
Abstract
Charcot–Marie–Tooth disease (CMT) is the most common inherited neuropathy with heterogeneous clinical presentation and genetic background. The axonal form (CMT2) is characterised by decreased action potentials indicating primary axonal damage. The underlying pathology involves axonal degeneration which is supposed to be related to axonal protein dysfunction caused by various gene mutations. The overlapping clinical manifestation of CMT2 with distal hereditary motor neuropathy (dHMN) and intermediate CMT causes further diagnostic difficulties. Aminoacyl-tRNA synthetases have been implicated in the pathomechanism of CMT2. They have an essential role in protein translation by attaching amino acids to their cognate tRNAs. To date six families have been reported worldwide with dominant missense alanyl-tRNA synthetase (AARS) mutations leading to clinically heterogeneous axonal neuropathies. The pathomechanism of some variants could be explained by impaired amino acylation activity while other variants implicating an editing defect need to be further investigated. Here, we report a cohort of six additional families originating from the United Kingdom and Ireland with dominant AARS-related neuropathies. The phenotypic manifestation was distal lower limb predominant sensorimotor neuropathy but upper limb impairment with split hand deformity occasionally associated. Nerve conduction studies revealed significant demyelination accompanying the axonal lesion in motor and sensory nerves. Five families have the c.986G>A, p.(Arg329His) variant, further supporting that this is a recurrent loss of function variant. The sixth family, of Irish origin, had a novel missense variant, c.2063A>G, p.(Glu688Gly). We discuss our findings and the associated phenotypic heterogeneity in these families, which expands the clinical spectrum of AARS-related neuropathies.
Similar content being viewed by others


Introduction
Charcot–Marie–Tooth disease (CMT) is a clinically and genetically heterogeneous group of peripheral neuropathies characterised by symmetric atrophy and weakness in the distal muscles, areflexia and variable range of sensory impairment. In respect of its worldwide prevalence of an estimated 1:2500, CMT is considered to be the most common cause of inherited peripheral neuropathies. Based on the electrophysiological and pathological characteristics it can be classified further into a demyelinating form (CMT1), where the nerve conduction velocity is reduced due to altered myelination, and to an axonal form (CMT2), where the primary axonal dysfunction leads to reduced amplitudes of evoked muscle action potentials. The intermediate form (CMTI) shares both features [1, 11, 13]. There have been more than 90 genes associated with CMT so far and, despite the overlapping phenotypes, some certain genes manifest with distinctive forms dependent on their contribution to Schwann cell and axonal functions [3].
Genetic diagnosis is particularly difficult to achieve in the axonal form (CMT2) which shows genotypic and phenotypic overlap with distal hereditary motor neuropathies (dHMN) or distal spinal muscular atrophy (dSMA), a subgroup of rare inherited motor axonal neuropathies with no significant sensory involvement [9]. Similarly, dominant intermediate CMT (DI-CMT) cases presenting with intermediate nerve conduction velocities of 25–45 m/s are also frequently misdiagnosed as CMT2 [6]. There have been 15 genes or loci described in relation to CMT2 [5]. Some of these genes have been already associated with axonal dysfunction, such as mitofusin 2 (MFN2) which is responsible for 5–10 % of CMT2 cases, neurofilament light chain (NEFL) in 1–5 % and ganglioside-induced differentiation-associated protein (GDAP1) in less than 1 % of the cases [1, 3, 4, 11, 13].
Aminoacyl-tRNA synthetases (ARS) are essential enzymes in the translation of the genetic code by attaching amino acids to their cognate tRNAs during protein synthesis [3, 5, 7]. There are 37 nuclear genes encoding ARSs for cytoplasmic or mitochondrial protein synthesis. Mutations in six genes encoding aminoacyl-tRNA synthetases (ARS) have been implicated in axonal pathology [3]. The majority of the mutations were described in glycyl-tRNA synthetase (GARS, MIM#600287) causing CMT2 type D (CMT2D) or distal spinal muscular atrophy type V (dSMA-V), both are autosomal dominant upper limb predominant motor axonal neuropathies. Mutations in tyrosyl-tRNA synthetase (YARS, MIM#603623) were reported in dominant intermediate CMT type C (CMTDIC). The relevance of methionyl-tRNA synthetase (MARS, MIM#156560) and histidyl-tRNA synthetase (HARS, MIM#142810) mutations for CMT is supported by strong functional and evolutionary evidence, yet incomplete segregation and absence of additional unrelated cases warrant future studies to substantiate this conclusion [2, 14]. Compound heterozygous lysyl-tRNA synthetase (KARS, MIM#601421) mutations were present in one patient with recessive intermediate CMT type B (CMTIRB) manifesting as part of a more complex neurological condition [3, 7, 15].
