
Abstract
Long QT(3) (LQT3) syndrome is associated with abnormal repolarisation kinetics, prolonged action potential durations (APD) and QT intervals and may lead to life-threatening ventricular arrhythmias. However, there have been few physiological studies of its effects on atrial electrophysiology. Programmed electrical stimulation and burst pacing induced atrial arrhythmic episodes in 16 out of 16 (16/16) wild-type (WT) and 7/16 genetically modified Scn5a+/Δ (KPQ) Langendorff-perfused murine hearts modelling LQT3 (P < 0.001 for both), and in 14/16 WT and 1/16 KPQ hearts (P < 0.001 for both; Fisher’s exact test), respectively. The arrhythmogenic WT hearts had significantly larger positive critical intervals (CI), given by the difference between atrial effective refractory periods (AERPs) and action potential durations at 90% recovery (APD90), compared to KPQ hearts (8.1 and 3.2 ms, respectively, P < 0.001). Flecainide prevented atrial arrhythmias in all arrhythmogenic WT (P < 0.001) and KPQ hearts (P < 0.05). It prolonged the AERP to a larger extent than it did the APD90 in both WT and KPQ groups, giving negative CIs. Quinidine similarly exerted anti-arrhythmic effects, prolonged AERP over corresponding APD90 in both WT and KPQ groups. These findings, thus, demonstrate, for the first time, inhibitory effects of the KPQ mutation on atrial arrhythmogenesis and its modification by flecainide and quinidine. They attribute these findings to differences in the CI between WT and mutant hearts, in the presence or absence of these drugs. Thus, prolongation of APD90 over AERP gave positive CI values and increased atrial arrhythmogenicity whereas lengthening of AERP over APD90 reduced such CI values and produced the opposite effect.
Similar content being viewed by others

Introduction
Atrial tachycardia (AT) is characterised by an abnormal rapid cardiac rhythm which can develop into atrial fibrillation (AF). Its clinical incidence is influenced by familial history and, therefore, genetic factors [9]. Re-entrant substrates have been implicated in such phenomena [2, 21]. In common with recent murine ventricular models for long QT syndromes (LQTS) [27, 34, 40], previous studies have attributed atrial arrhythmogenesis to decreased tissue refractoriness or alterations in action potential duration (APD) [1, 6, 24, 33, 48]. However, APD is determined by the balance between a number of inward and outward currents that flow during action potential restitution that could be specific for atrial tissue. Yet few studies have examined for the existence of or characterised atrial as opposed to ventricular arrhythmic phenotypes in LQTS models. Atrial arrhythmias were reported as uncommon in patients with the inherited LQTS who are susceptible to ventricular arrhythmias [43]. However, a recent study [14] reported that LQTS patients demonstrated abnormalities in atrial physiology similar to those found in the ventricle in the form of prolonged APD and refractory period (RP) durations and early after-depolarisations (EADs) triggering ‘atrial torsade de pointes’. This suggested that electrophysiological abnormalities seen in LQTS individuals could predispose not only to ventricular but also to atrial arrhythmias. However, this study included very few LQT1, LQT2, long QT(3) (LQT3) and unidentified carriers.
LQT3 is one of the known LQTS variants; the causes of LQT3 include ‘gain-of-function’ mutations in the inactivation domain between DIII and DIV of the Scn5a gene encoding the α-(pore-forming) subunit of the cardiac voltage-gated Na+ channel responsible for initiating the cardiac AP [38, 46, 47]. One such, ΔKPQ, mutation is a deletion of nine bases that code for Lys-1505, Pro-1506, Gln-1507, resulting in a prolonged APD. Scn5a+/Δ (KPQ) mutant mice modelling human LQT3 have been successfully used to demonstrate AP and QT prolongation along with torsade de pointes-like ventricular tachycardia, often observed in humans with LQT3 [25, 28, 40]. Furthermore, the cardiotropic Class I drugs, flecainide and quinidine respectively exerted anti- and pro-arrhythmic effects on KPQ hearts, but had precisely opposite effects in wild-type (WT) hearts [39, 40]. These actions correlated directly with the corresponding changes in endocardial and epicardial APDs as well as the corresponding RPs.
However, in contrast to the ventricles, atria are thin-walled structures less likely to generate these marked transmural heterogeneities that have been associated with ventricular arrhythmogenesis [3, 4, 40]. Yet, there are very few studies that have investigated atrial arrhythmogenesis in murine hearts [19, 42, 45]. Finally, to date, there has not been a study investigating atrial arrhythmogenicity specifically in KPQ mice modelling human LQT3. This investigation, therefore, tests the hypothesis that factors previously known to determine arrhythmias in ventricles do not necessarily determine atrial arrhythmogenicity. It demonstrated for the first time contrasting atrial electrophysiological properties of WT and KPQ Langendorff-perfused mouse hearts modelling LQT3 syndrome and that drugs causing ventricular arrhythmogenecity do not have identical effects in atrial tissue in WT and KPQ mouse hearts.
Methods
Experimental animals
WT and heterozygote Scn5a+/Δ (KPQ) mice breeding pairs both with a 129 genetic background were set up and their offspring genotyped, weaned and used for this study. Mice aged 5 to 9 months were used in all experiments. All mice were maintained in an animal house at room temperature, subjected to 12 h light–dark cycle, and had a free access to sterile rodent chow and water at all times. All procedures were in accordance with the UK Animals (Scientific Procedures) Act 1986.
Experimental preparations
Langendorff-perfused whole heart preparations were used for electrophysiological studies, as previously described by Balasubramaniam et al. [5] and Head et al. [12]. Animals were randomly selected and killed in accordance with Schedule 1: Animals (Scientific Procedures) Act 1986 by cervical dislocation. Whole hearts were then quickly excised and submerged in ice-cold bicarbonate-buffered Krebs–Henseleit solution prepared from (mM): 119 NaCl, 25 NaHCO3, 4.0 KCl, 1.2 KH2PO4, 1.0 MgCl2, 1.8 CaCl2, 10 glucose and 2.0 sodium pyruvate (pH 7.4). The solution was then bubbled with 95% O2–5% CO2 (British Oxygen Company, Manchester, UK). The aorta was cannulated and then sutured to a 21-gauge custom-made cannula prefilled with ice-cold buffer while the heart was fully submerged in Krebs–Henseleit solution. The preparation was then mounted onto a perfusion apparatus which allowed a warmed (37°C) Krebs–Henseleit buffer solution to pass through the heart via the aorta at a constant flow rate of 2.5–3.0 ml min−1 (Watson–Marlow Bredel Peristaltic pumps, model 505S, Falmouth, Cornwall, UK). Before entering the heart, the perfusate was passed through 200- and 5-μm filters (Millipore UK, Watford, UK). Hearts that regained a pink colouration and spontaneous rhythmic contractions upon warming were used in the experiments.
