y, Zhou Q, Xie C, Zhang J, Guo A, Tian X, Jones PP, O’Mara ML, Liu Y, Mi T, Zhang L, Bolstad J, Semeniuk L, Cheng H, Zhang J, Chen J, Tieleman DP, Gillis AM, Duff HJ, Fill M, Song L-S, Chen SRW (2014) The ryanodine receptor store-sensing gate controls Ca2+ waves and Ca2+-triggered arrhythmias. Nat Med 20:184–192. https://doi.org/10.1038/nm.3440
Chirasani VR, Pasek DA, Meissner G (2021) Structural and functional interactions between the Ca2+-, ATP-, and caffeine-binding sites of skeletal muscle ryanodine receptor (RyR1). J Biol Chem 297:101040. https://doi.org/10.1016/j.jbc.2021.101040
Colquhoun D, Sigworth FJ (1995) Fitting and statistical analysis of single-channel records. In: Sakmann B, Neher E (eds) Single-Channel Recording. Springer US, Boston, pp 483–587
Fan G, Baker ML, Wang Z, Baker MR, Sinyagovskiy PA, Chiu W, Ludtke SJ, Serysheva II (2015) Gating machinery of InsP3R channels revealed by electron cryomicroscopy. Nature 527:336–341. https://doi.org/10.1038/nature15249
Fan G, Baker MR, Wang Z, Seryshev AB, Ludtke SJ, Baker ML, Serysheva II (2018) Cryo-EM reveals ligand induced allostery underlying InsP3R channel gating. Cell Res 28:1158–1170. https://doi.org/10.1038/s41422-018-0108-5
Györke I, Hester N, Jones LR, Györke S (2004) The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J 86:2121–2128. https://doi.org/10.1016/S0006-3495(04)74271-X
Higo T, Hattori M, Nakamura T, Natsume T, Michikawa T, Mikoshiba K (2005) Subtype-specific and ER lumenal environment-dependent regulation of inositol 1,4,5-trisphosphate receptor type 1 by ERp44. Cell 120:85–98. https://doi.org/10.1016/j.cell.2004.11.048
Horn R (1991) Estimating the number of channels in patch recordings. Biophys J 60:433–439. https://doi.org/10.1016/S0006-3495(91)82069-0
Horne JH, Meyer T (1995) Luminal calcium regulates the inositol trisphosphate receptor of rat basophilic leukemia cells at a cytosolic site. Biochemistry 34:12738–12746. https://doi.org/10.1021/bi00039a033
Irvine RF (1990) ‘Quanta’ Ca 2+ release and the control of Ca 2+ entry by inositol phosphates - a possible mechanism. FEBS Lett 263:5–9. https://doi.org/10.1016/0014-5793(90)80692-C
Jones PP, Guo W, Chen SRW (2017) Control of cardiac ryanodine receptor by sarcoplasmic reticulum luminal Ca2+. J Gen Physiol 149:867–875. https://doi.org/10.1085/jgp.201711805
Kaftan EJ, Ehrlich BE, Watras J (1997) Inositol 1,4,5-trisphosphate (InsP3) and calcium interact to increase the dynamic range of InsP3 receptor-dependent calcium signaling. J Gen Physiol 110:529–538. https://doi.org/10.1085/jgp.110.5.529
Kobayashi T, Tsutsumi A, Kurebayashi N, Saito K, Kodama M, Sakurai T, Kikkawa M, Murayama T, Ogawa H (2022) Molecular basis for gating of cardiac ryanodine receptor explains the mechanisms for gain- and loss-of function mutations. Nat Commun 13:2821. https://doi.org/10.1038/s41467-022-30429-x
Li P, Chen SRW (2001) Molecular basis of Ca2+ activation of the mouse cardiac Ca2+ release channel (ryanodine receptor). J Gen Physiol 118:33–44. https://doi.org/10.1085/jgp.118.1.33
Maeda N, Kawasaki T, Nakade S, Yokota N, Taguchi T, Kasai M, Mikoshiba K (1991) Structural and functional characterization of inositol 1,4,5-trisphosphate receptor channel from mouse cerebellum. J Biol Chem 266:1109–1116
Mak D-OD, McBride S, Foskett JK (1998) Inositol 1,4,5-tris-phosphate activation of inositol tris-phosphate receptor Ca 2+ channel by ligand tuning of Ca 2+ inhibition. Proc Natl Acad Sci USA 95:15821–15825. https://doi.org/10.1073/pnas.95.26.15821
Mak D-OD, McBride S, Foskett JK (1999) ATP regulation of type 1 inositol 1,4,5-trisphosphate receptor channel gating by allosteric tuning of Ca2+ activation. J Biol Chem 274:22231–22237. https://doi.org/10.1074/jbc.274.32.22231
