
Abstract
Pathogenic variants in epilepsy genes result in a spectrum of clinical severity. One source of phenotypic heterogeneity is modifier genes that affect expressivity of a primary pathogenic variant. Mouse epilepsy models also display varying degrees of clinical severity on different genetic backgrounds. Mice with heterozygous deletion of Scn1a (Scn1a+/−) model Dravet syndrome, a severe epilepsy most often caused by SCN1A haploinsufficiency. Scn1a+/− mice recapitulate features of Dravet syndrome, including spontaneous seizures, sudden death, and cognitive/behavioral deficits. Scn1a+/− mice maintained on the 129S6/SvEvTac (129) strain have normal lifespan and no spontaneous seizures. In contrast, admixture with C57BL/6J (B6) results in epilepsy and premature lethality. We previously mapped Dravet Survival Modifier loci (Dsm1-Dsm5) responsible for strain-dependent differences in survival. Gabra2, encoding the GABAA α2 subunit, was nominated as a candidate modifier at Dsm1. Direct measurement of GABAA receptors found lower abundance of α2-containing receptors in hippocampal synapses of B6 mice relative to 129. We also identified a B6-specific single nucleotide deletion within Gabra2 that lowers mRNA and protein by nearly 50%. Repair of this deletion reestablished normal levels of Gabra2 expression. In this study, we used B6 mice with a repaired Gabra2 allele to evaluate Gabra2 as a genetic modifier of severity in Scn1a+/− mice. Gabra2 repair restored transcript and protein expression, increased abundance of α2-containing GABAA receptors in hippocampal synapses, and rescued epilepsy phenotypes of Scn1a+/− mice. These findings validate Gabra2 as a genetic modifier of Dravet syndrome, and support enhancing function of α2-containing GABAA receptors as treatment strategy for Dravet syndrome.
Similar content being viewed by others

Introduction
Dravet syndrome is a severe, infant-onset developmental and epileptic encephalopathy (DEE) with at least 80% of cases resulting from de novo pathogenic variants in SCN1A (Claes et al. 2001). Individuals with Dravet syndrome display multiple seizure types that are often refractory to standard therapeutics, an elevated risk for sudden unexpected death in epilepsy (SUDEP), cognitive and behavioral deficits, and motor system dysfunctions (de Lange et al. 2019; Dravet 2011). Phenotype heterogeneity is common in monogenic epilepsy syndromes, with a spectrum of clinical presentations, ranging from benign seizures to intractable epilepsy and increased SUDEP risk (Cetica et al. 2017; Escayg et al. 2000; Gardella et al. 2016; Goldberg-Stern et al. 2014; Helbig and Tayoun 2016; Møller et al. 2015; Nakamura et al. 2013; Patino et al. 2009; Syrbe et al. 2016; Weckhuysen et al. 2012). Modifier genes that affect penetrance and expressivity are likely contributors to this variability and may serve as effective therapeutic targets for epilepsy.
SCN1A haploinsufficiency is the major cause of Dravet syndrome; therefore, mice with heterozygous deletion of Scn1a were developed to model Dravet syndrome (Miller et al. 2014; Ogiwara et al. 2007; Yu et al. 2006). Scn1a+/− mice recapitulate several core features of Dravet syndrome, including spontaneous seizures, elevated risk of sudden death, and neurobehavioral and cognitive deficits (Han et al. 2012; Hawkins et al. 2017a; Ito et al. 2013; Kalume et al. 2013; Miller et al. 2014; Ogiwara et al. 2013). These overt phenotypes are highly penetrant on the C57BL/6 J (B6) genetic background, but are absent when the mutation is on inbred 129 strain backgrounds (Kang et al. 2018; Miller et al. 2014; Ogiwara et al. 2007; Yu et al. 2006). Although 129.Scn1a+/− mice do not exhibit spontaneous seizures, they have elevated susceptibility to acute seizures induced by hyperthermia or chemoconvulsants (Mistry et al. 2014; Rubinstein et al. 2015). Taking advantage of the strain difference, we previously performed genetic mapping and identified several Dravet survival modifier (Dsm) loci responsible for strain-dependent differences in incidence of sudden unexpected death (Dsm1-5) (Miller et al. 2014). Gabra2, encoding the α2 subunit of the GABAA receptor, was the highest ranked candidate gene at the Dsm1 locus (Hawkins et al. 2016; Miller et al. 2014). Notable differences in Gabra2 expression among several inbred mouse strains had been previously reported (Korostynski et al. 2006; Mulligan et al. 2012; Yeo et al. 2016; Yu et al. 2020). Further work investigating a Gabra2 expression quantitative trait locus (eQTL) identified a single nucleotide de novo deletion in a splice acceptor site that was only present in B6 genomes after 1976 (Mulligan et al. 2019, 2012). This B6-specific variant causes a global reduction of Gabra2 transcript and protein expression in brain compared to older B6 strains and to 16 other common and wild-derived inbred mouse strains (Mulligan et al. 2019). Repair of the single nucleotide deletion within the Gabra2 intron fully restores expression back to the level of other inbred strains (Mulligan et al. 2019). B6 mice with repair of the Gabra2 allele (Mulligan et al. 2019) provided an ideal platform for testing the modifier potential of Gabra2 in Scn1a+/− Dravet mice.
In the current study, we sought to determine if this single nucleotide deletion was responsible for the modifier effect associated with the Dsm1 locus on chromosome 5. We used B6 mice carrying a repaired (Edited) Gabra2 allele (Edited/B6 mice) and crossed them with 129.Scn1a+/− mice to ascertain effects on Dravet phenotypes. We observed rescue of seizure, survival and neuronal phenotypes with this single nucleotide repair, validating Gabra2 as an epilepsy modifier gene.
Methods
Ethics statement
All animal care and experimental procedures were approved by the Northwestern University Animal Care and Use Committee in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The principles outlined in the ARRIVE (Animal Research: Reporting of in vivo Experiments) guideline and Basel declaration (including the 3 R concept) were considered when planning experiments.