Electrophysiological procedures
On warming and regaining a healthy pink colour and a normal heart beat, hearts were allowed to spontaneously beat for at least 10 min before proceeding with programmed electrical stimulation (PES) [30, 31] and burst pacing (BP) as described below. Paired (1 mm inter-pole spacing) platinum stimulating and recording electrodes were positioned against the epicardial surfaces of the right and the left atrium, respectively, which allowed bipolar electrograms (BEGs) to be recorded from atria and ventricles. BEGs were amplified, band-pass filtered (30 Hz–1 kHz; Gould 2400S, Gould-Nicolet Technologies, Ilford, Essex, UK) and digitised (1401plus, Cambridge Electronic Design, Cambridge, UK) at a frequency of 5 kHz. In addition, a third electrode was placed on the basal epicardial surface of the left ventricle in order to obtain a simultaneous BEG signal from the left ventricle only. The procedure of simultaneous atrial and ventricular recording made it possible to separate contributions to electrical activity from each structure in the observed BEG recordings.
A paired platinum pacing electrode connected to Grass S48 stimulator (Grass-Telefactor, UK, Slough, UK) was used to pace the heart at the right atrium at 3.5 V. On positioning of all electrodes, each heart was briefly paced at a frequency of 8 Hz using 2 ms square-wave stimuli to ensure consistent capture and good quality signals. Upon acquiring a clear signal, the position of the stimulation and recording electrodes was fixed.
To assess tendencies to atrial arrhythmogenesis, two procedures were used. Firstly, each heart was subjected to PES. This consisted of drive trains of eight paced S1 beats at 8 or 10 Hz that were then followed by S2 extrastimuli every ninth beat. The S1–S2 interval was initially equal to the pacing interval and then reduced by 1 ms between successive drive trains until the preparation became refractory and no electrogram could be elicited by the S2 stimulus, or arrhythmic episode was initiated. If arrhythmia supervened, pacing was terminated in order to confirm its persistence or otherwise. Each heart was first subjected to PES stimulation conducted at 8 Hz; this was repeated three times and followed by PES stimulation conducted at 10 Hz that was also repeated three times.
Secondly, each heart was exposed to a modified BP protocol that was previously used to induce atrial arrhythmias in mice [19]. The alternative protocol involved delivering 25 trains of 50 stimuli imposed at high frequency (20 ms between each stimulus) to the right atrium of a spontaneously beating heart. Each successive train was delivered as soon as the heart had recovered from the preceding stimulation by returning to normal sinus rhythm.
In addition to BEGs, atrial monophasic action potentials (MAPs) were recorded from atrioventricular (AV)-node-ablated hearts. The left atrial appendage was pressed against the clip using an epicardial MAP electrode (Linton Instruments, Harvard Apparatus). This resulted in a recording of atrial MAP signals. The heart was then paced at 8 and 10 Hz stimulation frequencies using a pacing electrode described above. MAPs were amplified, band-pass filtered (0.5 Hz–1 kHz, Gould 2400S, Gould-Nicolet Technologies, Ilford, Essex, UK) and digitised (1401plus, Cambridge Electronic Design, Cambridge, UK) at a frequency of 5 kHz.
The abovementioned electrophysiological procedures were recorded in the presence and absence of pharmacological intervention. Only arrhythmias of more than six cycles in duration were included in the analysis. Atrial MAPs included in analysis satisfied previously described criteria including a rapid upstroke phase with consistent amplitude, a smooth contoured repolarisation phase and a stable baseline [8]. APDs were determined at 90% repolarisation level, where full repolarisation to baseline level was defined as 100% and the tip of each MAP upstroke as 0% repolarisation.
Data analysis and statistics
BEGs were captured and analysed using Spike5 software (Cambridge Electronic Design, Cambridge, UK). All data were imported into Microsoft EXCEL which was also used to construct graphs and figures. All data are expressed as means ± SEM. The sets of data were compared using ANOVA (SPSS software) and P values less than 0.05 were considered significant.
Reagents
Flecainide and quinidine (Sigma–Aldrich, Poole, UK) were dissolved in doubly distilled water to make 1.0-mM stock solutions. Final drug concentrations to be used in the experiments (1 and 3 μM for both flecainide and quinidine) were determined by testing five different concentrations (100 nM, 1, 3, 5 and 10 μM) of the drugs. Final drug concentrations were prepared by dilution of 1.0-mM stock solutions in buffer to 10 μM first and then further dilutions to the required concentrations. Flecainide was stored in the refrigerator at 4°C between experiments and quinidine at room temperature with aluminium foil wrapped around a tightly sealed container to prevent light degradation. Hearts were perfused with drugs for 15–20 min prior to PES and BP.
Results
KPQ hearts show lower incidences and shorter durations of atrial arrhythmias under both testing procedures
Two techniques were used to assess atrial arrhythmogenic tendency in each WT (n = 16; Figs. 1 and 2) and genetically modified Scn5a+/Δ (KPQ) murine heart (n = 16) using BEG recordings. First, three cycles of a PES procedure conducted at a pacing frequency of 8 Hz imposed trains of eight pacing (S1) stimuli each followed by a ninth S2 extra stimulus at progressively decreasing S1S2 intervals (exemplified by Fig. 1A) until either atrial refractoriness was reached (Fig. 1B) or atrial arrhythmogenesis took place (Fig. 1C). The procedure was then similarly repeated three times at a pacing frequency of 10 Hz. This procedure permitted both assessment of atrial arrhythmogenic tendency and determination of atrial RPs. Secondly, an atrial BP procedure delivered 25 trains of 50 S1 stimuli at a high frequency (corresponding to a S1S1 coupling interval of 20 ms) to the right atrium of a spontaneously beating heart (Fig. 2A). This was followed by an assessment for either an immediate return to sinus rhythm (Fig. 2B) or the onset of atrial arrhythmias (Fig. 2C). Each successive train was delivered once the heart had regained its normal sinus rhythm. The present protocols, thus, differed from our previous studies on ventricular arrhythmogenesis [12, 27, 39, 40] in requiring hearts to be paced from the atria rather than the ventricles. In some hearts, such higher pacing rates resulted in a gradual development of an AV block particularly at the higher (10 Hz) pacing rate. Nevertheless, withdrawal of the regular pacing allowed a resumption of intrinsic activity and permitted restoration of normal 1:1 AV conduction. Such AV block was also observed during both the PES and the BP procedures. These findings could relate to longer refractoriness in the AV node.