To date only six families have been described in the literature with dominant missense mutations in the alanyl-tRNA synthetase (AARS, MIM#601065) leading to clinically heterogeneous phenotypes (Table 1). The c.986G>A, p.(Arg329His) variant was reported as a recurrent mutation in two unrelated French families presenting with variable age of onset distal sensorimotor degeneration secondary to predominant axonal neuropathy and with absent or slight demyelination [5]. The same AARS variant was identified in a large Australian family with early onset axonal neuropathy, variable sensorineural deafness and foot deformities, accompanied by intermediate nerve conduction velocities [7]. Another Australian family with the c.2333A>C, p.(Glu778Ala) variant manifested as sensorimotor axonal neuropathy with rippling muscles and cramps in the proband, while only rippling muscles were present in three affected relatives [7]. In a Taiwanese pedigree pure axonal neuropathy with variable age of onset was associated with the c.211A>T, p.(Asn71Tyr) AARS variant [6]. In a three generation dominant Chinese family the c.2677G>A, p.(Asp893Asn) mutation was related to a distal motor neuropathy (dHMN) phenotype based on neurogenic EMG findings [15]. Additionally autosomal recessive loss of function AARS mutations (compound heterozygous p.Lys81Thr and p.Arg751Gly and homozygous p.Arg751Gly) were also described in two unrelated families, causing severe infantile epileptic encephalopathy with a central myelin defect and peripheral neuropathy [12].
Here, we report a cohort of multigenerational British and Irish families with autosomal dominant neuropathies and missense mutations in AARS, with the aim of expanding the clinical spectrum and providing further pheno-genotypic correlations in AARS-related neuropathies.
Methods
Patients
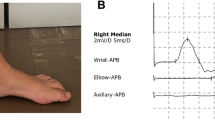
Clinical description and examination findings of the affected patients from 4 UK and 2 Irish families diagnosed with AARS-related neuropathy are provided in detail in Table 1 and Fig. 1 and in the Supplementary data.

a Pedigrees of UK and Irish families. Arrows the index patients of each family. b Image of patient II/4 in UK family 1 showing predominantly lower limb symptoms manifesting with bilateral pes cavus and severe feet drop. Index patient of the same UK1 family (III/1) representing moderate intrinsic hand muscles wasting accompanied by lower limb distal wasting and weakness. Image of patient (II.1) from UK family 3 showing severe bilateral foot drop and distal muscle wasting
Electrophysiology
Nerve conduction studies and needle electromyography were performed in the probands and in additional affected relatives from the UK and Irish families.
Genetic studies
Genetic testing was performed in the Bristol Genetics Laboratory, using the UK Genetic Testing Network approved approach; a multi-gene panel assay utilising next generation sequencing (NGS). Genomic DNA was extracted from the peripheral white blood cells obtained from probands of all families according to standard methods. Enrichment was performed using a custom-designed 330 kb SureSelect capture (Agilent Technologies) targeting 56 genes associated with inherited peripheral neuropathy (IPN). Libraries were prepared from genomic DNA according to the manufacturer’s protocol and sequenced on an Illumina MiSeq (2 × 150 bp). For data analysis and filtering, a bespoke open-source pipeline using BWA and GATK was used to align data to the reference human genome (UCSC hg19). Variant classification was based on Association for Clinical Genetic Science (ACGS) Practice Guidelines (2013). Candidate pathogenic variants were confirmed by Sanger Sequencing using an Applied Biosystems 3730 analyser.
Consent
All participants provided written informed consent to be involved in this study which was approved by local research ethics committees.