Mice
CRISPR/Cas9 editing was used to insert a single intronic nucleotide into Gabra2 on the C57BL/6J (B6) strain, B6.Gabra2em1Geh, as previously described (Mulligan et al. 2019). Heterozygous edited Gabra2 mice (Edited/B6) were maintained by continual backcrossing to C57BL/6J (Jackson Labs, #000664, Bar Harbor, ME).
Scn1atm1Kea mice with deletion of the first coding exon, were generated by homologous recombination in TL1 ES cells as previously described (Miller et al. 2014). This line has been maintained by continual backcrossing of heterozygotes (abbreviated as Scn1a+/−) to 129S6/SvEvTac inbred mice (129, Taconic, #129SVE, Rensselaer, NY).
To generate double mutant mice, heterozygous Edited/B6 Gabra2 mice were crossed to 129.Scn1a+/− mice. The resulting offspring had an overall F1 genome background with B6/129 or Edited/129 alleles at Gabra2 and WT (+ / +) or heterozygous deletion (+ /−) at Scn1a.
Mice were maintained in a Specific Pathogen Free (SPF) barrier facility with a 14-h light/10-h dark cycle and access to food and water ad libitum. Female and male mice were used for all experiments.
Genotyping
DNA was isolated from P14 tail biopsies using the Gentra Puregene Mouse Tail Kit according to the manufacturer’s instructions (Qiagen, Valencia, CA). For Gabra2, approximately 250 ng of DNA was digested with BAMHI-HF (R3136, New England Biolabs, Ipswich, MA) at 37 °C for 1 h. Digested DNA was then diluted 1:1 with nuclease-free water and used as template for droplet digital PCR (ddPCR) using ddPCR Supermix for Probes (No dUTP) (Bio-Rad, Hercules, CA, USA) and a custom TaqMan SNP Genotyping Assay (Life Technologies, Carlsbad, CA) to detect the insertion (Primer 1: 5'-GCTCATCTTTCCATTTTTGCCGAAA; Primer 2: 5'-GGCTTACTACTTCTAAAACATGTACTGTTTTCA; Edited Allele Probe: 5'-GGCTTACTACTTCTAAAACATGTACTGTTTTCA; B6 Allele Probe 2: 5'-FAM-CTATTGTATACTCCTAAATAT-NFQ). Reactions were partitioned into droplets in a QX200 droplet generator (Bio-Rad). PCR conditions were 95 °C for 10 min, then 44 cycles of 95 °C for 30 s and 60 °C for 1 min (ramp rate of 2 °C/sec) and a final inactivation step of 98 °C for 5 min. Following amplification, droplets were analyzed with a QX200 droplet reader with QuantaSoft v1.6.6 software (Bio-Rad). For Scn1a, the genotype was determined by multiplex PCR as previously described (Miller et al. 2014).
Transcript analysis
Whole brain total RNA was extracted from mice with the following Gabra2 alleles: B6/B6, Edited/B6, B6/129, Edited/129, and 129/129. Total RNA was isolated using TRIzol reagent according to the manufacturer’s instructions. First-strand cDNA was synthesized from 2 µg of RNA using oligo(dT) primer and Superscript IV reverse transcriptase (RT) according to the manufacturer’s instructions (Life Technologies). First-strand cDNA samples were diluted 1:10 and 5 μl was used as template. Quantitative ddPCR was performed using ddPCR Supermix for Probes (No dUTP) (Bio-Rad) and TaqMan Gene Expression Assays (Life Technologies) for mouse Gabra2 (FAM-MGB-Mm00433435_m1) and Tbp (VIC-MGB-Mm00446971_m1) as a normalization standard. Reactions were partitioned into a QX200 droplet generator (Bio-Rad). Thermocycling conditions and analysis were performed as described for genotyping. Both assays lacked detectable signal in no-RT and no template controls. Relative transcript levels were expressed as a ratio of Gabra2 to Tbp concentrations, with 6–13 biological replicates per group. Mice ranged in age from P23 to P41. Statistical comparison between groups was made using ANOVA with Dunnett’s post-hoc comparisons (GraphPad Prism, San Diego, CA). Data are presented as mean ± SEM.
Immunoblotting
Whole brain P3 membrane protein fractions were isolated from mice with the following Gabra2 alleles: B6/B6, Edited/B6, B6/129, Edited/129 and 129/129 mice (Hartshorne and Catterall 1984). Membrane fractions (50 μg/lane) were separated on a 7.5% SDS-PAGE gel and transferred to nitrocellulose. Blots were probed with rabbit polyclonal Gabra2 antibody (822-GA2CL, PhosphoSolutions; RRID:AB_2492101; 1:1000) and mouse monoclonal anti-mortalin/GRP75 antibody (NeuroMab N52A/42; RRID:2120479; 1 μg/mL), which served as a normalization control. Anti-rabbit Alexa Fluor 790 and anti-mouse 680 antibodies (Jackson ImmunoResearch, 1:20,000) were used to detect signal on an Odyssey imaging system (Li-COR). Relative protein levels were determined by densitometry with Image Studio software (Li-COR) and expressed as a ratio of Gabra2 to GRP75, with 5–6 biological replicates per group. Mice ranged in age from P21 to P42. Statistical comparison between groups was made using one-way ANOVA with Dunnett’s post-hoc comparisons (GraphPad Prism). Data are presented as mean ± SEM.