Assessment of atrial arrhythmogenesis using a programmed electrical stimulation procedure. Simultaneously recorded atrial (A, B and C, a) and ventricular (A, B and C, b) BEG traces obtained from a typical WT heart in response to pacing conducted at a 10 Hz stimulation frequency. The single vertical markers indicate the timing of S1 stimuli (stim S1), the double vertical markers represent S2 extra-stimuli (stim S2). A (a), Typical data obtained at the beginning of the PES procedure at a longer S1S2 interval. A succession of three S1 stimulation artefacts and their respective evoked atrial (“A”) and ventricular (“V”) electrograms are succeeded by the extrastimulus (S2) artefact and its resulting atrial electrogram (“A2”) which is then followed by the first of eight S1 stimulation artefacts and its corresponding “A” and “V” electrograms. A, b shows simultaneously recorded signals from the left ventricle and excludes the atrial contributions. B (a) Typical data obtained when atrial refractoriness is observed. The third S1 stimulation artefact is closely followed by a S1 evoked “A” electrogram and S2 stimulation artefact. It is clearly seen that the S2 premature stimulus failed to produce a corresponding “A2” atrial signal which indicates atrial refractoriness. The signal following the S2 stimulation artefact is a “V” electrogram which is confirmed by the corresponding “V” electrogram in the bottom trace (B, b). C (a), Typical data showing initiation of atrial arrhythmogenic episode (AA) in response to a premature S2 stimulus. The S1 stimulation artefact is closely followed by a S1 evoked “A” electrogram and S2 stimulation artefact which is in turn succeeded by a “V” electrogram and AT. The bottom trace (C, b) confirms the atrial origin of the tachycardia as no ventricular activity is observed at the time when atrium is tachycardic at the top trace (C, a)

Assessment of atrial arrhythmogenesis using atrial burst pacing. Typical baseline sinus rhythm recording from a WT heart, where each regularly occurring atrial (“A”) signal is followed by a corresponding ventricular (“V”) signal (A). A train of 50 high frequency S1 stimuli results in either an immediate return to a normal sinus rhythm (B) or initiation of an atrial arrhythmic episode (AA) with irregular ventricular activity (C)
Table 1 demonstrates lower levels of atrial arrhythmogenic tendency in KPQ (n = 16) compared to WT hearts (n = 16) with all three test procedures. Firstly, all 16 WT hearts showed at least one arrhythmic episode during PES conducted at stimulation frequencies of either 8 or 10 Hz. In contrast, only seven KPQ hearts displayed such arrhythmogenic tendency with the same procedures. Fisher’s exact test accordingly confirmed highly significant differences for atrial arrhythmogenicity between the two groups for both PES procedures (P < 0.001 in both cases). BP was less effective in inducing arrhythmogenesis in either group. Nevertheless, it also yielded clear-cut differences between numbers of WT (14 out of 16) and KPQ hearts (one out of 16) showing atrial arrhythmias, similarly giving highly significant differences between the two experimental groups (P < 0.001). Secondly, where arrhythmic episodes did occur, they showed longer durations in the WT than in the KPQ hearts through all three procedures, but with a larger spread in values particularly in the WT group, which is reflected in the SEM. Therefore, it was necessary to perform further breakdowns of the arrhythmic frequencies. These analyses that followed provided closer measures of the mean incidence of atrial arrhythmias lasting six or more cycles for each heart and their severity.
A third measure describing the mean overall incidence of atrial arrhthymogenesis was, therefore, obtained for each heart in the WT and mutant groups subject to each of the test procedures (Fig. 3). The incidence of atrial arrrhythmogenesis following each pulse train during the three cycles of PES at 8 and 10 Hz, respectively (Fig. 3A and B), as well as during the 25 burst pacing procedures were obtained for each heart (Fig. 3C). This permitted mean values to be obtained for each experimental group.

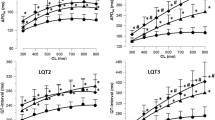
Overall and average incidence of atrial arrhythmic episodes (AAs) per heart in WT and KPQ preparations. Overall (a in A–C) and average (b in A–C) incidence of AAs per heart in WT (empty bars) and KPQ hearts (filled bars) for PES conducted at 8 Hz (A, a and b) and 10 Hz (B, a and b) stimulation frequencies and BP (C, a and b). The results of one-way ANOVA for correlated samples are shown (*P < 0.05, **P < 0.005 and ***P < 0.0005) and compare average and overall incidence of AA per heart between WT (n = 16) and KPQ (n = 16) groups
Figure 3A demonstrates the WT murine hearts (n = 16) showed significantly higher mean overall incidences of arrhythmic episodes per heart than KPQ (n = 16) hearts during PES whether at 8 Hz (P = 0.01; Fig. 3A, a) or 10 Hz (P < 0.0001; Fig. 3B, a) stimulation frequencies and during BP (P = 0.0007; Fig. 3C, a). Furthermore, arrhythmic episodes were significantly more frequent in WT hearts paced at the higher frequency during PES (P = 0.007); this was not the case for KPQ hearts (P > 0.05).
Finally, such arrhythmic episodes were sorted by duration (atrial arrhythmia lasting ≤ 1 s; 1.1–5.0 s; 5.1–10 s; and 10.1 s and longer). This provided mean incidences of atrial arrhythmias within each category within the two, WT (n = 16) and KPQ (n = 16) experimental groups.
Figure 3 also shows that for both, PES and BP, procedures, and both experimental groups, a significant proportion of atrial arrhythmias observed fell into a category with the lowest range of arrhythmic durations (episodes lasting ≤ 1 s; Fig. 3A–C, panel b). However, the mean incidences of arrhythmic episodes per heart for this category was significantly lower in the KPQ than the WT group whether PES was conducted at stimulation frequencies of 8 Hz (P = 0.03) and 10 Hz (P < 0.0001) and BP (P = 0.02). The mean incidence of arrhythmic episodes per heart for arrhythmias lasting between 1.1–5.0 and 5.1–10.0 s was also significantly lower in KPQ hearts for the BP procedure (P = 0.03 for both), and for arrhythmias lasting 10.1 s and longer for PES conducted at 8 Hz (P = 0.05). Finally, no KPQ hearts displayed arrhythmias lasting more than 5.1 s whereas we observed a total number of 24 separate episodes of atrial arrhythmias of this duration; these occurred in six WT hearts during both experimental procedures.
Flecainide and quinidine both inhibit atrial arrhythmogenesis
The experiments then went on to compare the effects of quinidine and flecainide on atrial arrhythmogenesis. This yielded patterns of action that contrasted with previous reports of these agents on ventricular arrhythmogenesis in the same murine system [39, 40].
Table 2 summarises the number of hearts in each group that showed one or more arrhythmic episodes during any one of the adopted test procedures. It shows that in WT hearts, 1 μM flecainide exerted a significant reduction in arrhythmogenic incidence during PES conducted at a frequency of 10 Hz and during BP (P < 0.01 in both cases; Fisher’s exact test); 3 μM flecainide produced significant effects through all the tests that were imposed (P < 0.001 for all three procedures). In KPQ hearts, 1 μM flecainide completely abolished arrhythmogenic phenomena through all the tests employed, with statistically significant effects obtained with PES conducted at an 8-Hz stimulation frequency (P < 0.05); 3 μM flecainide resulted in a loss of capture in the KPQ preparations which precluded detailed study.
Tables 3 and 4 summarise the effects of flecainide and quinidine, respectively, on mean overall incidences of atrial arrhythmic episodes per heart through the WT or KPQ hearts that were studied. In the WT preparations, flecainide (1 μM) reduced the average total incidences of arrhythmic episodes per heart whether these were undergoing PES at stimulation frequencies of both 8 Hz (P = 0.07) and 10 Hz (P = 0.002), and BP (P = 0.05) and higher (3 μM) concentrations completely abolished all such arrhythmic phenomena (P = 0.03, P < 0.0001 and P = 0.01, respectively). KPQ preparations required lower (1 μM) flecainide concentrations to achieve such a complete abolition of arrhythmias (P = 0.02, P = 0.06) with PES at 8 and 10 Hz; the BP protocol resulted in low mean total incidences of atrial arrhythmic episodes per heart in both control and treated groups, and this accounts for the lack of significance between the two results (P = 0.34).