Results
Patients
Due to overlapping and variable clinical phenotypes the referrals were occasionally misleading indicating CMT1, hereditary sensory neuropathy or intermediate CMT phenotypes. Multi-gene panel testing was performed in a total of 22 patients with ‘mixed/intermediate’ CMT and out of the 8 positives 5 were proved to be an AARS mutation.
Lower limb predominant sensorimotor neuropathy was characteristic in UK family 1. All family members developed pes cavus and foot drop bilaterally leading to progressive walking difficulties at different adult ages. They all remained ambulant with orthotic support. Various degree of upper limb involvement was observed. The proband manifested with fluctuating weakness of finger and intrinsic hand muscles at disease onset with progressive hand muscles wasting at later stage.
Slowly progressive various degree of motor weakness affecting the lower extremities and subsequent walking problems were the leading symptoms in UK family 2. Tiptoe walking and feet deformities were present. Sensory changes were mild or absent and no upper limb involvement was associated, but we note that the index patient is only 20 years of age.
In UK family 3, the clinical presentation was very heterogeneous. The proband manifested with young adult onset asymmetric upper and lower limb involvement equally affecting motor and sensory functions. His father had stable feet deformities since childhood which progressed rapidly in his late 50s exclusively and symmetrically affecting his lower limbs causing predominantly motor and milder sensory loss. Development of bilateral foot drop and walking difficulties were common symptoms within the family.
A slowly progressive disease course and predominant lower limb motor loss was the common presentation in UK family 4. Initial symptoms presented either in symmetric or asymmetric fashion and striking clinical features were bilateral foot and toe drop with subsequent walking difficulties. Various degrees of weakness and wasting of intrinsic hand muscles with split hand formation were associated. Mild to moderate sensory changes were present.
Lower limb motor symptoms progressed slowly in Irish family 1 leading to different impact on their walking abilities with ambulatory loss in some family members. Sensory loss in lower extremities accompanied and upper limb involvement manifested in later ages.
Early onset slowly progressive and predominantly motor impairment affecting both upper and lower extremities was observed in Irish family 2. Toe walking and bilateral foot drop were common features. Associated milder sensory changes became apparent at later ages. Wasting and weakness of intrinsic hand muscles resulted in split hand deformity (Fig. 1; Table 2).
Electrophysiology
A series of nerve conduction studies was performed in the proband (III.1) of UK family 1. Initially the multifocal patchy demyelinating changes with conduction block leading to the dispersion of the motor neuronal conduction velocities were more suggestive of CIDP, but CSF protein was normal and the patient did not improve on immunosuppressive therapy. Previous recordings indicated intermediate motor neuropathy both demyelinating and axonal in nature with multifocal patchy characteristics and severe sensory polyneuropathy. Family members (II.4 and II.6) had intermediate nerve conduction velocities indicating both demyelinating and axonal features. The proband (III.1) of UK family 2 showed both demyelinating and axonal sensorimotor neuropathy with nerve conduction velocities in the intermediate range. The index patient (III.1) of UK family 3 had intermediate nerve conduction studies underlying his progressive asymmetrical distal sensorimotor neuropathy symptoms. The proband (II.2) of UK family 4 had motor conduction velocities in the demyelinating range consistent with demyelinating CMT (CMT1). Nerve conduction studies in his father (I.1) also showed profound demyelinating neuropathy with significant axonal loss. Testing of both his affected brothers (II.3 and II.5) revealed severe predominantly axonal peripheral sensorimotor neuropathy with reduced conduction velocities. Neurophysiology in the Irish families confirmed length-dependent motor and sensory neuropathy with intermediate conduction velocities (Supplementary Table).
Genetic studies
Clinical characteristics and electrophysiology findings were directive in the candidate gene testing. Mutations in genes involved in axonal pathology (MPZ, MFN2, NEFL, GDAP1) and common demyelinating genes (PMP22, MPZ) were excluded in all patients. Upper limb involvement at disease onset and split hand deformity indicated exclusion of mutations in the GARS gene. Early onset lower extremity predominant motor symptoms jointly with Achilles contracture urged us to exclude the recently reported bicaudal D homolog 2 (BICD2)-related neuropathy. Prominent sensory manifestation in some patients indicated screening for SPTLC1 mutations.