Electrophysiology
Acute horizontal hippocampal slices (350 µm) were prepared from 22 to 31-day old male and female littermate mice with B6/B6 (n = 3 mice) or B6/Edited (n = 3 mice) alleles at Gabra2. All mice for this experiment were wild-type at Scn1a. Brains were quickly removed after decapitation and sections were made in ice-cold sucrose-slicing artificial cerebrospinal fluid (ACSF) containing the following (in mM): 85 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, 75 sucrose, 0.5 CaCl2 and 4 MgCl2 with 10 µM DL-APV and 100 µM kynurenate on a Leica Vibratome. Slices were incubated in the same sucrose ACSF for ∼30 min at 30 °C. The solution was gradually exchanged for a recovery ACSF containing the following (in mM): 125 NaCl, 2.4 KCl, 1.2 NaH2PO4, 25 NaHCO3, 25 glucose, 1 CaCl2, and 2 MgCl2 with 10 µM DL-APV and 100 µM kynurenate at room temperature. Slices were transferred to a recording chamber after a recovery period of at least 1.5 h. Recordings were made from CA1 pyramidal neurons in the hippocampus. Recording electrodes had tip resistances of 3–5 MΩ when filled with internal recording solution containing the following (in mM): 135 CsCl, 20 HEPES, 2 EGTA, 2 Mg-ATP, 0.5 Na-GTP, and 10 QX-314 (pH 7.25). Asynchronous IPSCs (aIPSCs) were recorded from perisomatic synapses where α2 GABAA receptor subunits are enriched (Nomura et al. 2019; Prenosil et al. 2006). Perisomatic aIPSCs were recorded in voltage clamp mode (− 70 mV) in the recording strontium-based ACSF containing the following (in mM): 125 NaCl, 2.4 KCl, 1.2 NaH2PO4, 25 NaHCO3, 25 glucose, 6 SrCl2, 0.5 CaCl2, and 2 MgCl2 with blockers of excitatory responses, CNQX (10 µM) and D-APV (50 µM), equilibrated with 95% O2 and 5% CO2. Perisomatic aIPSCs were evoked by electrical stimuli given through monopolar extracellular stimulating electrodes filled with ACSF and placed in stratum pyramidale and were analyzed within a 50–500 ms window following the stimulus artifact (Fernandes et al. 2015; Jurgensen and Castillo 2015; Nomura et al. 2019; Prenosil et al. 2006). Access resistance (Ra) was continuously monitored and experiments were omitted if the Ra changed > 20% during the recordings. Four to five cells were recorded per B6/B6 mouse; three to five cells were recorded per Edited/B6 mouse. Statistical comparison between genotype was made using Mann Whitney U-Test and paired data were compared using Wilcoxon signed rank test (GraphPad Prism). Data are presented as mean ± SEM.
8-Week survival monitoring
Scn1a+/− littermates with B6/129 or Edited/129 alleles at Gabra2 were weaned into standard vivarium holding cages containing four to five mice of the same age and sex. Survival was monitored until 8 weeks of age. During that time, all mice were monitored daily for general health. All recorded deaths were sudden and unexpected, occurring in otherwise healthy appearing animals (SUDEP-like). This was the same phenotype used for the original genetic mapping (Miller et al. 2014). Survival was first compared between sexes within genotypes. No sex difference was detected; therefore, groups were collapsed across sex for analysis of genotype effect. Survival statistics were calculated using time-to-event analysis with LogRank Mantel-Cox test (GraphPad Prism).
Seizure phenotyping
Scn1a+/− littermate mice carrying B6/129 or Edited/129 Gabra2 alleles were monitored for spontaneous generalized tonic–clonic seizures (GTCS), as previously described (Hawkins et al. 2017a). Briefly, at P18 or P19, mice were subjected to a single, brief (< 1 min) hyperthermia-induced GTCS and then immediately cooled to baseline temperature. If a GTCS did not occur, the mouse was excluded from the study (< 1%) in order to ensure all Scn1a+/− mice under study had a similar baseline with an initial ‘priming seizure’. Two to three mice of mixed genotype and sex were placed in a monitoring cage with ad libitium access to standard rodent chow and water. Spontaneous GTCS frequency was captured by continuous video monitoring as previously described (Hawkins et al. 2017a, 2016). Mice were monitored beginning at midnight (12–16 h post priming) for 12–14 consecutive days (278–336 h) or until sudden death occurred. This window captures the period of highest seizure frequency in Dravet mouse models (Cheah et al. 2013; Favero et al. 2018; Miller et al. 2014; Mistry et al. 2014; Oakley et al. 2009). Videos were scored offline by reviewers blinded to genotype to determine the frequency and severity of spontaneous GTCS. The total number of seizures for each mouse was divided by the total hours monitored and then converted to a seizure frequency per 24 h. GTCS events were scored using a modified Racine scale adapted for the Scn1a+/− model as follows: (1) Rearing and paddling, straub tail with no other movement; (2) Rearing and paddling, straub tail, loss of posture, short bursts of movement, often backwards; (3) Rearing and paddling with wild running and/or jumping without loss of posture; (4) Rearing and paddling with wild running and/or jumping with loss of posture; (5) Rearing and paddling with wild running and/or jumping with loss of posture progressing to tonic hindlimb extension (HLE); (6) Rearing and paddling with wild running and/or jumping with loss of posture progressing to tonic HLE ending in death. The number of mice in each score category was compared between groups using a chi-square test. The proportion of seizures with HLE (stage 5–6) was determined for each mouse based on presence or absence of tonic HLE phase for each GTCS event and proportions were averaged by genotype. Seizure frequency and average HLE proportions were first compared between sexes within genotypes. No sex difference was detected; therefore, groups were collapsed across sex for analysis of genotype effect. Seizure frequency and average HLE proportions were compared between genotypes using Mann Whitney U-Tests (GraphPad Prism). Data are presented as means ± SEM.