Table 3 also provides a summary of the effects of flecainide on the breakdown of arrhythmic episodes sorted by the four arrhythmic durations introduced above. In WT hearts, flecainide whether applied at 1 or 3 μM, significantly reduced mean incidences of arrhythmic episodes lasting ≤1.0 s, during PES whether conducted at stimulation frequencies of 8 Hz (P = 0.03 and P = 0.02, respectively) or 10 Hz (P = 0.002 and P = 0.0003, respectively). In the KPQ group, flecainide similarly reduced the incidence of such episodes lasting ≤1.0 s: 1 μM rather than 3 μM flecainide sufficed to abolish all atrial arrhytmias whether PES was conducted at 8 Hz (P = 0.04) and 10 Hz (P = 0.02) stimulation frequencies.
BP resulted in relatively low incidences of arrhythmic episodes per heart in both WT and KPQ whether in the presence or the absence of flecainide; the only one out of six hearts that displayed a short (0.24 s) atrial arrhythmic episode failed to show any with 1 μM flecainide.
In common with flecainide, quinidine was anti-arrhythmic in both KPQ and WT hearts. Table 5 confirms that in the absence of added agent WT hearts were more arrhythmogenic than KPQ preparations, whether for PES conducted at 8 Hz (P = 0.007), 10 Hz (P = 0.01) and BP (P = 0.008). In WT hearts, quinidine reduced the number of WT hearts showing at least one arrhythmic episode whatever the procedure, with 3 μM more potent than 1 μM, to give a significant difference between test and control conditions with PES conducted at 10 Hz (P = 0.002). KPQ hearts showed low incidences of arrhythmogenesis both before and after quinidine treatment, which made distinguishing the incidences of hearts showing atrial arrhythmias difficult. Nevertheless, quinidine tended to diminish this incidence, with none of the KPQ hearts displaying arrhythmias after addition of 3 μM quinidine. BP proved to be less effective in inducing arrhythmogenesis in both experimental groups.
Table 4 shows the mean overall incidence of atrial arrhythmic episodes per heart in WT and KPQ groups in the presence and absence of 1 and 3 μM quinidine. In WT hearts, the presence of 1 μM quinidine significantly reduced the average total incidence of arrhythmic episodes per heart only for PES performed at 10 Hz (P = 0.01), while 3 μM quinidine reduced the average total incidence of arrhythmic episodes per heart for PES conducted at stimulation frequencies of 8 Hz (P = 0.008), 10 Hz (P < 0.0001) and BP (P = 0.02). The control mean total incidence of arrhythmic episodes per heart in KPQ hearts for a PES protocol was very small when compared to that in WT preparations, and no atrial arrhythmogenesis was detected with a BP procedure. Addition of quinidine did not result in any significant changes in the mean total incidence of arrhythmic episodes per heart for either BP or PES.
Table 4 also summarises the effect of quinidine on mean incidence of arrhythmic episodes per heart for each of the four different categories of arrhythmic durations. Quinidine (3 μM) significantly reduced the appearance only of arrhythmic episodes lasting ≤1 s for PES conducted at both 8 Hz (P = 0.03) and 10 Hz (P = 0.0007) stimulation frequencies in WT hearts, but not for BP. Quinidine (1 μM) in WT hearts had no significant effect on the mean incidence of arrhythmic episodes per heart for any of the four different categories for either PES or BP. Quinidine (1 and 3 μM) in KPQ preparations did not have any significant effect on the mean incidence of arrhythmic episodes per heart for any of the four different categories for either PES or BP.
Atrial monophasic action potential durations are longer in KPQ than in WT hearts
Figure 4 illustrates the effects of flecainide and quinidine on atrial action potential durations at the 90% repolarisation level (APD90) in WT and KPQ hearts. In these experiments, hearts were regularly paced at 8 Hz; at the faster pacing frequency of 10 Hz, it was often not possible clearly to demonstrate full recovery of the action potential to baseline, which was necessary for a full characterisation of APD90 values. It displays action potential waveforms in WT (A) and KPQ hearts (B), before (a) and following addition of flecainide (b) and quinidine (c). Flecainide 3 μM (Ab and open bars in C) significantly prolonged APD90 in WT hearts (P < 0.0001) from 22.1 ± 0.4 (n = 13, seven hearts) to 27.3 ± 0.8 (n = 13, six hearts) but failed to ensure consistent capture in KPQ hearts. A lower concentration of 1 μM flecainide did not change APD90 in KPQ hearts, with values for mean APD90 being 24.5 ± 0.3 (n = 17, ten hearts) before and 23.9 ± 0.7 (n = 11, seven hearts) after addition of the drug (Bb and open bars in C). Monophasic action potential waveforms (MAPs) recorded during regular pacing at 8 Hz in WT and KPQ hearts both significantly (P < 0.0001 for both groups) increased in duration on addition of 3 μM quinidine (c, A and B, respectively, black bars in C) from 22.1 ± 0.4 (n = 13, seven hearts) to 31.0 ± 0.6 (n = 8, four hearts) and 24.5 ± 0.3 (n = 17, ten hearts) to 32.3 ± 0.6 (n = 7, four hearts), respectively. Figure 4C also shows that control mean APD90 was significantly (P < 0.0001 (#)) longer in KPQ (24.5 ± 0.3 (n = 17, ten hearts)) when compared to WT (22.1 ± 0.4 (n = 13, seven hearts)) hearts.

Effect of flecainide and quinidine on atrial APDs in WT and KPQ hearts. MAPs recorded during regular pacing at 8 Hz in WT (A) and KPQ (B) hearts before (a, A and B) and after addition of 1 μM (B, b) and 3 μM (A, b) flecainide and 3 μM quinidine (c, A and B). The bar chart (C) summarises effect of 1 μM flecainide (open bars; KPQ) and 3 μM flecainide (WT) and 3 μM quinidine (black bars) on mean APD90 of both experimental groups. Stars represent significantly different results (*P < 0.0001) between control (dashed bars) and drug-treated mean APD90 within each experimental group, while # indicates significantly different control mean APD90 between WT and KPQ hearts (#P < 0.0001)
KPQ hearts have longer atrial refractory periods than WT hearts
In contrast to previous studies of ventricular arrhythmogenesis [39, 40], the present experiments demonstrated that KPQ hearts showed a reduced propensity to atrial arrhythmogenesis compared to WT, and this tendency was reduced further by both flecainide and quinidine in both genetic variants. Yet KPQ hearts also showed increased atrial APD90 values compared to WT hearts. Furthermore, flecainide and quinidine exerted anti-arrhythmic effects in both KPQ and WT. Yet flecainide also increased APD90 in WT, and both flecainide and quinidine increased APD90 in KPQ hearts. However comparisons of refractory periods in KPQ and WT hearts provided possible correlates with their contrasting arrhythmogenic properties. Thus, Fig. 5 shows significantly longer mean AERPs in KPQ hearts compared to WT hearts when these were subject to PES conducted at either 8 or 10 Hz stimulation frequencies (P < 0.0001 for both). AERP observed at 8 Hz in WT hearts was 14.0 ± 1.0 ms (n = 30, eight hearts) and in KPQ hearts was equal to 21.3 ± 0.7 ms (n = 41, 16 hearts). At 10 Hz, these values were 13.7 ± 0.8 ms (n = 33, 15 hearts) and 21.2 ± 0.8 (n = 43, 15 hearts) in WT and KPQ experimental groups, respectively.