With the use of IPN 56 gene panel assay we identified the previously described pathogenic variant in exon 8 of the AARS gene (c.986G>A, p.Arg329His) in all 4 UK and in Irish family 1. We identified another AARS variant (c.2063A>G, p.Glu688Gly) which to date has not been reported in the literature, as the cause of the dominant sensorimotor neuropathy manifesting in the second family originated from Ireland. This variant is not recorded in dbSNP, 1000 genomes, Exome Variant Server or Exome Aggregation Consortium. Alignment of protein sequences from multiple species supported that glutamic acid 688 is highly conserved among all species from E.coli to H.sapiens. In silico prediction tools indicate this missense change as likely deleterious (SIFT: deleterious; Polyphen2: probably pathogenic; Mutation Taster disease causing). The variant segregated with disease in this family supporting pathogenicity (Fig. 2).

Sequenced c.986G>A missense mutation in AARS in affected UK and Irish index patients. Illustrated positions of AARS mutations and conservation of affected residues across species
Discussion
Aminoacyl-tRNA synthetases (ARSs) catalyse a two-step aminoacylation reaction by binding and activating amino acids and conjugate them with their cognate tRNA molecules [3]. Their essential role in keeping the fidelity of the genetic code during protein translation explains why ARSs are ubiquitous and highly conserved amongst species [5]. Several hypotheses have been proposed in the mechanisms of ARS-related CMT pathology. Most mutations are missense amino acid substitutions leading to dominant CMT phenotypes [3, 7]. In vitro aminoacylation assays indicate both quantitative error via impaired enzyme activation, and qualitative defects by non-cognate bindings. Yeast viability assays investigated in vivo functional consequences of the ARS mutations and support a loss of function effect. Protein localisation studies show altered distribution of some of the ARS proteins in cultured neurons suggesting spatially inappropriate protein synthesis [3, 7, 9].
Alanyl–aminoacyl-tRNA synthetase (AARS) consists of 21 exons and encodes 968 amino acids. From the N-terminal to the C-terminal it has an aminoacylation or catalytic domain (AD), helical or tRNA-binding domain (HD) and editing domain (ED). Unlike as in other ARSs, the acceptor arm of tRNAAla can be directly recognised by the catalytic site of AARS. The editing domain was evolutionarily integrated in the AARS with the aim of eliminating mischarged tRNAAla. Therefore, AARS has highly conserved sequence in all of its domains across species from E. coli to H. sapiens [5, 15].
In the literature, AARS mutations have been reported affecting all three domains. The p.(Asn71Tyr) AARS mutation, which was described in a Taiwanese pedigree, is located in the aminoacylation domain and both in vitro and in vivo assays revealed impaired aminoacylation activity result in reduced charging capacity [6, 7]. The recurrent, p.(Arg329His) mutation located in the middle helical domain of AARS has been largely investigated. Haplotype analysis in 2 French and 1 Australian family with Arg329His AARS mutation demonstrated a different founder for all three families. A methylation-mediated process gives rise to this recurrent mutation which was also found to associate with impaired enzyme activity. AARS exon 8, which contains the p.(Arg329His) variant, has a highly methylated CpG site and considered to be a mutational hot spot [7]. In the cohort reported here, this was the most common AARS mutation affecting one Irish and four UK families. A limited haplotype analysis of four polymorphic variants in the proximity of the causative AARS mutation showed identical haplotype for all four North UK families, and close similarity was suggested (3/4 identical variants) in the Irish family. These data imply that the p.