Results
In our prior genetic mapping studies, we nominated Gabra2 as a top Dsm1 candidate modifier gene influencing survival of Scn1a+/− mice and proposed that the observed differential expression of Gabra2 between 129 and B6 was the likely mechanism (Hawkins et al. 2016). We showed robust strain-dependent differential expression, which was unaffected by genotype (WT v. Scn1a+/−) on the same strain (Hawkins et al. 2019, 2016). Differential expression of Gabra2 was present prior to seizure onset (P14) and persisted following seizure onset (P24) (Supplemental Fig. S1) (Hawkins et al. 2019). Furthermore, Gabra2 expression was not altered by recent seizure events (< 24 h before RNA isolation) (Supplemental Fig. S1a) (Hawkins et al. 2019). Expression of other hippocampal GABAA α subunit transcripts (Gabra1, Gabra3, Gabra4, Gabra5) did not differ between 129 and B6 or between genotypes (WT v. Scn1a+/−) (Supplemental Fig. S1b) (Hawkins et al. 2019). In parallel work, we attributed lower expression in B6 to a single nucleotide intronic deletion present only in the current B6 genome (Mulligan et al. 2019). Repair of the B6-specific variant by re-insertion of the deleted nucleotide via CRISPR/Cas9 editing restored expression of Gabra2 to again match that of other mouse strains, including 129. (Fig. 1a) (Mulligan et al. 2019). The B6 mice with restored expression of Gabra2 (Edited) provide a definitive resource to test the hypothesis that strain-dependent differences in Gabra2 expression are responsible for the Dsm1 modifier effect and have even enabled us to localize the effect to specific nucleotide differences among the parental strains.

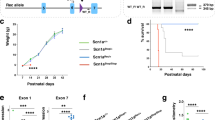
Editing of Gabra2 B6 allele normalizes expression relative to 129. A The B6 Gabra2 intronic deletion is located on Chr 5 at 71,014,638 bp (GRCm38.p6) (yellow). Sequence of the B6 allele (black) is compared to the Edited (grey) and 129S6/SvEvTac alleles (blue). B Breeding scheme for mice used in the study. For expression and electrophysiology experiments, isogenic B6.Gabra2 Edited mice were crossed with B6 to generate offspring with Edited/B6 (grey) or B6/B6 (black) alleles at Gabra2. For expression and seizure experiments, B6.Gabra2 mice were crossed with isogenic 129.Scn1a+/− mice to generate F1.Scn1a+/− or F1. Scn1a+/+ mice with Edited/129 (purple) or B6/129 (red) alleles at Gabra2. C Relative expression of Gabra2 transcript assayed by quantitative RT-ddPCR on whole brain samples from mice with B6/B6, Edited/B6, B6/129, Edited/129 and 129/129 alleles at Gabra2. Transcript expression differed between genotypes (F4, 35 = 104.5, p < 0.0001; one-way ANOVA). Relative to B6/B6 (0.85 ± 0.04), Gabra2 transcript expression was ~ 1.8-fold higher in Edited/B6 (1.78 ± 0.05) and B6/129 (2.00 ± 0.08), and ~ threefold higher in Edited/129 (3.11 ± 0.14) and 129/129 (2.85 ± 0.06). **** p < 0.0001, Dunnett’s post-hoc comparisons to B6/B6. Symbols represent samples from individual mice, horizontal lines represent group averages, and error bars represent SEM with 6–13 mice per genotype. D Quantification of GABRA2 protein expression determined from western blots of whole brain membrane protein from mice with B6/B6, Edited/B6, B6/129, Edited/129 and 129/129 alleles at Gabra2 assayed by western blot. Relative GABRA2 protein differed between genotypes (F4, 22 = 5.349, p = 0.0037; One-way ANOVA). Relative to B6/B6 (1.0 ± 0.1), GABRA2 protein expression was ~ 2.2-fold higher in Edited/B6 (2.2 ± 0.3) and B6/129 (2. ± 0.2), and ~ threefold higher in Edited/129 (2.4 ± 0.3) and 129/129 (2.9 ± 0.4). *p < 0.03, **p < 0.01, *** p < 0.0009, Dunnett’s post-hoc comparisons to B6/B6. E Representative immunoblot probed for GABRA2 (green) and GPR75/Mortalin (red), which served as a loading control
Single nucleotide repair of Gabra2 increases transcript and protein expression
We first confirmed that repair of Gabra2 (Edited/B6) altered allele-specific expression when crossed with the 129 strain (Fig. 1b). We evaluated transcript and protein expression in wild-type (WT) mice carrying the following alleles at Gabra2: B6/B6, Edited/B6, B6/129, Edited/129 or 129/129 (Fig. 1b). Gabra2 transcript expression differed between genotypes (F4, 35 = 104.5, p < 0.0001, One-way ANOVA) (Fig. 1c). Allele-specific expression differences between B6/B6, 129/B6 and 129/129 were consistent with prior reports (Hawkins et al. 2019, 2016; Miller et al. 2014; Yu et al. 2020). Expression of Gabra2 transcript in Edited/B6 and B6/129 was elevated ~ 1.8-fold relative to B6/B6 (p < 0.0001, Dunnett’s), while Gabra2 expression levels in Edited/129 and 129/129 were elevated by ~ threefold relative to B6/B6 (Fig. 1c) (p < 0.0001, Dunnett’s). GABRA2 protein expression followed the same pattern as transcript and differed between genotypes, (F4, 22 = 5.349, p = 0.0037, One-way ANOVA) (Fig. 1d, e). GABRA2 expression in Edited/B6 and B6/129 was approximately 2.2-fold higher relative to B6/B6 (p < 0.03, Dunnett’s), while expression in Edited/129 and 129/129 was ~ 2.6-fold higher relative to B6/B6 (p < 0.01, Dunnett’s) (Fig. 1d, e). These results demonstrate that repair of the B6 Gabra2 allele normalized transcript and protein expression to levels that were comparable to 129, consistent with our prior report of normalized expression (Mulligan et al. 2019).