Effect of flecainide on mean atrial effective refractory periods (AERP) in WT and KPQ hearts. AERPs observed during 8 Hz (A) and 10 Hz (B) PES before (dashed bars) and after addition of 1 μM flecainide (open bars) and 3 μM flecainide (black bars) to WT and KPQ hearts. The results of one-way ANOVA for correlated samples are shown (***P < 0.0001) and compare AERP between control and 1 μM flecainide and control and 3 μM flecainide for KPQ and WT groups, respectively. X marking represents loss of capture with 3 μM flecainide for KPQ preparations. The results comparing control AERP between WT and KPQ groups are also shown (P < 0.0001 (#))
Flecainide and quinidine both prolong the refractory periods in both WT and KPQ hearts
Figure 5 also shows effect of 1 and 3 μM flecainide on AERP in WT and KPQ hearts obtained during PES conducted at 8 Hz (A) and 10 Hz (B) stimulation frequencies. A highly significant increase in AERP from 14.0 ± 1.0 ms (n = 30, eight hearts) to 29.9 ± 2.5 (n = 14, five hearts) and from 13.7 ± 0.8 ms (n = 33, 15 hearts) to 45.3 ± 4.7 (n = 17, six hearts) was observed in WT hearts at 8 and 10 Hz, respectively, on addition of 1 μM flecainide (P < 0.0001 for both frequencies). Flecainide (3 μM) further significantly increased AERP to 58.4 ± 4.3 (n = 9, three hearts) and 75.0 ± 5.9 (n = 8, three hearts) at stimulation frequencies of 8 and 10 Hz, respectively (P < 0.0001). In addition, the differences in AERP between results from using 1 and 3 μM concentrations of the drug were also significant whether at stimulation frequencies of 8 Hz (P < 0.0001) or 10 Hz (P = 0.001). However, KPQ hearts showed a failure of consistent stimulus capture with 3 μM flecainide. AERP significantly increased in this group from 21.3 ± 0.7 ms (n = 41, 16 hearts) to 45.0 ± 2.7 (n = 17, six hearts) at 8 Hz, and from 21.2 ± 0.8 (n = 43, 15 hearts) to 55.5 ± 4.8 (n = 11, five hearts) at a 10 Hz stimulation frequency (P < 0.0001 for both frequencies) with 1 μM flecainide.
Figure 6 illustrates effects of 1 and 3 μM quinidine on AERP in WT and KPQ hearts obtained during PES conducted at 8 Hz (A) and 10 Hz (B) stimulation frequencies. In WT hearts, a significant increase in AERP from 13.7 ± 0.8 ms (n = 33, 15 hearts) to 19.4 ± 1.2 (n = 13, five hearts) on addition of 1 μM quinidine was only observed at 10 Hz (P < 0.001). AERP further increased to 53.7 ± 2.7 (n = 17, six hearts) in addition of 3 μM quinidine at this frequency (P < 0.0001). The same concentration of quinidine also significantly increased AERP from 14.0 ± 1.0 ms (n = 30, eight hearts) to 35.6 ± 3.5 (n = 19, six hearts) at an 8 Hz stimulation frequency in this experimental group (P < 0.0001). In addition, AERPs obtained at 1 and 3 μM concentrations of the drug were significantly different whether at 8 Hz (P = 0.03) and 10 Hz (P < 0.0001) stimulation frequencies. In KPQ hearts a highly significant increase in AERP from 21.3 ± 0.7 ms (n = 41, 16 hearts) to 37.2 ± 4.0 (n = 14, four hearts) and from 21.2 ± 0.8 ms (n = 43, 15 hearts) to 51.3 ± 8.6 (n = 12, four hearts) was observed at 8 and 10 Hz, respectively, in addition of quinidine 1 μM (P < 0.0001 for both frequencies). Quinidine (3 μM) also significantly increased AERP to 35.4 ± 1.8 (n = 15, five hearts) and 47.7 ± 3.7 (n = 15, five hearts) at 8 and 10 Hz, respectively when compared to control (P < 0.0001) in mutant hearts, doing so to similar extents as obtained with 1 μM quinidine for either frequency (P > 0.05).

Effect of quinidine on mean AERP in WT and KPQ hearts. AERPs observed during 8 Hz (A) and 10 Hz (B) PES before (dashed bars) and after addition of 1 μM quinidine (open bars) and 3 μM quinidine (black bars) to WT and KPQ hearts. The results of one-way ANOVA for correlated samples are shown (***P < 0.0001, **P < 0.001) and compare AERP between control and 1 μM quinidine and control and 3 μM quinidine for KPQ and WT groups, respectively. The results comparing control AERP between WT and KPQ groups are also shown (#P < 0.0001)
Arrhythmogenic tendency correlates with increased local critical intervals
Previous work on murine ventricular arrhythmogenecity [27] demonstrated increased risks of re-excitation when the APD90 exceeded the ventricular effective refractory period (VERP). We tested such a hypothesis for the atria studied here by comparing the above values of atrial APD90 and AERP in both WT and KPQ groups in the presence and the absence of flecainide and quinidine. We hypothesised that the risk of local re-excitation might be reflected in a critical interval (CI) given by the relevant difference (APD90–AERP).
Figure 7 illustrates typical action potential waveforms during regular pacing at 8 Hz recorded from WT (A) and KPQ (B) hearts with the corresponding APD90s (horizontal broken lines and clear shading), AERPs (horizontal solid lines and black shading), and local CIs (shading and hashing) shown below each one. Under control conditions, APD90 in WT (22.1 ± 0.4 ms) and KPQ (24.5 ± 0.3 ms) were significantly larger than the corresponding AERP (14 ± 1.0 and 21.3 ± 0.7 ms) (A and B, a). However, this difference was more significant in the WT group than in the KPQ group (P < 0.0001 and P = 0.005, respectively). This is reflected in WT hearts having significantly larger positive CI values when compared to KPQ hearts (8.1 and 3.2 ms, respectively; P < 0.001) which is clearly demonstrated in a larger shaded area on the WT group AP waveform (A and B, a). Flecainide significantly prolonged APD90 in WT but not in KPQ hearts and significantly increased AERP in both experimental groups (see findings above). The resulting negative CIs were −31.1 and −21.1 ms in WT and KPQ preparations, respectively (P < 0.0001 for both), indicating that the drug prolonged AERP to a much larger extent than APD90 in both groups. Accordingly, the solid vertical line (AERP) moved further to the right on action potential waveform while the position of the dashed vertical line (APD90) moved slightly to the right (WT) or remained virtually unchanged (KPQ). Quinidine significantly prolonged APD90 in both genetic variants (P < 0.0001 for both) moving the dashed line to the right. However, the drug also significantly prolonged AERP in the two experimental groups (P < 0.0001 for both), also moving the solid vertical line to the right. The resulting negative CIs were −4.6 (WT) and −3.1 ms (KPQ), and were significantly different from the control data (P < 0.001 for both groups). In contrast to flecainide, quinidine prolonged both APD90 and AERP similarly in the two groups, which resulted in smaller CI values that were still negative because the AERPs were longer than APD90.