(Arg329His) mutation is potentially a founder mutation in the five reported families (data not shown). The phenotypic manifestation was rather heterogeneous within the families despite the same genotypic background. However, lower limb predominance was characteristic in all families; significant upper limb involvement with split hand (selective atrophy of thenar muscles) could not be ignored in some patients similar to GARS-related pathology. Acute or sub-acute episodes of worsening, resembling acquired neuropathies were also reported. Compared to the previously reported pedigrees, nerve conduction studies in our families showed greater demyelination process accompanying the always present axonal dysfunction. Sensory responses were frequently severely impaired along with occasional clinically significant sensory loss. Similar to the French pedigree, we observed asymmetric distribution of the symptoms in some of the patients, but in contrast to the Australian cohort, sensorineural deafness was not present in either of the families (Tables 1, 2). There have been two different phenotypes described so far in relation to the AARS editing domain mutations. The p.(Glu778Ala) variant manifesting in an Australian family with dominant rippling muscle disease, did not involve an evolutionarily conserved amino acid within the AARS editing domain and impairment in the editing capacity also could not be proven [7]. In a Chinese pedigree, the p.(Asp893Asn) mutation led to a dominant pure motor pathology causing dHMN. It was found to reside in a highly conserved sequence of the C-terminal editing domain and prediction programs indicated deleterious pathogenicity [15]. In our cohort, we identified a novel AARS variant which also locates to the AARS editing domain. The clinical phenotype related to p.(Glu688Gly) is of an early onset length-dependent motor and sensory neuropathy. Neurophysiology, similar to our families with the p.Arg329His mutation, showed intermediate conduction velocities. In the proband, there was also more marked involvement of FDIO and APB than ADM, suggesting a split hand phenotype. Functional studies would be necessary to further analyse the pathogenicity of this variant and its impact on the editing capacity of the enzyme.
Nothing indicates better the variability of AARS-related pathology than an utmost recent paper, published during the revision of our manuscript, which describes a novel heterozygous missense c.304G>C (p.Gly102Arg) AARS variant manifesting with a novel myeloneuropathy phenotype in a large family [8]. This warrants that genetic screening toward AARS and other aminoacyl-tRNA synthetase mutations might be always considered in axonal neuropathology.
Conclusions
AARS-related neuropathy was identified in a cohort of dominant UK and Irish families by multi-gene panel approach. Our cohort supports that the p.(Arg329His) AARS variant is a recurrent mutation presenting worldwide. The associated phenotypic spectrum is heterogeneous and may cause difficulties in achieving diagnosis based only on clinical examination, underlying the valuable contribution of next generation sequencing.
References
Gess B, Schirmacher A, Boentert M, Young P (2013) Charcot-Marie-Tooth disease: frequency of genetic subtypes in a German neuromuscular center population. Neuromuscul Disord 23(8):647–651
Gonzalez M, McLaughlin H, Houlden H, Guo M, Yo-Tsen L, Hadjivassilious M, Speziani F, Yang XL, Antonellis A, Reilly MM, Züchner S, Inherited Neuropathy Consortium (2013) Exome sequencing identifies a significant variant in methionyl-tRNA synthetase (MARS) in a family with late-onset CMT2. J Neurol Neurosurg Psychiatry 84(11):1247–1249
Griffin LB, Sakaguchi R, McGuigan D, Gonzalez MA, Searby C, Züchner S, Hou YM, Antonellis A (2014) Impaired function is a common feature of neuropathy-associated glycyl-tRNA synthetase mutation. Hum Mutat 35(11):1363–1371
Houlden H, Reilly MM (2006) Molecular genetics of autosomal-dominant demyelinating Charcot-Marie-Tooth disease. Neuromol Med 8(1–2):43–62
Latour P, Thauvin-Robinet C, Baudelet-Méry C, Soichot P, Cusin V, Faivre L, Locatelli MC, Mayençon M, Sarcey A, Broussolle E, Camu W, David A, Rousson R (2010) A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic Alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet 86(1):77–82
