
Abstract
SARS-CoV-2, which led to the 2020 global pandemic, is responsible for the Coronavirus Disease 2019 (COVID-19), a respiratory illness, and presents a tropism for the central nervous system. Like most members of this family, the virus is composed of structural and non-structural proteins (NSPs). The non-structural proteins are critical elements of the replication and transcription complex (RTC), as well as immune system evasion. Through hijacking the endoplasmic reticulum (ER) membrane, NSPs help the virus establish the RTC, inducing ER stress after membrane rearrangement and causing severe neuronal disturbance. In this review, we focus on the role of Nsp3, 4, and 6 in intracellular membrane rearrangement and evaluate the potential disruption of the central nervous system and the neurodegeneration which it could trigger. Studies of these NSPs will not only bring to light their specific role in viral infection but also facilitate the discovery of novel targeted drugs.
Similar content being viewed by others

Background
Since the discovery of the first coronaviruses (CoV) by Tyrrell and Bynoe in 1965 [1], more strains have emerged. Only two circulating human coronaviruses (HCoVs), HCoV-229E [2] and HCoV-OC43 [3], were described as the frequent cause of the common cold until 2002–2003 when the severe acute respiratory syndrome (SARS)-CoV spread into the human population in China [4, 5]. Then, two new coronaviruses, CoV NL63 in 2004 (alphacoronavirus) [6] and HKU1 in 2005 (betacoronavirus) [7], were discovered. Another coronavirus, the MERS-CoV, emerged in the Middle East and engenders lower respiratory tract infections with high mortality [8].
HCoVs were considered relatively harmless until the SARS outbreak in 2003, where they appeared clinically for the first time as a severe respiratory illness with high mortality and morbidity. Coronaviruses have been identified to infect a wide variety of animals, birds, and mammals, including mice, cats, bats, dogs, pangolins, as well as humans, with a diversity of clinical presentation including respiratory tract infection and gastroenteritis. The outbreaks of SARS and MERS and the frequent re-introduction of coronaviruses in the human population increased the awareness of the danger of emergence of other zoonotic coronaviral infections. These epidemics indicate the need to develop antiviral compounds to combat coronavirus infections. However, gaps still exist in the knowledge of the molecular pathogenesis and transmissivity of this family of viruses.
CoVs belong to the Nidovirales order—Coronaviridae family—and received their names from the unique “crown-like” spikes that are visible on the outer membrane of the virions. Coronaviruses are large enveloped, single-stranded positive-sense RNA viruses that are broken down into four classes: Alpha, Beta, Gamma, and Deltacoronavirus. Coronaviruses are the largest RNA viruses known with a genome ranging between 27 and 32 kb. Like all the Nidovirales members, Coronaviruses have a highly conserved genomic organization, unique enzymatic activities, and express non-structural genes by ribosomal frameshifting.
Viral replication and structure
SARS-CoV-2 contains a 30 kb genome encoding 14 open reading frames (Orfs) where each encodes several proteins (structural and non-structural) (Fig. 1). The first non-structural proteins (NSPs) encoded by Orf1a/Orf1ab are Papain-like proteinase (PL proteinase, Nps3) and 3-chymotrypsin-like proteinase (3CLPro protease) [9]. The PL proteinase Nsp3 cleaves NSPs 1 to 3 [10] and the 3CLPro proteinase slices the C-terminus from Nsp4 to Nsp16 in all coronaviruses [11].

Structure of SARS-CoV-2. Schematic representation of SARS-CoV-2 structure where all the open reading frames (Orfs) and non-structural proteins (NSPs) positions are indicated. Cleavage sites processed by the papain-like proteinase are indicated by blue arrowheads and the 3C-like proteinase are indicated by red arrowheads
Orf1a gene is expressed early and translated into a polyprotein that will be cleaved into 11 non-structural proteins (Nsp1-11) by the papain-like activity of Nsp3 [12] or by Nsp5, which possess a chymotrypsin-like protease activity [13]. Orf1ab is in a different frame from Orf1a and the virus uses programmed-1 ribosomal frameshifting (-1 PRF) to synthesize it.
Replication and transcription of the virus happen within a replication/transcription complex (RTC) encoded by the virus with NSPs as primary constituents. Nearly all RTC elements are encoded by the large replicase gene that consists of Orf1a and Orf1ab. The early formation of the RTC is an essential step in the SARS-CoV-2 life cycle to safeguard viral genome replication and to synthesize subgenomic mRNA.
The RTC comprises multiple proteins; the non-structural proteins Nsp3, Nsp4, and Nsp6 [14, 15] that participate in the formation of sites for viral RNA synthesis, the principal protease (Nsp5), the Nsp7–Nsp8 primase complex, the main RNA-dependent RNA polymerase (Nsp12), the helicase/triphosphatase (Nsp13), the exoribonuclease (Nsp14), the endonuclease (Nsp15), and the N7- and 2′O- methyltransferases (Nsp10/Nsp16). The replicase-transcriptase proteins are expected to be great targets for anti-coronaviral drugs.
Nsp3 is the largest element of the RTC. In addition to cleaving, Nsp3 alters cytokine expression to decrease the host innate immune response [10]. Nsp5 is a 3CLPro protease crucial for RNA replication. Nsp3, Nsp4, and Nsp6 form a complex to induce double-membrane vesicles [16]. Nsp7 and Nsp8 form an RNA polymerase complex performing de novo initiation and primer extension [17]. Nsp8 interacts with the replicase Nsp9 [18], and with the Orf6 accessory protein. In addition, four structural proteins: Spike (S), Envelope (E), Membrane (M), and Nucleocapsid (N) are encoded at the 3′-end. Eleven Orfs and 8 postulated accessory factors are also part of the virus structure.