Single nucleotide repair of Gabra2 alters neuronal phenotype
We previously demonstrated that perisomatic inhibitory synapses of hippocampal CA1 neurons had a greater abundance of α2-containing GABAA receptors in 129 mice compared to B6 mice (Nomura et al. 2019). α2 GABAA receptor mediated currents can be distinguished by the use of the selective α2/α3 positive allosteric modulator (PAM) AZD7325, which has a larger effect on slowing the decay kinetics of inhibitory postsynaptic currents (IPSCs) enriched in α2 subunits (Nomura et al. 2019). To determine whether the Edited allele affected synaptic GABAA receptors, we recorded perisomatic IPSCs in CA1 neurons in B6/B6 and Edited/B6 mice. Evoked IPSCs were desynchronized by application of extracellular Sr2+ so that asynchronous quantal GABAergic events (aIPSCs) could be recorded. aIPSCs from perisomatic synapses were recorded by stimulating in Stratum Pyramidale (SP) during a control period and after application of AZD7325 (100 nM) and current decay times were measured (Fig. 2). aIPSCs from B6/B6 mice exhibited an average baseline decay of 7.07 ± 0.28 ms during the control period, and aIPSCs from Edited/B6 mice had an average decay of 6.89 ± 0.51 ms during the baseline period. AZD7325 application prolonged aIPSC decay times in slices from both B6/B6 mice (8.76 ± 0.35 ms, p = 0.0002, Wilcoxon) and Edited/B6 mice (9.576 ± 0.64 ms, p = 0.0010, Wilcoxon). However, the effect of AZD7325 was significantly greater in recordings from Edited/B6 slices, which exhibited a 140 ± 5.3% increase compared to B6/B6 which had a125 ± 2.6% increase (Fig. 2d, p = 0.047, Mann–Whitney). This suggests that perisomatic CA1 GABAergic synapses in Edited/B6 mice are enriched in α2-containing receptors compared to those in B6/B6 mice. Consistent with our previous report (Nomura et al. 2019), the amplitude of aIPSC was unaffected by genotype (p = 0.12, Mann Whitney) or AZD7325 administration (B6/B6: p = 0.94; B6/Edited: p = 0.70; Wilcoxon) (Fig. 2c) (B6/B6 baseline: 58.1 ± 3.1 pA, n = 13; B6/B6 post: 102 ± 3.6%; and Edited/B6 baseline: 68.5 ± 4.7 pA, n = 11; Edited/B6 post: 100.6 ± 4.2%). Furthermore, aIPSC frequency (B6/B6 baseline: 6.56 ± 0.56 Hz; B6/B6 post: 6.92 ± 0.44 Hz; and Edited/B6 baseline: 7.10 ± 0.87 Hz; Edited/B6 7.37 ± 0.59 Hz) was unaffected by genotype (p > 0.999, Mann–Whitney) or AZD7325 administration (B6/B6 p = 0.33, Edited/B6 p = 0.50, Wilcoxon). These results demonstrate that repair of the B6-specific Gabra2 allele normalized functional expression of α2-containing GABAA receptors in perisomatic CA1 GABAergic synapses, likely by altering subunit composition rather than altering the number of perisomatic GABAA receptors.

AZD7325 has a larger effect on inhibitory synapses in Edited/B6 mice. A Representative traces of perisomatic aIPSCs in CA1 neurons before (baseline) and after AZD7325 (post) treatment in slices from B6/B6 mice and B slices from Edited/B6 mice. C Effect of AZD7325 on amplitudes of perisomatic aIPSC in CA1 of B6/B6 and Edited/B6 mice. D Effect of AZD7325 on the decay kinetics of aIPSC in B6/B6 and Edited/B6 mice. AZD7325 had a greater effect on decay kinetics in Edited/B6 mice (140 ± 5.3%) compared to B6/B6 mice (125 ± 2.6%). *p = 0.047, Mann–Whitney. For panels C, D, symbols represent samples from individual cells, horizontal lines represent group averages, and error bars represent SEM
Single nucleotide repair of Gabra2 improves phenotype of F1.Scn1a +/− mice
129.Scn1a+/− mice have no overt unprovoked seizures or premature lethality phenotype, whereas F1.Scn1a+/− mice have spontaneous seizures and premature lethality (Kang et al. 2018; Miller et al. 2014; Ogiwara et al. 2007; Yu et al. 2006). Similarly, mapping with interval-specific congenic (ISC) strains demonstrated that homozygosity for 129 alleles in the Gabra2 region could rescue seizure and sudden death phenotypes in otherwise F1.Scn1a+/− mice (Hawkins et al. 2016). To further refine the genetic mechanism, we investigated whether repair of the B6-specific Gabra2 intronic variant could improve survival of F1.Scn1a+/− mice. We crossed B6 mice with heterozygous repair of Gabra2 (B6/Edited) to 129.Scn1a+/− to generate F1.Scn1a+/− mice carrying Edited/129 or B6/129 alleles at Gabra2 (Fig. 1b). Survival was improved in F1.Scn1a+/− mice with Edited/129 versus B6/129 alleles at Gabra2 (p < 0.0001, Logrank Mantel-Cox). Mice with Edited/129 alleles had 97% (32 of 33) survival to 8 weeks of age compared to only 43% (13 of 30) of mice with B6/129 alleles (Fig. 3a), supporting Gabra2 as the modifier gene at the Dsm1 locus, which was originally mapped using 8-week survival as the primary phenotype (Miller et al. 2014).