Effect of flecainide and quinidine on mean critical interval in WT and KPQ hearts. Changes in atrial action potential duration at 90% repolarization (APD 90 ), atrial effective refractory period (AERP), and local critical interval (CI) in WT (A) and KPQ (B) hearts before (a, A and B) and after exposure to flecainide (b, A and B) and quinidine (c, A and B). MAP waveforms during regular stimulation at 8 Hz frequency in hearts before (a, A and B) and after exposure to flecainide (b, A and B) and quinidine (c, A and B) comparing APD90 (vertical dashed lines), AERP (vertical solid lines), and local CI (shading). Asterisks indicate CI values that are significantly (**P < 0.001; ***P < 0.0001) different before and after addition of a drug, and # those that are significantly (#P < 0.001) different between WT and KPQ hearts at baseline
Discussion
Human LQT3 syndrome results from ‘gain-of-function’ mutations in the SCN5A gene encoding the pore-forming α-subunit of the cardiac voltage-gated Na+ channel, producing incomplete Na+ channel inactivation [47]. This prolongs the cardiac APD in turn predisposing the myocardium to early EADs that can trigger life-threatening polymorphic ventricular tachyarrhythmias known as torsade de pointes [41, 44]. Patients with different LQTS variants demonstrated abnormalities in atrial physiology similar to those found in the ventricle in the form of prolonged APD and RP durations, and EADs triggering ‘atrial torsade de pointes’ [14]. However, atrial arrhythmias are uncommon in LQT3 patients [43].
The present experiments accordingly compared atrial arrhythmogenicity and its physiological basis in isolated perfused WT and Scn5a+/Δ (KPQ) murine hearts modelling human LQT3 for the first time. Experimental conditions thus differed from those used in some recent reports on atrial arrhythmogenesis of electrocardiographic studies in intact anaesthetised animals [29, 32, 37] as opposed to isolated perfused hearts. This may account for some of the differences in arrhythmogenecity seen in the control situations between the present and those previous studies. KPQ mice have previously been shown to provide useful cardiac models for the investigation of ventricular arrhythmogenesis in LQT3. Their hearts showed a marked susceptibility to such arrhythmias, in agreement with clinical findings [12]. Furthermore, pharmacological intervention using the cardiotropic agents flecainide and quinidine, respectively, alleviated and exacerbated such arrhythmogenicity [39, 40] likely through actions on inhomogeneities of APD through the thickness of the ventricular wall.
However, unlike the ventricle, the murine atrium is a thin-walled structure unlikely to generate such spatial heterogeneities between epicardial and endocardial layers. This difference would be consistent with distinct mechanisms for atrial and ventricular arrhythmogenesis. We initially assessed atrial arrhythmic tendency in WT and genetically modified KPQ Langendorff-perfused heart preparations in the presence and absence of the same pharmacological interventions as cited above using both PES and BP protocols. These techniques both showed that a significantly greater proportion of WT hearts were arrhythmogenic compared to KPQ hearts. In addition, where it occurred, the average duration of atrial arrhythmia was longer in WT when compared to KPQ hearts. Therefore, it appears that the KPQ mutation exerts a surprising ‘protective’ effect against atrial arrhythmogenesis. This is in sharp contrast to the findings concerning ventricular arrhythmogenecity.
Furthermore, we demonstrated that the pharmacological agents described above exerted differing actions on arrhythmogenecity in the atria when these were compared to their known ventricular effects whether in WT and KPQ mouse hearts. Thus, both flecainide and quinidine reduced or completely eliminated atrial arrhythmogenesis in KPQ and WT groups. This was in contrast to the action of flecainide in reducing and that of quinidine in increasing ventricular arrhythmogencity in KPQ hearts while having the opposite effects in WT preparations [39, 40].
To account for the differences observed in atria and ventricles of the same mouse model of LQT3, we went on to compare particular electrophysiological properties of the WT and KPQ hearts. Firstly, previous reports have implicated the ERP in atrial as opposed to ventricular arrhythmogenicity. Thus, decreases in atrial tissue refractoriness and APD are associated with atrial arrhythmogenesis [1, 6, 7, 22, 24, 33, 45, 48]. The present experiments similarly associated atrial arrhythmogenicity with short ERPs. Thus, arrhythmogenic WT hearts had significantly shorter ERPs when compared to the significantly less arrhythmogenic KPQ hearts. This is in contrast to findings in the ventricles in which non-arrhythmogenic WT hearts had shorter ventricular ERPs when compared to arrhythmogenic KPQ preparations [40]. Furthermore, we observed that flecainide and quinidine significantly increased the ERP in both WT and KPQ groups in parallel with their reducing or completely eliminating atrial arrhythmic episodes in both genetic variants. Again, this contrasts with the findings in the ventricles, in which the repolarisation gradient across the ventricular wall was a more consistent predictor of ventricular arrhythmogeneity than differences in ERP.
However, despite a number of studies showing that ventricular arrhythmogenicity in LQTS is associated with changes in electrical gradient across the ventricular wall [20, 35, 36], a recent study by Sabir et al. established another predictor of such arrhythmias in hypokaleimic mouse hearts [27]. It demonstrated increased tendencies to local re-excitation and arrhythmogenicity when recovery of the action potential, measured by its duration at 90% recovery (APD90), exceeded the corresponding recovery of excitability, as measured by the VERP. The difference between the two (APD90−VERP) was represented as a CI. Positive shifts in CI reflected a relatively proarrhythmic state, whereas negative shifts reflected an antiarrhythmic state. We tested this hypothesis in murine atria. Accordingly, we measured atrial effective refractory periods (AERPs) and APD90s both before and during pharmacological intervention to obtain atrial CIs for each test condition in both experimental groups. Our study demonstrated that in the absence of pharmacological agents the atria of arrhythmogenic WT hearts had larger APD90 values when compared to their AERP, thus giving a positive CI. Furthermore, such baseline recordings showed that WT hearts had a significantly larger positive CI value (P < 0.001) when compared to KPQ hearts. This correlated with greater atrial arrhythmic tendency of the WT preparations.
Furthermore, both flecainide and quinidine were anti-arrhythmic in either experimental group and produced significant negative shifts in CI, especially with flecainide. These findings fulfil expectations from similar reports of altered ventricular as opposed to atrial arrhythmogenecity under particular circumstances. Under normal conditions, local VERP closely corresponds to APD. Thus, previous studies in ventricles from a range of species reported that manoeuvres that alter APD also result in corresponding changes in VERPs [10, 17, 18]. During PES, a premature extrastimulus shortens RP to a larger extent than APD, which in turn can induce ventricular arrhythmias [18]. Conversely, Class I cardiotropic drugs could either suppress or initiate ventricular arrhythmogenesis by altering the ERP/APD ratio through selective changes in one or other parameter thereby modifiying post-repolarisation refractoriness (PRR) [13, 16]. They then would also be expected to exert corresponding effects on atrial arrhythmogenecity.