Formation of double-membrane vesicles by Nsp3-4-6
To evade detection by host innate immune sensors, many viruses that replicate in the cytoplasm compartmentalize their genome transcription in organelle-like structures, thus protecting the virus against host cell defenses and increasing the replication efficiency [19, 20].
Coronaviruses are enveloped positive-sense single-stranded RNA viruses and, as such, arrogate intracellular membranes of different cellular organelles [21]. The membrane rearrangements can be an invagination towards the lumen of the endoplasmic reticulum (ER) or other organelles, or an extrusion of the ER membrane forming double-membrane vesicles (DMVs) [22, 23]. Outer membranes of DMVs interconnect with the ER and other virus-induced structures called convoluted membranes (CM). The ER is probably the main donor for this elaborate Reticulovesicular Network (RVN), but autophagosomes and late endosomes can also be a membrane source of DMVs [24, 25]. The formation of replication sites most likely emerges from the ER membrane, while the major budding sites of SARS- CoV use the membranes in the ERGIC and Golgi region [26].
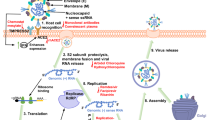
The co-expression of all three SARS-CoV NSPs (Nsp3, Nsp4, and Nsp6) is essential to induce DMVs that are comparable to those described in SARS-CoV-infected cells [16] (Fig. 2). These proteins contain particular multiple transmembrane domains that help the virus replication complex via the recruitment of intracellular membranes. The rough ER outer membrane forms an RVN of membranes that includes convoluted membranes [22] and DMVs [27]. DMVs are found in hepatitis C, poliovirus, coxsackievirus, and SARS-CoV infected cells. Therefore, knowing how the virus uses the host cell to hide in DMVs and replicate will lead to the development of therapies, not only against human CoV but against other pathogens that use the same mechanism, as well. In this regard, K22 was identified as a small compound targeting membrane-bound coronaviral RNA synthesis inhibiting DMV formation, viral replication, and infectivity in a wide range of coronaviruses HCoV-229E, FCoV, MHV, IBV, MERS, and SARS-CoV [28].

Nsp3/4/6 domain similarities. Transmembrane domains were determined by CCTop software (http://cctop.enzim.ttk.mta.hu/). The 1D and 2D panels show the amino acid sequence colored by the consensus topology. Colors are based on the localization: gray, black, blue, red, yellow, and orange for transit sequence, signal peptide, extra-cytosolic, cytosolic, membrane, and re-entrant loop regions, respectively
NSP3 is a 215-kDa glycosylated protein with transmembrane domains interacting with several proteins involved in transcription and replication, and also serves as a scaffolding protein for these mechanisms [29]. Nsp3 contains two conserved transmembrane regions [30] that insert into the ER membrane. These transmembrane domains are required for the DMVs’ pairing [31] (Fig. 3a). Exogenous Nsp3, both full length and/or the C-terminal transmembrane-containing region, induces disordered membrane bodies (DMB) as well as multilamellar and giant vesiculation (MGV), showing the ability for the C-terminus to participate in membrane production or expansion of existing membranes. In SARS-infected cells, Nsp3 stimulates membrane buildups that grow larger in size with time supporting. Expression of Nsp3 with either Nsp4 or Nsp6 reduces the formation of MGV but not DMB, indicating that Nsp4 and Nsp6 have a regulatory role on Nsp3′s membrane proliferation ability. Co-transfection of Nsp3 with Nsp4 provokes a particular membrane conformation, generating a perinuclear double-membrane walled maze-like body (MLB) [16], suggesting a significant interaction between Nsp3 and Nsp4 and supporting published data. The C-terminal one-third of Nsp3 (nsp3C) is sufficient to interact with Nsp4 [32] and Nsp6 [33].

Phylogenetic trees. Based on sequence alignment, we built a phylogenetic trees of Nsp3 (a), Nsp4 (b), and Nsp6 (c) for coronaviruses strains OC43, 229E, MERS, SARS-CoV, and SARS-CoV-2 acquired with Clustal Omega and MView visualization
NSP4
Glycosylated Nsp4 is necessary for normal DMV formation [34, 35], as described in SARS-CoV and MHV [36]. The N-terminal region of Nsp4 comprising the first three transmembrane regions necessary for DMV membrane pairing [37]. We observe in Fig. 3b that they are highly conserved among all coronaviruses as well as in SARS-CoV-2 Nsp4.
The interaction between Nsp3 and Nsp4 was shown to be necessary for the formation of convoluted membranes (CM) and DMVs in MERS [38] and SARS-CoV [16], probably through the luminal loops of Nsp3 and Nsp4 [22] positioned in the TM1 and TM2 regions, respectively. The interaction of these domains with their counterparts on the opposite side of the ER lumen induces membrane pairing. The N-terminal region of Nsp3 is required for complete DMVs formation and its TM1 region along with Nsp4 is sufficient to induce membrane pairing.
NSP6 is a membrane protein of approximately 34 kDa with six transmembrane helices and a highly conserved C-terminus [39] (Fig. 3c). Inserted in the ER membrane [15], it associates with Nsp3 and Nsp4 multi-pass transmembrane proteins during the assembly of coronavirus replication complex to form DMVs [16]. Transfection of Nsp6 produces single-membrane small vesicle clustering around the microtubule-organizing center (MTOC). Interestingly, its co-transfection with Nsp4 has a negative effect on this phenotype and Nsp6 is relocalized outside the MTOC area. The microtubule scaffolding does not seem necessary for productive infection [40]; however, several members of the RTC circulate in the cell through the microtubules.