Mak D-OD, McBride S, Foskett JK (2001) Atp-dependent adenophostin activation of inositol 1,4,5-trisphosphate receptor channel gating. J Gen Physiol 117:299–314. https://doi.org/10.1085/jgp.117.4.299
Mak D-OD, McBride SMJ, Petrenko NB, Foskett JK (2003) Novel regulation of calcium inhibition of the inositol 1,4,5-trisphosphate receptor calcium-release channel. J Gen Physiol 122:569–581. https://doi.org/10.1085/jgp.200308808
Mignery GA, Südhof TC, Takei K, De Camilli P (1989) Putative receptor for inositol 1,4,5-trisphosphate similar to ryanodine receptor. Nature 342:192–195. https://doi.org/10.1038/342192a0
Mikoshiba K (2007) IP 3 receptor/Ca 2+ channel: from discovery to new signaling concepts: IP 3 receptor/Ca 2+ channel. J Neurochem 102:1426–1446. https://doi.org/10.1111/j.1471-4159.2007.04825.x
Mikoshiba K, Furuichi T, Miyawaki A (1994) Structure and function of IP3 receptors. Semin Cell Biol 5:273–281. https://doi.org/10.1006/scel.1994.1033
Miyakawa T (2001) Ca2+-sensor region of IP3 receptor controls intracellular Ca2+ signaling. EMBO J 20:1674–1680. https://doi.org/10.1093/emboj/20.7.1674
Nakai J, Imagawa T, Hakamata Y, Shigekawa M, Takeshima H, Numa S (1990) Primary structure and functional expression from cDN A of the cardiac ryanodine receptor/calcium release channel. FEBS Lett 271:169–177. https://doi.org/10.1016/0014-5793(90)80399-4
Newton CL, Mignery GA, Südhof TC (1994) Co-expression in vertebrate tissues and cell lines of multiple inositol 1,4,5-trisphosphate (InsP3) receptors with distinct affinities for InsP3. J Biol Chem 269:28613–28619
Otsu K, Willard HF, Khanna VK, Zorzato F, Green NM, MacLennan DH (1990) Molecular cloning of cDNA encoding the Ca2+ release channel (ryanodine receptor) of rabbit cardiac muscle sarcoplasmic reticulum. J Biol Chem 265:13472–13483
Peng W, Shen H, Wu J, Guo W, Pan X, Wang R, Chen SRW, Yan N (2016) Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science 354:aah5324. https://doi.org/10.1126/science.aah5324
Putney JW, Bird GStJ (1993) The inositol phosphate-calcium signaling system in nonexcitable cells. Endocr Rev 14:610–631. https://doi.org/10.1210/edrv-14-5-610
Ramos-Franco J, Fill M, Mignery GA (1998) Isoform-specific function of single inositol 1,4,5-trisphosphate receptor channels. Biophys J 75:834–839. https://doi.org/10.1016/S0006-3495(98)77572-1
Ramos-Franco J, Caenepeel S, Fill M, Mignery G (1998) Single channel function of recombinant type-1 inositol 1,4,5-trisphosphate receptor ligand binding domain splice variants. Biophys J 75:2783–2793. https://doi.org/10.1016/S0006-3495(98)77721-5
Ramos-Franco J, Gomez AM, Nani A, Liu Y, Copello JA, Fill M (2010) Ryanodol action on calcium sparks in ventricular myocytes. Pflugers Arch - Eur J Physiol 460:767–776. https://doi.org/10.1007/s00424-010-0839-8
Schug ZT, da Fonseca PCA, Bhanumathy CD, Wagner L, Zhang X, Bailey B, Morris EP, Yule DI, Joseph SK (2008) Molecular characterization of the inositol 1,4,5-trisphosphate receptor pore-forming segment. J Biol Chem 283:2939–2948. https://doi.org/10.1074/jbc.M706645200
Seo M-D, Enomoto M, Ishiyama N, Stathopulos PB, Ikura M (2015) Structural insights into endoplasmic reticulum stored calcium regulation by inositol 1,4,5-trisphosphate and ryanodine receptors. Biochim Biophys Acta (BBA) - Mol Cell Res 1853:1980–1991. https://doi.org/10.1016/j.bbamcr.2014.11.023
Seo M-D, Velamakanni S, Ishiyama N, Stathopulos PB, Rossi AM, Khan SA, Dale P, Li C, Ames JB, Ikura M, Taylor CW (2012) Structural and functional conservation of key domains in InsP3 and ryanodine receptors. Nature 483:108–112. https://doi.org/10.1038/nature10751
Shinohara T, Michikawa T, Enomoto M, Goto J-I, Iwai M, Matsu-ura T, Yamazaki H, Miyamoto A, Suzuki A, Mikoshiba K (2011) Mechanistic basis of bell-shaped dependence of inositol 1,4,5-trisphosphate receptor gating on cytosolic calcium. Proc Natl Acad Sci USA 108:15486–15491. https://doi.org/10.1073/pnas.1101677108