Survival and seizure burden improved in F1.Scn1a+/− with Edited/129 versus B6/129 alleles at Gabra2. A Kaplan Meier survival plot comparing 8 week survival of B6/129 and Edited/129 F1.Scn1a+/− mice. Survival was worse in B6/129 mice (43%) compared to Edited/129 (97%) with n = 30–33 per genotype. ****p < 0.0001, Logrank Mantel-Cox. B The proportion of mice exhibiting spontaneous GTCS and average GTCS frequency differed between F1.Scn1a+/− mice with B6/129 (81% with seizures; 3.4 ± 0.7 GTCS/day) versus Edited/129 (35% with seizures; 0.8 ± 0.4 GTCS/day) alleles at Gabra2. Symbols represent samples from individual mice, horizontal lines represent the group average, and error bars represent SEM with n = 20–21 per genotype. ***p = 0.0008, Mann–Whitney. C Percentage of seizures in each category of an adapted modified Racine scale (colors defined below) differed between F1.Scn1a+/− mice with B6/129 or Edited/129 alleles at Gabra2. The majority of seizures in Edited/129 mice scored ≤ 4 compared to 63% in B6/129 mice. B6/129 n = 246 seizures total; Edited/129 n = 94 seizures total. p < 0.0006, chi-square test. D Among mice with GTCS, the average proportion of GTCS that progressed to HLE differed between mice with B6/129 (0.50 ± 0.07) versus Edited/129 (0.16 ± 0.04) alleles at Gabra2. Symbols represent samples from individual mice, and error bars represent SEM with n = 7–17 per genotype. ***p = 0.0007, Mann–Whitney. E Spontaneous GTCS diary plots for individual F1.Scn1a+/− mice with B6/129 or Edited/129 alleles at Gabra2. Each row represents a single F1.Scn1a+/− mouse (n = 20–21 per genotype) over the 336-h monitoring period or until occurrence of sudden, unexpected death indicated by a black triangle. Green circles represent subjects with no GTCS events. Tick marks represent individual seizure events with color indicating severity score using an adapted modified Racine scale. Colors used for the modified Racine scale adapted for Scn1a+/− mice in c and e are as follows: Green = 1, rearing and paddling, straub tail with no other movement; Orange = 2, rearing and paddling, straub tail, loss of posture, short bursts of movement, often backwards; Grey = 3, rearing and paddling with wild running and/or jumping without loss of posture; Teal = 4, rearing and paddling with wild running and/or jumping with loss of posture; Pink = 5, rearing and paddling with wild running and/or jumping with loss of posture progressing to tonic hindlimb extension (HLE); Black = 6, rearing and paddling with wild running and/or jumping with loss of posture progressing to tonic hindlimb extension (HLE) and death
Next, we investigated if the Gabra2 variant altered seizure frequency and/or severity. At P18-19, F1.Scn1a+/− mice with Edited/129 and B6/129 alleles at Gabra2 were subjected to a single hyperthermia-induced priming seizure and quickly cooled to baseline temperature (Hawkins et al. 2017a). We previously demonstrated that this paradigm enhances seizure incidence in F1.Scn1a+/− mice and improves discrimination power for ascertaining seizure reduction (Hawkins et al. 2017a, 2017b). Importantly, the temperature for seizure onset did not differ between Edited/129 (41.1 ± 0.2 °C) and B6/129 (41.2 ± 0.2 °C) groups (p = 0.9127, Student’s t-test), as expected since 129.Scn1a+/− mice also have a similar temperature threshold for hyperthermia-induced seizures (Supplemental Fig. S2). All mice in both the Edited/129 and B6/129 groups had a hyperthermia-induced priming seizure. Following the priming seizure, mice were continuously monitored for subsequent spontaneous generalized tonic–clonic seizures (GTCS) for 12–14 days or until sudden death occurred. This window coincides with the period of highest seizure frequency in Dravet mouse models (Cheah et al. 2013; Favero et al. 2018; Miller et al. 2014; Mistry et al. 2014; Oakley et al. 2009). The proportion of F1.Scn1a+/− exhibiting GTCS during the monitoring period differed between Gabra2 genotypes (p < 0.0026, Fisher’s exact). Only 35% of mice with Edited/129 alleles exhibited seizures, while 81% of mice with B6/129 alleles had seizures (Fig. 3b). Seizure frequency in F1.Scn1a+/− mice with Edited/129 alleles was lower (0.8 ± 0.4 GTCS/day) relative to mice with B6/129 alleles (3.4 ± 0.7 GTCS/day) (Fig. 3b) (p < 0.0018, Mann–Whitney).
While previous studies suggested a correlation between seizure frequency and survival, additional studies have shown that the relationship between total seizure frequency and survival is more complex, and that seizure severity is a critical factor (Kalume et al. 2013; Teran et al. 2019). Recorded/witnessed sudden death events in Scn1a+/− mice occur exclusively following seizures that progress to tonic HLE, while high frequencies of non-HLE seizures are tolerated (Hawkins et al. 2017a, 2017b; Kalume et al. 2013; Kang et al. 2019; Teran et al. 2019). Therefore, we also assessed seizure severity using a modified Racine scale adapted for the Scn1a+/− Dravet model. Across the population of mice with Edited/129 alleles, 84% of seizures scored 4 or below, while 63% of seizures in mice with B6/129 alleles scored similarly. Importantly, the proportion of GTCS events progressed to HLE in individual mice with Edited/129 alleles was lower (16 ± 4% of events) relative to mice with B6/129 alleles (50 ± 7% of events) (p = 0.0007, Mann–Whitney) (Fig. 3c, d). Seizure diary plots show individual mice and their respective scored seizure events (Fig. 3e). Together, these data demonstrate that repair of the B6 Gabra2 allele lessened seizure burden of F1.Scn1a+/− mice, supporting Gabra2 as the gene responsible for the modifier effect associated with the Dsm1 locus.
Discussion
In the present study, we demonstrated that Gabra2 is the modifier gene Dsm1 responsible for the strain-dependent difference in survival between F1 and 129 Scn1a+/− mice attributed to Dsm1 (Hawkins et al. 2016; Miller et al. 2014). Furthermore, we defined the responsible nucleotide difference underlying the modifier effect. Editing of the B6-specific single nucleotide intronic deletion in Gabra2 normalized brain transcript and protein expression relative to the 129 allele, elevated enrichment of α2-containing GABAA receptors in hippocampal synapses, and dramatically improved seizure and sudden unexpected death (SUDEP-like) phenotypes in the F1.Scn1a+/− Dravet mouse model. This work has clear therapeutic implications and suggests that interventions that increase CNS expression or function of GABRA2 should improve outcomes in Dravet syndrome. Moreover, Gabra2 was also recently nominated as a modifier of survival based on genetic mapping in a mouse model of SCN8A-associated DEE (Yu et al. 2020). Thus, the therapeutic implications may apply more broadly to other DEEs.