The avian coronavirus infectious bronchitis virus (IBV) Nsp6 has been shown to activate autophagosome formation, inducing vesicles containing Atg5 and LC3-II [41], and it shares this property with Nsp6 of the mouse hepatitis virus (MHV) and SARS-CoV. All the Nsp6 orthologs throughout the Nidovirales order preserved the ability to engender autophagosomes directly from the ER. However, the autophagosomes formed by Nsp6 have smaller sizes compared with those induced by starvation, suggesting that Nsp6 might also inhibit the enlargement of autophagosomes [42]. The mechanisms behind Nsp6-induced autophagy are poorly described, even though involvement of mTOR and the ER stress transcription factor CHOP are neutralized [41]. Nsp6 may modify adaptive immune responses by sending immunomodulatory proteins synthesized by the ER into autophagosomes for degradation. The SARS-CoV-2 Nsp6 protein also interacts with the sigma receptor, which is known to participate in ER stress response [43].
Effect of coronaviruses on neurons
SARS-CoV
The histological examination of brain tissue harvested during the 2003 SARS epidemic has shown degeneration and necrosis of neurons, extensive glial cell hyperplasia, edema, and cellular infiltration [44, 45]. Electron microscopy studies detected viral genomes and viral particles in the cytoplasm of neurons, mostly in the cortex and the hippocampus [45, 46], confirming that the virus can cross the blood–brain barrier. Interestingly, in patients infected with SARS-CoV, the virus localized almost exclusively to neurons. This observation is congruent with studies showing a distinct neuronal tropism in infected hACE2-Tg mice [47, 48]. Moreover, human neurons are susceptible to SARS-CoV [49] and express ACE2 [50]. Note that at least four known coronaviruses (HCoV-229E, HCoV-OC43, SARS-CoV, and MERS-CoV) possess the property to penetrate the central nervous system (CNS) [51, 52].
SARS-CoV-2
The great structural similarity between SARS-CoV-2 and betacoronaviruses brings the hypothesis that SARS-CoV-2 also possesses similar neurotrophic and neuro-invasive properties. Furthermore, SARS-CoV and SARS-CoV-2 use the same host receptor, the human angiotensin-converting enzyme 2 (ACE2) [53]. It is proposed that the virus enter the CNS through different routes including the olfactory and trigeminal nerves, the cerebrospinal fluid, the vasculature, and the lymphatic system [54].
The central respiratory failure observed with some patients may be partially due to the neuro-invasive potential of the virus [55]. It has the ability to damage the brainstem where the pneumotaxic center is located, and hence, endotracheal intubation and mechanical respiratory support may be needed. A recent study registered neurological manifestations in 214 patients, of which 78 patients (36.4%) had neurologic manifestations, showing a neurotropic potential for the SARS-CoV-2 [56]. Furthermore, about 8% of patients from Wuhan-China reported headaches and 5% nausea and vomiting. Edema and partial neuronal degeneration were observed in brain tissues in China and many cases of viral encephalitis in different countries were registered.
It is too premature to know the long-term neurological complications of the SARS-CoV-2 infection, and coronaviruses have not been linked so far with particular long-term neurological sequelae. However, the long-term neurocognitive consequences of SARS-CoV-2 should not be ignored. Additionally, the presence of anosmia and ageusia is intriguing, since hyposmia is a feature of early Parkinson’s disease and the accumulation of alpha-synuclein in the olfactory system is often seen in the prodrome. Several studies suggest that HCoV RNA frequently persists in human brains [57], as was observed for murine coronaviruses [58]. The neurotropism of HCoV could lead to persistence within the central nervous system.
Consequences of interfering with intracellular membranes
Coronavirus can provoke ER stress
A healthy and functional ER is vital for the synthesis of new plasma membrane proteins, lysosomal enzymes, proteins for the Golgi apparatus, and proteins for extracellular secretion. Any alteration in its function can provoke ER stress and accumulation of misfolded proteins that could lead to cellular deregulation and diseases. In this regard, global proteomic studies revealed that several genes related to ER stress are overexpressed in cells infected with SARS- CoV, such as glucose-regulated protein 78 (GRP78 or BiP) and glucose-regulated protein 94 (GRP94) [59]. ER stress can also be activated by pro-inflammatory cytokines [60]. Several SARS-CoV proteins have been shown to induce the ER stress response such as the Spike protein [59, 61], upregulating GRP78/94 and the PERK pathway, while 3a and 8ab proteins were shown to induce lysosomal damage [62, 63]. Orf6 and Orf7a were also described to induce ER stress, where Orf6 activates caspase-3 through JNK-dependent apoptosis pathway [64]. However, nothing is known about SARS-CoV-2 proteins. Some of these events could be observed, because at least one domain (N- or C-terminal) of BiP, CHOP, or other involved ER factors belong to the group of intrinsically disordered proteins. Any deregulation of one of the domains can alter that protein function and contributes to ER stress. This phenomenon is mainly assured by SARS-CoV-2 nucleocapsid protein that associates with the other viral proteins to induce these deregulations.