Sienaert I, De Smedt H, Parys JB, Missiaen L, Vanlingen S, Sipma H, Casteels R (1996) Characterization of a cytosolic and a luminal Ca2+ binding site in the type I inositol 1,4,5-trisphosphate receptor. J Biol Chem 271:27005–27012. https://doi.org/10.1074/jbc.271.43.27005
Stathopulos PB, Seo M, Enomoto M, Amador FJ, Ishiyama N, Ikura M (2012) Themes and variations in ER/SR calcium release channels: structure and function. Physiology 27:331–342. https://doi.org/10.1152/physiol.00013.2012
Sun B, Ni M, Tian S, Guo W, Cai S, Sondergaard MT, Chen Y, Mu Y, Estillore JP, Wang R, Chen J, Overgaard MT, Fill M, Ramos-Franco J, Nyegaard M, Wayne Chen SR (2022) A gain-of-function mutation in the ITPR1 gating domain causes male infertility in mice. J Cell Physiol 237:3305–3316. https://doi.org/10.1002/jcp.30783
Taylor CW, Laude AJ (2002) IP3 receptors and their regulation by calmodulin and cytosolic Ca2+. Cell Calcium 32:321–334. https://doi.org/10.1016/S0143416002001859
Thrower EC, Mobasheri H, Dargan S, Marius P, Lea EJA, Dawson AP (2000) Interaction of luminal calcium and cytosolic ATP in the control of type 1 inositol (1,4,5)-trisphosphate receptor channelS. J Biol Chem 275:36049–36055. https://doi.org/10.1074/jbc.M000970200
Vais H, Wang M, Mallilankaraman K, Payne R, McKennan C, Lock JT, Spruce LA, Fiest C, Chan MY, Parker I, Seeholzer SH, Foskett JK, Mak D-OD (2020) ER-luminal [Ca2+] regulation of InsP3 receptor gating mediated by an ER-luminal peripheral Ca2+-binding protein. eLife 9:e53531. https://doi.org/10.7554/eLife.53531
Vais H, Foskett JK, Ullah G, Pearson JE, Daniel Mak D-O (2012) Permeant calcium ion feed-through regulation of single inositol 1,4,5-trisphosphate receptor channel gating. J Gen Physiol 140:697–716. https://doi.org/10.1085/jgp.201210804
Watras J, Bezprozvanny I, Ehrlich BE (1991) Inositol 1,4,5-trisphosphate-gated channels in cerebellum: presence of multiple conductance states. J Neurosci 11:3239–3245. https://doi.org/10.1523/JNEUROSCI.11-10-03239.1991
Yuan Q, Dridi H, Clarke OB, Reiken S, Melville Z, Wronska A, Kushnir A, Zalk R, Sittenfeld L, Marks AR (2021) RyR1-related myopathy mutations in ATP and calcium binding sites impair channel regulation. Acta Neuropathol Commun 9:186. https://doi.org/10.1186/s40478-021-01287-3
Yule DI, Betzenhauser MJ, Joseph SK (2010) Linking structure to function: Recent lessons from inositol 1,4,5-trisphosphate receptor mutagenesis. Cell Calcium 47:469–479. https://doi.org/10.1016/j.ceca.2010.04.005
Funding
The authors received the support of research grants from the National Institutes of Health grant/award numbers R01GM111397 to S. R. W. Chen, M. Fill, and J. Ramos‐Franco, R01HL057832 to M. Fill and by the Canadian Institutes of Health Research, grant/award number PJT‐173352. S. R. W. Chen holds the Heart and Stroke Foundation Chair in Cardiovascular Research (END611955). A. Tambeaux was supported by the Graduate College of Rush University Medical Center.
Author information
Authors and Affiliations
Contributions
Conceived of or designed study: RMA, MF, SRWC, and JRF. Performed research: AT, YAS, MM, KMD, RMA, and JRF. Analyzed data: AT, YAS, DJS, RMA, MF, SRWC, and JRF. Wrote the paper: AT, YAS, DJS, RMA, MF, SRWC, and JRF.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
Mouse cerebellar tissues were obtained and approved by the Institutional Animal Care and Use Committee at the University of Calgary and performed in accordance with the NIH guidelines.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tambeaux, A., Aguilar-Sánchez, Y., Santiago, D.J. et al. Ligand sensitivity of type-1 inositol 1,4,5-trisphosphate receptor is enhanced by the D2594K mutation. Pflugers Arch - Eur J Physiol 475, 569–581 (2023). https://doi.org/10.1007/s00424-023-02796-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-023-02796-x