Lin KP, Soong BW, Yang CC, Huang LW, Chang MH, Lee IH, Antonellis A, Lee YC (2011) The mutational spectrum in a cohort of Charcot-Marie-Tooth disease type 2 among the Han Chinese in Taiwan. PLoS One 6(12):e29393
McLaughlin HM, Sakaguchi R, Giblin W, Wilson TE, Biesecker L, Lupski JR, Talbot K, Vance JM, Züchner S, Lee YC, Kennerson M, Hou YM, Nicholson G, Antonellis A (2012) A recurrent loss-of-function alanyl-tRNA synthetase (AARS) mutation in patients with Charcot-Marie-Tooth disease type 2N (CMT2N). Hum Mutat 33(1):244–253
Motley WW, Griffin LB, Mademan I, Baets J, De Vriendt E, De Jonghe P, Antonellis A, Jordanova A, Scherer SS (2015) A novel AARS mutation in a family with dominant myeloneuropathy. Neurology 84(20):2040–2047
Motley WW, Talbot K, Fischbeck KH (2010) GARS axonopathy: not every neuron’s cup of tRNA. Trends Neurosci 33(2):59–66
Murphy SM, Herrmann DN, McDermott MP, Scherer SS, Shy ME, Reilly MM, Pareyson D (2011) Reliability of the CMT neuropathy score (second version) in Charcot-Marie-Tooth disease. J Peripher Nerv Syst 16(3):191–198
Murphy SM, Laura M, Fawcett K, Pandraud A, Liu YT, Davidson GL, Rossor AM, Polke JM, Castleman V, Manji H, Lunn MP, Bull K, Ramdharry G, Davis M, Blake JC, Houlden H, Reilly MM (2012) Charcot-Marie-Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. J Neurol Neurosurg Psychiatry 83(7):706–710
Simons C, Griffin LB, Helman G, Golas G, Pizzino A, Bloom M, Murphy JL, Crawford J, Evans SH, Topper S, Whitehead MT, Schreiber JM, Chapman KA, Tifft C, Lu KB, Gamper H, Shigematsu M, Taft RJ, Antonellis A, Hou YM, Vanderver A (2015) Loss-of-function alanyl-tRNA synthetase mutations cause an autosomal-recessive early-onset epileptic encephalopathy with persistent myelination defect. Am J Hum Genet 96(4):675–681
Sivera R, Sevilla T, Vílchez JJ, Martínez-Rubio D, Chumillas MJ, Vázquez JF, Muelas N, Bataller L, Millán JM, Palau F, Espinós C (2013) Charcot-Marie-Tooth disease: genetic and clinical spectrum in a Spanish clinical series. Neurology 81(18):1617–1625
Vester A, Velez-Ruiz G, McLaughlin HM, Comparative Sequencing Program NISC, Lupski JR, Talbot K, Vance JM, Züchner S, Roda RH, Fischbeck KH, Biesecker LG, Nicholson G, Beg AA, Antonellis A (2013) A loss-of-function variant in the human histidyl-tRNA synthetase (HARS) gene is neurotoxic in vivo. Hum Mutat 34(1):191–199
Zhao Z, Hashiguchi A, Hu J, Sakiyama Y, Okamoto Y, Tokunaga S, Zhu L, Shen H, Takashima H (2012) Alanyl-tRNA synthetase mutation in a family with dominant distal hereditary motor neuropathy. Neurology 78(21):1644–1649
Acknowledgments
RH is supported by the Medical Research Council (UK) (G1000848) and the European Research Council (309548). HL is supported by a grant from the Medical Research Council UK (reference G1002274, Grant ID 98482). HL receives funding from the European Union Seventh Framework Programme (FP7/2007–2013) under grant agreement No. 305444 (RD-Connect) and 305121 (Neuromics). PFC is a Wellcome Trust Senior Fellow in Clinical Science and an NIHR Senior Investigator who also receives funding from the Medical Research Council (UK), the UK Parkinson’s Disease Society, and the UK NIHR Biomedical Research Centre for Ageing and Age-related disease award to the Newcastle upon Tyne Foundation Hospitals NHS Trust. We are grateful to the Medical Research Council (MRC) Centre for Neuromuscular Diseases Newcastle for supporting this project. SMM receives funding from Ataxia Ireland and has received educational grant funding from Novartis, Bayer-Schering, Teva and Lundbeck. The gene panel assay was set up with the support of the North Bristol Trust Showering Fund awarded to TA.
Conflicts of interest
The authors have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
B. Bansagi, T. Antoniadi contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bansagi, B., Antoniadi, T., Burton-Jones, S. et al. Genotype/phenotype correlations in AARS-related neuropathy in a cohort of patients from the United Kingdom and Ireland. J Neurol 262, 1899–1908 (2015). https://doi.org/10.1007/s00415-015-7778-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-015-7778-4