The ER-associated degradation (ERAD) tuning vesicles (or EDEMosomes) controls the degradation of misfolded glycoproteins [65]. EDEMosomes are large vesicles budding from the ER that fuse in an Atg5-dependent manner with lysosomes for degradation of their contents. Coronavirus hijacks the machinery of EDEMososme formation for the production of DMVs. Viral NSPs associate with an EDEMosome cargo receptor that usually organizes the sorting of EDEMososmes from the ER. Furthermore, the unfolded protein response (UPR) maintains ER homeostasis by shutting down the translation and increasing the ER protein folding capacity. However, under sustained ER stress, the UPR can induce apoptotic cell death and cytokine production [66]. The UPR also stimulates transcription of genes encoding proteins that mediate ERAD. Chronic UPR activation has been described in many diseases, including diabetes, cancer, and neurodegeneration.
Coronavirus might promote Golgi fragmentation
Proteins synthesized in the ER are packaged into vesicles that will fuse with the Golgi apparatus. These cargo proteins are modified and destined for secretion via exocytosis or for use in the cell. The Golgi is also involved in lipid transport and lysosome formation. Therefore, provoking its fragmentation can cause the cell to collapse. In this regard, it has been shown that SARS-CoV and MHV infections trigger the rearrangement and fragmentation of the Golgi apparatus [67, 68]. The Golgi complex usually forms a continuous ribbon in mammalian cells and its alteration causes a stress response resulting in cell death.
Coronavirus may alter autophagy machinery
Viruses can use autophagy machinery for their own replication and have also developed strategies to escape autophagic degradation. Recent evidence suggests an inhibitory effect of SARS-CoV and MERS on the autophagy process. MERS blocks the autophagosome/lysosome fusion and causes the suppression of the autophagic flux. However, it was not elucidated if this blockage is mandatory for viral replication. Three MERS proteins are described to limit autophagy, Nsp6, p4b, and p5, with Nsp6 also inhibiting the expansion of autophagosomes as shown for other betacoronaviruses [69]. The actual induction of autophagy in infected cells could be due to other factors, such as the presence of double-stranded RNA within the cell, activation of toll receptor signaling during cell entry, and/or induction of ER stress during replication and envelopment. Therefore, it is important to study these events in SARS-CoV-2-infected cells and to determine a) which SARS-CoV02 proteins are involved in ER stress, b) what is the impact of this stress on cell function, and c) can these events be reversed to prevent viral replication.
Potential long-term consequences of coronaviruses on the central nervous system
Several studies have linked the presence and persistence of coronavirus in human brains to long-term sequelae with the initiation or exacerbation of chronic neurological diseases [51] such as multiple sclerosis [70, 71] or Parkinson’s disease [72].
The neurotropic mouse hepatitis virus (MHV) strain JHM RNA has been found in both the rodent models and human CNS for extended periods after the initial infection [73]. Oligodendrocytes and most likely neurons are potential reservoirs for the coronavirus infection.
Human respiratory pathogens have been recognized to be associated with the initiation or aggravation of neurological diseases, but the etiology is still poorly understood [74]. A persistent HCoV- 229E infection was observed in the brains from different age groups after autopsy and may play a role in further chronic pathologies witnessed with coronoviral infections [75, 76]. HCoV-OC43 was shown to establish a persistent infection in human cells [77, 78], and was detected in the CNS of mice a year after an infection that triggered acute encephalitis [58].
Based on studies using SARS-CoV and/or other viruses, researchers suggest two pathways to explain how SARS-CoV-2 can enter the human CNS: i—the hematogenous entry, and/or ii—the neuronal retrograde dissemination [54, 74, 79].
Hematogenous entry
In the hematogenous entry, endothelial cells of the blood brain barrier (BBB) or leukocytes are infected by the virus to infiltrate the CNS [54, 74, 79]. SARS-CoV infects endothelial cells of the BBB “channeling a passage” through the BBB into the CNS [80]. In the same way, HCoV-229E infects monocytes/macrophages [81], peritoneal macrophages, and dendritic cells [82]. Similarly, SARS-CoV-2 may infect monocytes and macrophages through ACE2-dependent and ACE2-independent pathways or attach to ACE2 expressed in the endothelium of BBB to obtain access into the CNS [83]. The angiotensin-converting enzyme type 2 receptor is widely distributed in the lungs, heart, liver, kidney, and intestine. After the infection, monocytes migrate to the tissues where they turn into infected resident macrophages, permitting viruses to reach different organs and tissues. Cerebral cytokine storms can result in the breakdown of the BBB and allow monocytes to traverse the BBB physically.
In support of this pathway, studies showed a significant heterogeneity in the range of estimates for the presence of SARS-CoV-2 RNA in blood, from 0% in numerous studies [84], and even up to 76% in a critical care setting. Pooled data estimated that SARS-CoV-2 RNA may be detected at low copy numbers in about 10% of blood samples from patients with COVID-19 [84].
Neuronal retrograde dissemination
The second major access to the CNS for SARS-CoV-2 could be via a nerve terminal. By this means, some viruses can infect peripheric neurons and enter the CNS via a retrograde axonal transport machinery to the soma [74, 79, 85]. ACE2 is broadly expressed on the epithelial cells of the oral mucosa [86]. SARS-CoV penetrates the brains of hACE2 mice mostly through the olfactory bulb, before spreading to the brain [87]. Olfactory sensory neurons extend dendrites into the nasal cavity and spread out axons across the cribriform plate into the olfactory bulb of the brain [88]. From the lungs, SARS-CoV-2 could spread into the CNS by trans-neural route, through the vagus nerve and the dorsal root ganglia. The trans‐synaptic transfer has been well described for other coronaviruses, such as HEV67 [89] and influenza a [90].
These pathways and their impacts confirm the need for a long-term assessment of neurological damage in the care of SARS-CoV-2 patients.