The present studies demonstrated that flecainide exerted anti-arrhythmic effects. That could also be correlated with the present findings concerning changes in CI. Thus, flecainide only significantly increased APD90 in WT but increased AERP in both WT and KPQ. The result was a negative CI, These results are in agreement with previous findings on the effect of flecainide in human atria. For example, Kirchholf et al (2005) [15] demonstrated that flecainide induced post recovery refractoriness by prolonging AERP more than APD90 in fibrillating human atrium and prevented arrhythmia reoccurrence after electrical cardioversion.
Quinidine also decreased arrhythmogenic tendency in both WT and KPQ hearts, although to a smaller extent than did flecainide. Quinidine increased both APD90 and AERP, but exerted a greater effect on AERP, thereby resulting in a negative CI value, again to a lesser extent than shown in the presence of flecainide. These findings parallel the effects of quinidine on ventricular arrhythmogenesis. Thus, quinidine can promote development of human ventricular arrhythmia especially during bradycardia by prolonging repolarisation (APD) and not refractoriness [23, 26]. On the other hand, once arrhythmia has been initiated after quinidine administration, it can often be suppressed by pacing the heart at high rates [26]. This can be attributed to it having a more marked effect on VERP than on APD with increasing heart rate resulting in a protective effect [23, 11].
In summary, this study compared for the first time the effects of flecainide and quinidine on the electrophysiological properties of WT and genetically modified (Scn5a+/Δ) mice lacking the same KPQ residues as LQT3 patients [25]. It appears that KPQ mutation, while predisposing to ventricular arrhythmias, has a ‘protective’ effect for atrial arrhythmogenicity. Therefore, our investigation shows that factors previously known to determine arrhythmias in ventricles do not necessarily determine atrial arrhythmogenic tendency. Nevertheless, the present findings bearing on atrial arrhythmogenecity could be correlated with corresponding criteria developed for ventricular arrhythmogenesis in one important respect: prolonging RP beyond APD may be beneficial for prevention of atrial arrhythmias. Therefore, agents that promote post recovery refractoriness and shift CI in the negative direction may be a good target for development of drugs aimed at preventing atrial arrhythmogenesis.
References
Allessie MA, Boyden PA, Camm AJ, Kléber AG, Lab MJ, Legato MJ, Rosen MR, Schwartz PJ, Spooner PM, Van Wagoner DR, Waldo AL (2001) Pathophysiology and prevention of atrial fibrillation. Circulation 103:769–777
Antzelevitch C (2001) Basic mechanisms of re-entrant arrhythmias. Curr opin cardiol 16:1–7
Antzelevitch C, Oliva A (2006) Amplification of spatial dispersion of repolarisation underlies sudden cardiac death associated with catecholaminergic polymorphic VT, long QT, short QT and Brugada syndromes. J Inter Med 259:48–58
Antzelevitch C, Shimizu W (2002) Cellular mechanisms underlying the long QT syndrome. Curr Opin Cardiol 17:43–51
Balasubramaniam R, Grace AA, Saumarez RC, Vandenberg JI, Huang CLH (2003) Electrogram prolongation and nifedipine-suppressible ventricular arrhythmias in mice following targeted disruption of KCNE1. J Physiol 552:535–546
Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang Y, Xu WY, Jin HW, Sun H, Su XY, Zhuang QN, Yang YQ, Li YB, Liu Y, Xu HJ, Li XF, Ma N, Mou CP, Chen Z, Barhanin J, Huang W (2003) KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science 299:251–254
Euler DE, Scanlon PJ (1987) Acetylcholine release by a stimulus train lowers atrial fibrillation threthold. Am J Physiol 253(4 Pt 2):H863–868
Fabritz L, Kirchhof P, Franz MR, Nuyens D, Rossenbacker T, Ottenhof A, Haverkamp W, Gunter B, Carmeliet E, Carmeliet P (2003) Effect of pacing and mexiletine on dispersion of repolarisation and arrhythmias in ΔKPQ SCN5A (long QT3) mice. Cardiovasc Res 57:1085–1093
Fox CS, Parise H, D’Agostino RB Sr, Lloyd-Jones DM, Vasan RS, Wang TJ, Levy D, Wolf PA, Benjamin EJ (2004) Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. JAMA 291:2851–2855
Franz MR, Chin MC, Sharkey HR, Griffin JC, Scheinman MM (1990) A new single catheter technique for simultaneous measurement of action potential duration and refractory period in vivo. J Am Coll Cardiol 16:878–886
Franz MR, Costard A (1988) Frequency-dependent effects of quinidine on the relationship between action potential duration and refractoriness in the canine heart in situ. Circulation 77(5):1177–1184
Head CE, Balasubramaniam R, Thomas G, Goddard CA, Lei M, Colledge WH, Grace AA, Huang CLH (2005) Paced electrogram fractionation analysis of arrhythmogenic tendency in ΔKPQ Scn5a mice. J Cardiovasc Electrophysiol 16:1329–1340
Kirchhof P, Degen H, Franz M, Eckardt L, Fabritz L, Milberg P, Laer S, Neumann J, Breithardt G, Haverkamp W (2003) Amiodarone-induced post-repolarization refractoriness suppresses induction of ventricular fibrillation. J Pharmacol Exp Ther 305:257–263
Kirchhof P, Eckardt L, Franz MR, Mönnig G, Loh P, Wedekind H, Schulze-Bahr E, Breithardt G, Haverkamp W (2003) Prolonged atrial action potential durations and polymorphic atrial tachyarrhythmias in patients with long QT syndrome. J Cardiovasc Electrophysiol 14:1034–1035
Kirchhof P, Engelen M, Franz MR, Ribbing M, Wasmer K, Breithardt G, Haverkamp W, Eckardt L (2005) Electrophysiological effects of flecainide and sotalol in the human atrium during persistent atrial fibrillation. Basic Res Cardiol 100(2):112–121
Kirchhof P, Fabritz L, Franz MR (1998) Post-repolarization refractoriness versus conduction slowing caused by class I antiarrhythmic drugs – antiarrhythmic and proarrhythmic effects. Circulation 97:2567–2574
Knollmann BC, Katchman AN, Franz MR (2001) Monophasic action potential recordings from intact mouse heart: Validation, regional heterogeneity, and relation to refractoriness. J Cardiovasc Electrophysiol 12:1286–1294
Koller BS, Karasik PE, Solomon AJ, Franz MR (1995) Relation between repolarization and refractoriness during programmed electrical stimulation in the human right ventricle. Implications for ventricular tachycardia induction. Circulation 91:2378–2384
Kovoor P, Wickman K, Maguire CT, Pu W, Gehrmann J, Berul CI, Clapham DE (2001) Evaluation of the role of I(KACh) in atrial fibrillation using a mouse knockout model. J Am Coll Cardiol 37:2136–2143