In previous work we and others demonstrated robust allele-specific expression of Gabra2 (Hawkins et al. 2019, 2016; Mulligan et al. 2019, 2012; Yu et al. 2020). Also, we established that unusually low GABRA2 protein expression is caused by a non-coding single nucleotide deletion in the C57BL/6J genome that alters splicing efficiency (Hawkins et al. 2017b; Korostynski et al. 2006; Mulligan et al. 2019, 2012; Yeo et al. 2016). B6 mice are the most commonly used laboratory mouse strain and was the first to be sequenced (GRCm38). With drastic improvements in next-generation sequencing techniques, it has become evident the GRCm38 reference genome, generated from filial (F) generation 204–207 mice, may differ from the current (~ F226) B6 mouse strain (Fairfield et al. 2015; Sarsani et al. 2019). Recently, Sarani et al. sequenced C57BL/6JEve, the “mother” (F223) of the current laboratory B6 animals sourced from the Jackson Laboratory (F226) (Sarsani et al. 2019). The group identified 59 indels and inversions between B6-Eve and GRCm38, many located within noncoding intronic regions (Sarsani et al. 2019). Furthermore, recent efforts from the Jackson Laboratory identified 1083 quality control-filtered variants between the GRCm38 reference sequence and sequencing from the most recent inbreeding generations of C57BL/6J (Fairfield et al. 2015). These variants can provide insight into the private de novo variants in the B6 genome compared to other inbred strains. This is of particular interest when investigating genetic modifiers of diseases, including epilepsy. Another recent example of a B6-specific de novo variant that acts as a genetic modifier of epilepsy is the C50T variant in neuronal tRNA n-Tr20. The C50T variant results in lower n-Tr20 levels that are associated with enhanced seizure susceptibility in WT mice and elevated spike wave discharge incidence in the Gabrg2R43Q absence epilepsy mouse model (Ishimura et al. 2014; Kapur et al. 2020).
While editing of the C57BL/6J-specific Gabra2 allele improved many aspects of the Dravet-like phenotype, F1.Scn1a+/− characteristics were not diminished into a benign 129.Scn1a+/− phenotype, as spontaneous seizures were still observed. This was not unexpected as our initial genetic mapping of the Dravet survival modifiers (Dsm) identified multiple loci (Dsm1-5), with the protective effect of 129 alleles at the Dsm1 locus accounting for 4–10% of the phenotypic variance (Miller et al. 2014). In terms of Dsm1, the magnitude of Gabra2 allele effects was similar to our prior fine mapping study using interval-specific congenic mice, with survival improving to 98 or 92%, respectively, and 55 or 42% reduction in occurrence of severe HLE seizures (Hawkins et al. 2016). This suggests that Gabra2 is the major modifier at the Dsm1 locus. Three additional modifier loci of the Scn1a+/− phenotype (Dsm2, Dsm3, Dsm5) have yet to be investigated (Miller et al. 2014). Preliminary examination of B6-specific private variants may suggest additional candidate modifier genes of survival in the mouse model of Dravet syndrome. Furthermore, our prior RNA-seq study demonstrated that strains differ in their homeostatic response to the Scn1a+/− mutation, and we and others have shown that seizures themselves can induce additional homeostatic remodeling (Dutton et al. 2017; Favero et al. 2018; Hawkins et al. 2017a, 2019; Salgueiro-Pereira et al. 2019).
A potential limitation of this study is that the single nucleotide deletion in B6 is a mouse-specific variant, which is not present in humans. However, studies on human tissue have identified multiple eQTLs for GABRA2, where expression level varies in association with genetic variation. For example, the single nucleotide polymorphism rs279829 has an overall minor allele frequency (MAF) of 0.2325, varying among populations with a MAF of 0.5259 in East Asian populations compared to a MAF of 0.05706 in African populations (Karczewski et al. 2020). Homozygous carriers of the minor allele have elevated GABRA2 expression relative to individuals that are heterozygous or homozygous for the major allele (Fig. 4) (GTEx Consortium 2015). Thus, natural variation in GABRA2 expression may contribute to variable expressivity of Dravet syndrome and other epilepsy phenotypes, and possibly contribute to treatment response for drugs that target GABAA receptors. Although we did not define the precise mechanism whereby restoration of α2 containing receptors alters inhibitory balance, it is likely that enhanced perisomatic phasic inhibition is predominantly mediated by α2-containing receptors, which is supported by the differential effect of AZD7325 in B6/edited mice. The lack of difference in the baseline properties of IPSC in B6/edited mice does not exclude this possibility. IPSC amplitude and kinetics are not altered even when the α2 subunit is completely knocked out (Panzanelli et al. 2011). This indicates that the majority of GABAA receptors at these synapses are heteromeric combinations of the α1 and α2 subunits, and the α1 subunit is dominant in dictating the properties of synaptic currents. Consistent with our previous study (Nomura et al. 2019), AZD7325 treatment had a greater effect in prolonging the decay kinetics of perisomatic IPSCs without affecting the amplitude, demonstrating a differential contribution of α2 activity in these synapses in B6/edited mice.