Multiple sclerosis is a disease of unidentified etiology associated with idiopathic inflammatory demyelinating disease of the central nervous system. It is interesting to note that contact with infectious pathogens before puberty seems to increase the risk of developing MS and relapses of the disease are often preceded with respiratory tract infection [91, 92]. Coronavirus-like particles [93], HCoV RNA [94], and intrathecal anti-HCoV antibody synthesis indicative of a CNS infection [95], were detected in autopsied brain tissues from MS patients. In rodents, mouse hepatitis virus (MHV) infection of the CNS triggers neurological symptoms similar to MS [96]. A cross-reactivity between myelin and coronavirus antigens was noticed in T-cell lines isolated from MS patients [97].
Parkinson’s disease
A potential relationship or at least an epidemiological association [98] between viral infections and PD was first mentioned after the 1920–1930s influenza epidemic, which was associated with Encephalitis Lethargica (EL) [99]. The EL patients presented a type of ‘sleeping sickness’ which included uncontrollable sleepiness but also headache, nausea, fever, catatonia, and occasionally coma [100]. Compared to normal age-matched controls, PD patients have an elevated cerebrospinal fluid antibody response to coronaviruses [72]. Furthermore, reduction or blocking of the ER- to-Golgi trafficking leads to the accumulation of α-Synuclein [100] that contributes to ER stress as observed in PD disease model. Furthermore, Golgi fragmentation observed in PD results in reduced delivery of dopamine transporter [101] and impaired axonal synapses and dendrite trafficking. Interestingly, Golgi fragmentation was described in human neurons infected with MHV and SARS-CoV.
Discussion and future studies
In this review, we described the potential effect of coronaviruses on cells in general and on neurons in general and the ability of viral proteins to cause failure of several cell organelles (ER stress, Golgi fragmentation, and Autophagy machinery alteration). These events start with the formation of double-membrane vesicles (DMVs) that the virus will use to hide and to translate its so-called accessory proteins. DMVs or membrane rearrangements can be an invagination towards the lumen of the endoplasmic reticulum (ER) or other organelles, or an extrusion of the ER membrane. Since DMVs originate from the endoplasmic reticulum, it is normal to observe a stressed and dysfunctional ER that could lose its functional interaction with the mitochondria and Golgi apparatus as displayed in Fig. 4. This domino effect of altered organelle traffic lead to accumulation of aggregated proteins that could activate the autophagy machinery in vain, since the lysosomes membranes are going to be affected also [102]. It is not clear yet whether SARS-CoV-2 can cause all these events, since most of the studies focus on drug discovery and what are the protein(s) involved. However, phylogenetic trees presented in Fig. 3 gave the rationale that the function of SARS-CoV-2 Nsp3, 4, and 6 should be similar to their counterpart from other coronaviruses. In support of this idea, using human neuronal and lung cells in vitro, we demonstrated that these proteins can promote ER stress and Golgi apparatus fragmentation. Therefore, a thorough investigation of these events should be conducted, and patients recovering from COVID-19 should be enrolled in clinical studies to determine whether they will develop organ failure due to ER stress and the contribution of these events in ischemic strokes, lungs collapse, and long-term organ damage. Therefore, to avoid future problems and to be able to develop inhibitors of SARS-CoV-2 proteins, a systematic study should be established using all affected cell types.

Schematic presentation of ER stress and its consequences. Model displays impact of non-structural proteins 3, 4, and 6 (Nsp3/4 and 6) on the ER after double-membrane vesicle formation and the consequence of this formations on several cellular organelles as indicated
Abbreviations
- COVID-19:
-
Coronavirus disease 2019
- DMV:
-
Double-membrane vesicle
- MSA:
-
Multiple sequence alignment
- SARS:
-
Severe acute respiratory syndrome
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
References
Tyrrell DA, Bynoe ML (1965) Cultivation of a novel type of common-cold virus in organ cultures. Br Med J 1(5448):1467–1470
Hamre D, Procknow JJ (1966) A new virus isolated from the human respiratory tract. Proc Soc Exp Biol Med 121(1):190–193
McIntosh K, Dees JH, Becker WB, Kapikian AZ, Chanock RM (1967) Recovery in tracheal organ cultures of novel viruses from patients with respiratory disease. Proc Natl Acad Sci USA 57(4):933–940
Tsang KW, Ho PL, Ooi GC et al (2003) A cluster of cases of severe acute respiratory syndrome in Hong Kong. N Engl J Med 348(20):1977–1985
Chinese SMEC (2004) Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science 303(5664):1666–1669
van der Hoek L, Pyrc K, Jebbink MF et al (2004) Identification of a new human coronavirus. Nat Med 10(4):368–373
Woo PC, Lau SK, Chu CM et al (2005) Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol 79(2):884–895