Milberg P, Reinsch N, Wasmer K, Monnig G, Stypmann J, Osada N, Breithardt G, Haverkamp W, Eckardt L (2005) Transmural dispersion of repolarization as a key factor of arrhythmogenicity in a novel intact heart model of LQT3. Cardiovasc Res 65:397–404
Moe GK (1962) On the multiple wavelet hypothesis of atrial fibrillation. Int Pharmacodyn Ther 140:183–188
Nadeau RA, Roberge FA, Billette J (1970) Role of the sinus node in the mechanism of cholinergic atrial fibrillation. Circ Res 27:129–138
Nademanee K, Stevenson WG, Weiss JN, Frame VB, Antimisiaris MG, Suithichaiyakul T, Pruitt CM (1990) Frequency-dependent effects of quinidine on the ventricular action potential and QRS duration in humans. Circulation 81(3):790–796
Nattel S (2002) New ideas about atrial fibrillation 50 years on. Nature 415:219–226
Nuyens D, Stengl M, Dugarmaa S, Rossenbacker T, Compernolle V, Rudy Y, Smits JF, Flameng W, Clancy CE, Moons L, Vos MA, Dewerchin M, Benndorf K, Collen D, Carmeliet E, Carmeliet P (2001) Abrupt rate accelerations or premature beats cause life-threatening arrhythmias in mice with long-QT3 syndrome. Nat Med 7:1021–1027
Roden DM, Thompson KA, Hoffman BF, Woosley RL (1986) Clinical features and basic mechanisms of quindine-induced arrhythmias. J Am Coll Cardiol 81:73A–78A
Sabir IN, Fraser JA, Killeen MJ, Grace AA, Huang CLH (2007) The contribution of refractoriness to arrhythmic substrate in hypokalemic Langendorff-perfused murine hearts. Pflugers Arch 454(2):209–222
Salama G, London B (2007) Mouse models of long QT syndrome. J Physiol 578:43–53
Sampson KJ, Terrenoire C, Cervantes DO, Kaba RA, Peters NS, Kass RS (2008) Adrenergic regulation of a key cardiac potassium channel can contribute to atrial fibrillation: evidence from an IKs transgenic mouse. J Physiol 586(2):627–637
Saumarez RC, Chojnowska L, Derksen R, Pytkowski M, Sterlinski M, Huang CL, Sadoul N, Hauer RN, Ruzyllo W, Grace AA (2003) Sudden death in noncoronary heart disease is associated with delayed paced ventricular activation. Circulation 107:2595–2600
Saumarez RC, Grace AA (2000) Paced ventricular electrogram fractionation and sudden death in hypertrophic cardiomyopathy and other non-coronary heart diseases. Cardiovasc Res 47:11–22
Sawaya SE, Rajawat YS, Rami TG, Szalai G, Price RL, Sivasubramanian N, Mann DL, Khoury DS (2007) Downregulation of connexin40 and increased prevalence of atrial arrhythmias in transgenic mice with cardiac-restricted overexpression of tumor necrosis factor. Am J Physiol Heart Circ Physiol 292(3):H1561–1567
Schuessler RB, Grayson TM, Bromberg BI, Cox JL, Boineau JP (1992) Cholinergically mediated tachyarrhythmias induced by a single extrastimulus in the isolated canine right atrium. Circ Res 71:1254–1267
Shimizu W, Antzelevitch C (1999) Cellular and ionic basis for T-wave alternans under long-QT conditions. Circulation 99:1499–1507
Shimizu W, Antzelevitch C (1998) Cellular basis for the ECG features of the LQT1 form of the long-QT syndrome: effects of beta-adrenergic agonists and antagonists and sodium channel blockers on transmural dispersion of repolarization and torsade de pointes. Circulation 98:2314–2322
Shimizu W, Antzelevitch C (1997) Sodium channel block with mexiletine is effective in reducing dispersion of repolarization and preventing torsade des pointes in LQT2 and LQT3 models of the long-QT syndrome. Circulation 96:2038–2047
Sood S, Chelu MG, van Oort RJ, Skapura D, Santonastasi M, Dobrev D, Wehrens XH (2008) Intracellular calcium leak due to FKBP12.6 deficiency in mice facilitates the inducibility of atrial fibrillation. Heart Rhythm 5(7):1047–1054
Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, Moss AJ, Schwartz PJ, Towbin JA, Vincent GM, Keating MT (2000) Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 102:1178–1185
Stokoe KS, Balasubramaniam R, Goddard CA, Colledge WH, Grace AA, Huang CLH (2007) Effects of flecainide and quinidine on arrhythmogenic properties of Scn5a+/- murine hearts modelling the Brugada syndrome. J Physiol 581(Pt 1):255–275
Stokoe KS, Thomas G, Goddard CA, Colledge WH, Grace AA, Huang CLH (2007) Effects of flecainide and quinidine on arrhythmogenic properties of Scn5a+/Δ murine hearts modelling long QT syndrome 3. J Physiol 578(Pt 1):69–84
Tan HL, Hou CJ, Lauer MR, Sung RJ (1995) Electrophysiologic mechanisms of the long QT interval syndromes and torsade de pointes. Ann Intern Med 122:701–714
Temple J, Frias P, Rottman J, Yang T, Wu Y, Verheijck EE, Zhang W, Siprachanh C, Kanki H, Atkinson JB, King P, Anderson ME, Kupershmidt S, Roden DM (2005) Atrial fibrillation in KCNE1-null mice. Circ Res 97:62–69
Vincent GM (2003) Atrial arrhythmias in the inherited long QT syndrome: Laboratory quirk or clinical arrhythmia. J Cardiovasc Electrophysiol 14:1034–1035
Volders PG, Vos MA, Szabo B, Sipido KR, de Groot SH, Gorgels AP, Wellens HJ, Lazzara R (2000) Progress in the understanding of cardiac early afterdepolarizations and torsades de pointes: Time to revise current concepts. Cardiovasc Res 46:376–392
Wakimoto H, Maguire CT, Kovoor P, Hammer PE, Gehrmann J, Triedman JK, Berul CI (2001) Induction of atrial tachycardia and fibrillation in the mouse heart. Cardiovasc Res 50:463–473
Wang Q, Shen J, Li Z, Timothy K, Vincent GM, Priori SG, Schwartz PJ, Keating MT (1995) Cardiac sodium channel mutations in patients with long QT syndrome, an inherited cardiac arrhythmia. Hum Mol Genet 4:1603–1607
Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT (1995) SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 80:805–811
Wijffels MCEF, Kirchhof CJHJ, Dorland R, Allessie MA (1995) Atrial fibrillation begets atrial fibrillation: A study on awake chronically instrumented goats. Circulation 92:1954–1968
Acknowledgements
We thank the James Baird Fund, the Frank Elmore Fund, the Medical Research Council, the Wellcome Trust and the British Heart Foundation, UK, and the National Natural Science Foundation of China (NSFC 30371571 and 30672209), PRC for their generous support.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Dautova, Y., Zhang, Y., Sabir, I. et al. Atrial arrhythmogenesis in wild-type and Scn5a+/Δ murine hearts modelling LQT3 syndrome. Pflugers Arch - Eur J Physiol 458, 443–457 (2009). https://doi.org/10.1007/s00424-008-0633-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-008-0633-z