
Abstract
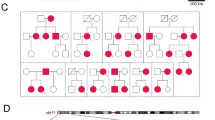
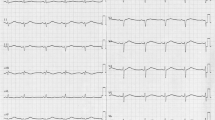
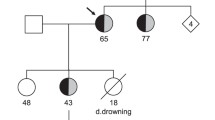
Patients with high-risk long QT syndrome (LQTS) mutations may experience life-threatening cardiac events. The present study sought to characterize a novel pathogenic mutation, KCNQ1p.Thr312del, in a Chinese LQT1 family. Clinical and genetic analyses were performed to identify this novel causative gene mutation in this LQTS family. Autosomal dominant inheritance of KCNQ1p.T312del was demonstrated in the three-generation pedigree. All mutation carriers presented with prolonged QT intervals and experienced recurrent syncope during exercise or emotional stress. The functional consequences of the mutant channel were investigated by computer homology modeling as well as whole-cell patch-clamp, western-blot and co-immunoprecipitation techniques using transfected mammalian cells. T312 is in the selectivity filter (SF) of the pore region of the KCNQ1-encoded channel. Homology modeling suggested that secondary structure was altered in the mutant SF compared with the wild-type (WT) SF. There were no significant differences in Kv7.1 expression, membrane trafficking or physical interactions with KCNE1-encoded subunits between the WT and mutant transfected channels. However, the KCNQ1p.T312del channels expressed in transfected cells were non-functional in the absence or presence of auxiliary KCNE1-subunits. Dominant-negative suppression of current density and decelerated activation kinetics were observed in cells expressing KCNQ1WT and KCNQ1p.T312del combined with KCNE1 (KCNQ1WT/p.T312del + KCNE1 channels). Those electrophysiological characteristics underlie the pathogenesis of this novel mutation and also suggest a high risk of cardiac events in patients carrying KCNQ1p.T312del. Although protein kinase A-dependent current increase was preserved, a significant suppression of rate-dependent current facilitation was noted in the KCNQ1WT/p.T312del + KCNE1 channels compared to the WT channels during 1- and 2-Hz stimulation, which was consistent with the patients’ phenotype being triggered by exercise. Overall, KCNQ1p.Thr312del induces a loss of function in channel electrophysiology, and it is a high-risk mutation responsible for LQT1.





Similar content being viewed by others
References
Schwartz PJ, Crotti L, Insolia R (2012) Long-QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol 5:868–877
Obeyesekere MN, Antzelevitch C, Krahn AD (2015) Management of ventricular arrhythmias in suspected channelopathies. Circ Arrhythm Electrophysiol 8:221–231
Jons C, Moss AJ, Lopes CM, McNitt S, Zareba W, Goldenberg I, Qi M, Wilde AA, Shimizu W, Kanters JK, Towbin JA, Ackerman MJ, Robinson JL (2009) Mutations in conserved amino acids in the KCNQ1 channel and risk of cardiac events in type-1 long-QT syndrome. J Cardiovasc Electrophysiol 20:859–865
Goldenberg I, Horr S, Moss AJ, Lopes CM, Barsheshet A, McNitt S, Zareba W, Andrews ML, Robinson JL, Locati EH, Ackerman MJ, Benhorin J, Kaufman ES, Napolitano C, Platonov PG, Priori SG, Qi M, Schwartz PJ, Shimizu W, Towbin JA, Vincent GM, Wilde AA, Zhang L (2011) Risk for life-threatening cardiac events in patients with genotype-confirmed long-QT syndrome and normal-range corrected QT intervals. J Am Coll Cardiol 57:51–59
Ruwald MH, Xu Parks X, Moss AJ, Zareba W, Baman J, McNitt S, Kanters JK, Shimizu W, Wilde AA, Jons C, Lopes CM (2016) Stop-codon and C-terminal nonsense mutations are associated with a lower risk of cardiac events in patients with long QT syndrome type 1. Heart Rhythm 13:122–131
Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT (1996) Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 384:80–83
Burashnikov A, Antzelevitch C (2000) Block of IKs does not induce early afterdepolarization activity but promotes β-adrenergic agonist-induced delayed afterdepolarization activity. J Cardiovasc Electrophysiol 11:458–465
Heijman J, Spatjens RL, Seyen SR, Lentink V, Kuijpers HJ, Boulet IR, de Windt LJ, David M, Volders PG (2012) Dominant-negative control of cAMP-dependent IKs upregulation in human long-QT syndrome type 1. Circ Res 110:211–219
Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, Denjoy I, Guicheney P, Breithardt G, Keating MT, Towbin JA, Beggs AH, Brink P, Wilde AA, Toivonen L, Zareba W, Robinson JL, Timothy KW, Corfield V, Wattanasirichaigoon D, Corbett C, Haverkamp W, Schulze-Bahr E, Lehmann MH, Schwartz K, Coumel P, Bloise R (2001) Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation 103:89–95