Al-Tawfiq JA, Zumla A, Memish ZA (2014) Coronaviruses: severe acute respiratory syndrome coronavirus and Middle East respiratory syndrome coronavirus in travelers. Curr Opin Infect Dis 27(5):411–417
Chen Y, Liu Q, Guo D (2020) Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol 92(4):418–423
Serrano P, Johnson MA, Chatterjee A et al (2009) Nuclear magnetic resonance structure of the nucleic acid-binding domain of severe acute respiratory syndrome coronavirus nonstructural protein 3. J Virol 83(24):12998–13008
Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R (2003) Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 300(5626):1763–1767
Yang X, Chen X, Bian G et al (2014) Proteolytic processing, deubiquitinase and interferon antagonist activities of Middle East respiratory syndrome coronavirus papain-like protease. J Gen Virol 95(Pt 3):614–626
Lin PY, Chou CY, Chang HC, Hsu WC, Chang GG (2008) Correlation between dissociation and catalysis of SARS-CoV main protease. Arch Biochem Biophys 472(1):34–42
Perlman S, Netland J (2009) Coronaviruses post-SARS: update on replication and pathogenesis. Nat Rev Microbiol 7(6):439–450
Oostra M, Hagemeijer MC, van Gent M et al (2008) Topology and membrane anchoring of the coronavirus replication complex: not all hydrophobic domains of nsp3 and nsp6 are membrane spanning. J Virol 82(24):12392–12405
Angelini MM, Akhlaghpour M, Neuman BW, Buchmeier MJ (2013) Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio 4(4):e00524
te Velthuis AJ, van den Worm SH, Snijder EJ (2012) The SARS-coronavirus nsp7 + nsp8 complex is a unique multimeric RNA polymerase capable of both de novo initiation and primer extension. Nucleic Acids Res 40(4):1737–1747
Sutton G, Fry E, Carter L et al (2004) The nsp9 replicase protein of SARS-coronavirus, structure and functional insights. Structure 12(2):341–353
Miller S, Krijnse-Locker J (2008) Modification of intracellular membrane structures for virus replication. Nat Rev Microbiol 6(5):363–374
Belov GA, van Kuppeveld FJ (2012) (+)RNA viruses rewire cellular pathways to build replication organelles. Curr Opin Virol 2(6):740–747
den Boon JA, Diaz A, Ahlquist P (2010) Cytoplasmic viral replication complexes. Cell Host Microbe 8(1):77–85
Knoops K, Kikkert M, Worm SH et al (2008) SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol 6(9):e226
de Wilde AH, Raj VS, Oudshoorn D et al (2013) MERS-coronavirus replication induces severe in vitro cytopathology and is strongly inhibited by cyclosporin A or interferon-alpha treatment. J General Virol 94(Pt 8):1749–1760
van der Meer Y, Snijder EJ, Dobbe JC et al (1999) Localization of mouse hepatitis virus nonstructural proteins and RNA synthesis indicates a role for late endosomes in viral replication. J Virol 73(9):7641–7657
Verheije MH, Raaben M, Mari M et al (2008) Mouse hepatitis coronavirus RNA replication depends on GBF1-mediated ARF1 activation. PLoS Pathog 4(6):e1000088
Stertz S, Reichelt M, Spiegel M et al (2007) The intracellular sites of early replication and budding of SARS-coronavirus. Virology 361(2):304–315
Gosert R, Kanjanahaluethai A, Egger D, Bienz K, Baker SC (2002) RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J Virol 76(8):3697–3708
Rappe JCF, de Wilde A, Di H et al (2018) Antiviral activity of K22 against members of the order Nidovirales. Virus Res 246:28–34
Barretto N, Jukneliene D, Ratia K et al (2005) The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J Virol 79(24):15189–15198
Neuman BW (2016) Bioinformatics and functional analyses of coronavirus nonstructural proteins involved in the formation of replicative organelles. Antiviral Res 135:97–107
Hagemeijer MC, Monastyrska I, Griffith J et al (2014) Membrane rearrangements mediated by coronavirus nonstructural proteins 3 and 4. Virology 458–459:125–135
Hagemeijer MC, Ulasli M, Vonk AM et al (2011) Mobility and interactions of coronavirus nonstructural protein 4. J Virol 85(9):4572–4577
Hagemeijer MC, Rottier PJ, de Haan CA (2012) Biogenesis and dynamics of the coronavirus replicative structures. Viruses 4(11):3245–3269
Clementz MA, Kanjanahaluethai A, O’Brien TE, Baker SC (2008) Mutation in murine coronavirus replication protein nsp4 alters assembly of double membrane vesicles. Virology 375(1):118–129
Gadlage MJ, Sparks JS, Beachboard DC et al (2010) Murine hepatitis virus nonstructural protein 4 regulates virus-induced membrane modifications and replication complex function. J Virol 84(1):280–290
Oostra M, te Lintelo EG, Deijs M et al (2007) Localization and membrane topology of coronavirus nonstructural protein 4: involvement of the early secretory pathway in replication. J Virol 81(22):12323–12336
Sparks JS, Lu X, Denison MR (2007) Genetic analysis of Murine hepatitis virus nsp4 in virus replication. J Virol 81(22):12554–12563
Oudshoorn D, Rijs K, Limpens R et al (2017) Expression and cleavage of middle east respiratory syndrome coronavirus nsp3-4 polyprotein induce the formation of double-membrane vesicles that mimic those associated with coronaviral RNA replication. mBio 8(6):e01658
Baliji S, Cammer SA, Sobral B, Baker SC (2009) Detection of nonstructural protein 6 in murine coronavirus-infected cells and analysis of the transmembrane topology by using bioinformatics and molecular approaches. J Virol 83(13):6957–6962