Wu J, Naiki N, Ding WG, Ohno S, Kato K, Zang WJ, Delisle BP, Matsuura H, Horie M (2014) A molecular mechanism for adrenergic-induced long QT syndrome. J Am Coll Cardiol 63:819–827
Bernèche S, Roux B (2005) A gate in the selectivity filter of potassium channels. Structure 13:591–600
Bichet D, Grabe M, Jan YN, Jan LY (2006) Electrostatic interactions in the channel cavity as an important determinant of potassium channel selectivity. Proc Natl Acad Sci USA 103:14355–14360
Priori SG, Blomstrom-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R, Hindricks G, Kirchhof P, Kjeldsen K, Kuck KH, Hernandez-Madrid A, Nikolaou N, Norekval TM, Spaulding C, Van Veldhuisen DJ (2015) 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC)Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Europace 17:1601–1687
Chen X, Yu L, Shi S, Jiang H, Huang C, Desai M, Li Y, Barajas-Martinez H, Hu D (2016) Neuronal Nav1.8 channels as a novel therapeutic target of acute atrial fibrillation prevention. J Am Heart Assoc 5:e004050
Moss AJ, Shimizu W, Wilde AA, Towbin JA, Zareba W, Robinson JL, Qi M, Vincent GM, Ackerman MJ, Kaufman ES, Hofman N, Seth R, Kamakura S, Miyamoto Y, Goldenberg I, Andrews ML, McNitt S (2007) Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation 115:2481–2489
Ruscic KJ, Miceli F, Villalba-Galea CA, Dai H, Mishina Y, Bezanilla F, Goldstein SA (2013) IKs channels open slowly because KCNE1 accessory subunits slow the movement of S4 voltage sensors in KCNQ1 pore-forming subunits. Proc Natl Acad Sci USA 110:E559–E566
Ikrar T, Hanawa H, Watanabe H, Okada S, Aizawa Y, Ramadan MM, Komura S, Yamashita F, Chinushi M, Aizawa Y (2008) A double-point mutation in the selectivity filter site of the KCNQ1 potassium channel results in a severe phenotype, LQT1, of long QT syndrome. J Cardiovasc Electrophysiol 19:541–549
Kobori A, Sarai N, Shimizu W, Nakamura Y, Murakami Y, Makiyama T, Ohno S, Takenaka K, Ninomiya T, Fujiwara Y, Matsuoka S, Takano M, Noma A, Kita T, Horie M (2004) Additional gene variants reduce effectiveness of beta-blockers in the LQT1 form of long QT syndrome. J Cardiovasc Electrophysiol 15:190–199
Ikrar T, Hanawa H, Watanabe H, Aizawa Y, Ramadan MM, Chinushi M, Horie M, Aizawa Y (2009) Evaluation of channel function after alteration of amino acid residues at the pore center of KCNQ1 channel. Biochem Biophys Res Commun 378:589–594
Zhou M, MacKinnon R (2004) A mutant KcsA K(+) channel with altered conduction properties and selectivity filter ion distribution. J Mol Biol 338:839–846
Morais-Cabral JH, Zhou Y, MacKinnon R (2001) Energetic optimization of ion conduction rate by the K + selectivity filter. Nature 414:37–42
Treptow W, Tarek M (2006) K + conduction in the selectivity filter of potassium channels is monitored by the charge distribution along their sequence. Biophys J 91:L81–L83
Goldenberg I, Moss AJ, Peterson DR, McNitt S, Zareba W, Andrews ML, Robinson JL, Locati EH, Ackerman MJ, Benhorin J, Kaufman ES, Napolitano C, Priori SG, Qi M, Schwartz PJ, Towbin JA, Vincent GM, Zhang L (2008) Risk factors for aborted cardiac arrest and sudden cardiac death in children with the congenital long-QT syndrome. Circulation 117:2184–2191
Shimizu W, Horie M, Ohno S, Takenaka K, Yamaguchi M, Shimizu M, Washizuka T, Aizawa Y, Nakamura K, Ohe T, Aiba T, Miyamoto Y, Yoshimasa Y, Towbin JA, Priori SG, Kamakura S (2004) Mutation site-specific differences in arrhythmic risk and sensitivity to sympathetic stimulation in the LQT1 form of congenital long QT syndrome: multicenter study in Japan. J Am Coll Cardiol 44:117–125
Jons C, Jin OU, Moss AJ, Reumann M, Rice JJ, Goldenberg I, Zareba W, Wilde AA, Shimizu W, Kanters JK, McNitt S, Hofman N, Robinson JL, Lopes CM (2011) Use of mutant-specific ion channel characteristics for risk stratification of long QT syndrome patients. Sci Transl Med 3:76ra28
Nakano Y, Shimizu W (2016) Genetics of long-QT syndrome. J Hum Genet 61:51–55
Funding
This work was supported by National Natural Science Foundation of China (no. 81270258), the State Key Program of National Natural Science Foundation of China (no. 81530015), Shanghai City Committee of Science and Technology Research Projects (nos. 12411951900, 13140903801, and 14441902502) and Doctoral Innovation Fund Projects from Shanghai Jiao Tong University School of Medicine (BXJ201427).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
About this article

Cite this article
Chen, XM., Guo, K., Li, H. et al. A novel mutation KCNQ1p.Thr312del is responsible for long QT syndrome type 1. Heart Vessels 34, 177–188 (2019). https://doi.org/10.1007/s00380-018-1223-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00380-018-1223-4