Hagemeijer MC, Verheije MH, Ulasli M et al (2010) Dynamics of coronavirus replication-transcription complexes. J Virol 84(4):2134–2149
Cottam EM, Maier HJ, Manifava M et al (2011) Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 7(11):1335–1347
Cottam EM, Whelband MC, Wileman T (2014) Coronavirus NSP6 restricts autophagosome expansion. Autophagy 10(8):1426–1441
Gordon DE, Jang GM, Bouhaddou M et al (2020) A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583:459–468
Ding Y, He L, Zhang Q et al (2004) Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV) in SARS patients: implications for pathogenesis and virus transmission pathways. J Pathol 203(2):622–630
Gu J, Gong E, Zhang B et al (2005) Multiple organ infection and the pathogenesis of SARS. J Exp Med 202(3):415–424
Xu J, Zhong S, Liu J et al (2005) Detection of severe acute respiratory syndrome coronavirus in the brain: potential role of the chemokine mig in pathogenesis. Clin Infect Dis 41(8):1089–1096
Ng ML, Tan SH, See EE, Ooi EE, Ling AE (2003) Proliferative growth of SARS coronavirus in Vero E6 cells. J General Virol 84(Pt 12):3291–3303
Tseng CT, Huang C, Newman P et al (2007) Severe acute respiratory syndrome coronavirus infection of mice transgenic for the human Angiotensin-converting enzyme 2 virus receptor. J Virol 81(3):1162–1173
Yamashita M, Yamate M, Li GM, Ikuta K (2005) Susceptibility of human and rat neural cell lines to infection by SARS-coronavirus. Biochem Biophys Res Commun 334(1):79–85
Harmer D, Gilbert M, Borman R, Clark KL (2002) Quantitative mRNA expression profiling of ACE 2, a novel homologue of angiotensin converting enzyme. FEBS Lett 532(1–2):107–110
Arbour N, Day R, Newcombe J, Talbot PJ (2000) Neuroinvasion by human respiratory coronaviruses. J Virol 74(19):8913–8921
Li K, Wohlford-Lenane C, Perlman S et al (2016) Middle east respiratory syndrome coronavirus causes multiple organ damage and lethal disease in mice transgenic for human dipeptidyl peptidase 4. J Infect Dis 213(5):712–722
Hamming I, Timens W, Bulthuis ML et al (2004) Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 203(2):631–637
Desforges M, Le Coupanec A, Dubeau P et al (2019) Human coronaviruses and other respiratory viruses: underestimated opportunistic pathogens of the central nervous system? Viruses 12(1):14
Li YC, Bai WZ, Hashikawa T (2020) The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J Med Virol 92(6):552–555
Mao L, Jin H, Wang M et al (2020) Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol 77(6):683–690
Phares TW, DiSano KD, Stohlman SA, Bergmann CC (2014) Progression from IgD + IgM + to isotype-switched B cells is site specific during coronavirus-induced encephalomyelitis. J Virol 88(16):8853–8867
Jacomy H, Fragoso G, Almazan G, Mushynski WE, Talbot PJ (2006) Human coronavirus OC43 infection induces chronic encephalitis leading to disabilities in BALB/C mice. Virology 349(2):335–346
Yeung YS, Yip CW, Hon CC et al (2008) Transcriptional profiling of Vero E6 cells over-expressing SARS-CoV S2 subunit: insights on viral regulation of apoptosis and proliferation. Virology 371(1):32–43
Kharroubi I, Ladriere L, Cardozo AK et al (2004) Free fatty acids and cytokines induce pancreatic beta-cell apoptosis by different mechanisms: role of nuclear factor-kappaB and endoplasmic reticulum stress. Endocrinology 145(11):5087–5096
Chan CP, Siu KL, Chin KT et al (2006) Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J Virol 80(18):9279–9287
Shi CS, Nabar NR, Huang NN, Kehrl JH (2019) SARS-coronavirus open reading frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov 5:101
Sung SC, Chao CY, Jeng KS, Yang JY, Lai MM (2009) The 8ab protein of SARS-CoV is a luminal ER membrane-associated protein and induces the activation of ATF6. Virology 387(2):402–413
Ye Z, Wong CK, Li P, Xie Y (2008) A SARS-CoV protein, ORF-6, induces caspase-3 mediated, ER stress and JNK-dependent apoptosis. Biochem Biophys Acta 1780(12):1383–1387
Cali T, Galli C, Olivari S, Molinari M (2008) Segregation and rapid turnover of EDEM1 by an autophagy-like mechanism modulates standard ERAD and folding activities. Biochem Biophys Res Commun 371(3):405–410
Schroder M, Kaufman RJ (2005) The mammalian unfolded protein response. Annu Rev Biochem 74:739–789
Lavi E, Wang Q, Weiss SR, Gonatas NK (1996) Syncytia formation induced by coronavirus infection is associated with fragmentation and rearrangement of the Golgi apparatus. Virology 221(2):325–334
Freundt EC, Yu L, Goldsmith CS et al (2010) The open reading frame 3a protein of severe acute respiratory syndrome-associated coronavirus promotes membrane rearrangement and cell death. J Virol 84(2):1097–1109
Gassen NC, Niemeyer D, Muth D et al (2019) SKP2 attenuates autophagy through Beclin1-ubiquitination and its inhibition reduces MERS-Coronavirus infection. Nature communications 10(1):5770
Cristallo A, Gambaro F, Biamonti G et al (1997) Human coronavirus polyadenylated RNA sequences in cerebrospinal fluid from multiple sclerosis patients. New Microbiol 20(2):105–114
Murray RS, Brown B, Brian D, Cabirac GF (1992) Detection of coronavirus RNA and antigen in multiple sclerosis brain. Ann Neurol 31(5):525–533
Fazzini E, Fleming J, Fahn S (1992) Cerebrospinal fluid antibodies to coronavirus in patients with Parkinson’s disease. Mov Disord 7(2):153–158
Sorensen O, Coulter-Mackie MB, Puchalski S, Dales S (1984) In vivo and in vitro models of demyelinating disease. IX. Progression of JHM virus infection in the central nervous system of the rat during overt and asymptomatic phases. Virology 137(2):347–357
Desforges M, Le Coupanec A, Stodola JK, Meessen-Pinard M, Talbot PJ (2014) Human coronaviruses: viral and cellular factors involved in neuroinvasiveness and neuropathogenesis. Virus Res 194:145–158
Stewart JN, Mounir S, Talbot PJ (1992) Human coronavirus gene expression in the brains of multiple sclerosis patients. Virology 191(1):502–505
Lavi E, Gilden DH, Highkin MK, Weiss SR (1984) Persistence of mouse hepatitis virus A59 RNA in a slow virus demyelinating infection in mice as detected by in situ hybridization. J Virol 51(2):563–566
Arbour N, Cote G, Lachance C et al (1999) Acute and persistent infection of human neural cell lines by human coronavirus OC43. J Virol 73(4):3338–3350
Arbour N, Ekande S, Cote G et al (1999) Persistent infection of human oligodendrocytic and neuroglial cell lines by human coronavirus 229E. J Virol 73(4):3326–3337
Swanson PA 2nd, McGavern DB (2015) Viral diseases of the central nervous system. Curr Opin Virol 11:44–54
Guo Y, Korteweg C, McNutt MA, Gu J (2008) Pathogenetic mechanisms of severe acute respiratory syndrome. Virus Res 133(1):4–12
Desforges M, Miletti TC, Gagnon M, Talbot PJ (2007) Activation of human monocytes after infection by human coronavirus 229E. Virus Res 130(1–2):228–240
Patterson S, Macnaughton MR (1982) Replication of human respiratory coronavirus strain 229E in human macrophages. J General Virol 60(Pt 2):307–314
Baig AM, Khaleeq A, Ali U, Syeda H (2020) Evidence of the COVID-19 virus targeting the CNS: tissue distribution, host-virus interaction, and proposed neurotropic mechanisms. ACS Chem Neurosci 11(7):995–998
Andersson M, Arancibia Carcamo CV, Auckland K et al. (2020) SARS-CoV-2 RNA detected in blood samples from patients with COVID-19 is not associated with infectious virus. medRxiv:2020.2005.2021.20105486
Li YC, Bai WZ, Hirano N, Hayashida T, Hashikawa T (2012) Coronavirus infection of rat dorsal root ganglia: ultrastructural characterization of viral replication, transfer, and the early response of satellite cells. Virus Res 163(2):628–635
Xu H, Zhong L, Deng J et al (2020) High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int J Oral Sci 12(1):8
Netland J, Meyerholz DK, Moore S, Cassell M, Perlman S (2008) Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J Virol 82(15):7264–7275
van Riel D, Verdijk R, Kuiken T (2015) The olfactory nerve: a shortcut for influenza and other viral diseases into the central nervous system. J Pathol 235(2):277–287
Li YC, Bai WZ, Hirano N et al (2013) Neurotropic virus tracing suggests a membranous-coating-mediated mechanism for transsynaptic communication. J Comp Neurol 521(1):203–212
Matsuda K, Park CH, Sunden Y et al (2004) The vagus nerve is one route of transneural invasion for intranasally inoculated influenza a virus in mice. Vet Pathol 41(2):101–107
Marrie RA, Wolfson C, Sturkenboom MC et al (2000) Multiple sclerosis and antecedent infections: a case-control study. Neurology 54(12):2307–2310
Andersen O, Lygner PE, Bergstrom T, Andersson M, Vahlne A (1993) Viral infections trigger multiple sclerosis relapses: a prospective seroepidemiological study. J Neurol 240(7):417–422
Tanaka R, Iwasaki Y, Koprowski H (1976) Intracisternal virus-like particles in brain of a multiple sclerosis patient. J Neurol Sci 28(1):121–126
Burks JS, DeVald BL, Jankovsky LD, Gerdes JC (1980) Two coronaviruses isolated from central nervous system tissue of two multiple sclerosis patients. Science 209(4459):933–934
Salmi A, Ziola B, Hovi T, Reunanen M (1982) Antibodies to coronaviruses OC43 and 229E in multiple sclerosis patients. Neurology 32(3):292–295
Lane TE, Buchmeier MJ (1997) Murine coronavirus infection: a paradigm for virus-induced demyelinating disease. Trends Microbiol 5(1):9–14
Talbot PJ, Paquette JS, Ciurli C, Antel JP, Ouellet F (1996) Myelin basic protein and human coronavirus 229E cross-reactive T cells in multiple sclerosis. Ann Neurol 39(2):233–240
Ravenholt RT, Foege WH (1982) 1918 influenza, encephalitis lethargica, parkinsonism. Lancet 2(8303):860–864
Hunter C (1931) The late sequelae of encephalitis lethargica and of influenza. Can Med Assoc J 24(6):828–830
Cooper AA, Gitler AD, Cashikar A et al (2006) Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 313(5785):324–328
Lashuel HA, Hirling H (2006) Rescuing defective vesicular trafficking protects against alpha-synuclein toxicity in cellular and animal models of Parkinson’s disease. ACS Chem Biol 1(7):420–424
Hetz C, Saxena S (2017) ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol 13(8):477–491
Acknowledgements
This work is supported by an NIH-NIA (R01-AG054411) awarded to BES.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
None.
Rights and permissions
About this article

Cite this article
Santerre, M., Arjona, S.P., Allen, C.N. et al. Why do SARS-CoV-2 NSPs rush to the ER?. J Neurol 268, 2013–2022 (2021). https://doi.org/10.1007/s00415-020-10197-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-020-10197-8