Human GABRA2 eQTL. Genetic variation at SNP rs279829 (chr4:46,333,638 in GRCh38.p12) is associated with variation in GABRA2 transcript expression (p = 0.0000025; n = 205 donor samples). Individuals that are homozygous for the minor allele have elevated expression relative to individuals that are heterozygous or homozygous for the major allele. Data used for this figure was obtained from: the GTEx Portal on 07/16/2020 (dbGaP Accession phs000424.v8.p2) [https://www.gtexportal.org/home/gene/GABRA2#eQTLBlock]
Interestingly, although there is strong strain-dependence for spontaneous seizures and premature death, strain-dependence does not extend to hyperthermia-induced seizures as Scn1a+/− mice on F1 and 129 strains have similar threshold temperatures. This suggests that 129 alleles shift seizure risk, rather than providing complete protection, and that hyperthermia is a strong provocateur in the context of an Scn1a mutation. This may have implications for preclinical screening assays, as hyperthermia may be a more demanding condition relative to spontaneous seizure monitoring.
This study highlights the importance of continued efforts in identifying modifier genes in mouse models. Several epilepsy modifier genes, including CACNA1G and GABRA2, were first identified in the Scn1a+/− mouse model of Dravet syndrome and later confirmed as epilepsy risk genes in humans (Bergren et al. 2005, 2009; Butler et al. 2018; Calhoun et al. 2016, 2017; Chemin et al. 2018; Feng et al. 2019; Hawkins and Kearney 2012, 2016; Hawkins et al. 2011, 2016; Hernandez et al. 2016; Kearney et al. 2006; Miller et al. 2014). Identifying modifier genes can provide refined insights into the molecular basis of genetic disease. Furthermore, it may provide the basis for improving predictions about disease course and clinical management. Finally, modifier genes and pathways can provide novel targets for therapeutics. Previously, we used AZD7325, a GABAA α2/α3-selective PAM, to modulate the neuronal phenotype of Scn1a+/− mice and demonstrated protective effects against hyperthermia-induced seizures (Nomura et al. 2019). The current study confirms that AZD7325 can be used to distinguish the GABAA receptor type in perisomatic synapses and directly demonstrates that the Edited allele contributes to an enriched α2 subunit content. Future studies assessing efficacy of treatment with AZD7325 or other GABRA2-selective PAMs on spontaneous seizures and survival would provide further support for targeting α2-containing GABAA receptors for the treatment of Dravet syndrome and, potentially, other DEEs that share reduced GABAergic signaling as a common pathogenic mechanism.
Data availability
All data and material are available upon reasonable request.
References
Bergren SK, Chen S, Galecki A, Kearney JA (2005) Genetic modifiers affecting severity of epilepsy caused by mutation of sodium channel Scn2a. Mamm Genome 16:683–690
Bergren SK, Rutter ED, Kearney JA (2009) Fine mapping of an epilepsy modifier gene on mouse Chromosome 19. Mamm Genome 20:359–366
Butler KM, Moody OA, Schuler E, Coryell J, Alexander JJ, Jenkins A, Escayg A (2018) De novo variants in GABRA2 and GABRA5 alter receptor function and contribute to early-onset epilepsy. Brain 141:2392–2405. https://doi.org/10.1093/brain/awy171
Calhoun JD, Hawkins NA, Zachwieja NJ, Kearney JA (2016) Cacna1g is a genetic modifier of epilepsy caused by mutation of voltage-gated sodium channel Scn2a. Epilepsia 57:e103-107. https://doi.org/10.1111/epi.13390
Calhoun JD, Hawkins NA, Zachwieja NJ, Kearney JA (2017) Cacna1g is a genetic modifier of epilepsy in a mouse model of Dravet syndrome. Epilepsia 58:e111–e115. https://doi.org/10.1111/epi.13811
Cetica V, Chiari S, Mei D, Parrini E, Grisotto L, Marini C, Pucatti D, Ferrari A, Sicca F, Specchio N, Trivisano M, Battaglia D, Contaldo I, Zamponi N, Petrelli C, Granata T, Ragona F, Avanzini G, Guerrini R (2017) Clinical and genetic factors predicting Dravet syndrome in infants with SCN1A mutations. Neurology 88:1037–1044. https://doi.org/10.1212/wnl.0000000000003716
Cheah CS, Westenbroek RE, Roden WH, Kalume F, Oakley JC, Jansen LA, Catterall WA (2013) Correlations in timing of sodium channel expression, epilepsy, and sudden death in Dravet syndrome. Channels (austin) 7:468–472. https://doi.org/10.4161/chan.26023
Chemin J, Siquier-Pernet K, Nicouleau M, Barcia G, Ahmad A, Medina-Cano D, Hanein S, Altin N, Hubert L, Bole-Feysot C, Fourage C, Nitschké P, Thevenon J, Rio M, Blanc P, Vidal C, Bahi-Buisson N, Desguerre I, Munnich A, Lyonnet S, Boddaert N, Fassi E, Shinawi M, Zimmerman H, Amiel J, Faivre L, Colleaux L, Lory P, Cantagrel V (2018) De novo mutation screening in childhood-onset cerebellar atrophy identifies gain-of-function mutations in the CACNA1G calcium channel gene. Brain 141:1998–2013. https://doi.org/10.1093/brain/awy145
Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P (2001) De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet 68:1327–1332
de Lange IM, Gunning B, Sonsma ACM, van Gemert L, van Kempen M, Verbeek NE, Sinoo C, Nicolai J, Knoers NVAM, Koeleman BPC, Brilstra EH (2019) Outcomes and comorbidities of SCN1A-related seizure disorders. Epilepsy Behav 90:252–259. https://doi.org/10.1016/j.yebeh.2018.09.041
Dravet C (2011) The core Dravet syndrome phenotype. Epilepsia 52(Suppl 2):3–9. https://doi.org/10.1111/j.1528-1167.2011.02994.x
Dutton SBB, Dutt K, Papale LA, Helmers S, Goldin AL, Escayg A (2017) Early-life febrile seizures worsen adult phenotypes in Scn1a mutants. Exp Neurol 293:159–171. https://doi.org/10.1016/j.expneurol.2017.03.026
Escayg A, MacDonald BT, Meisler MH, Baulac S, Huberfeld G, An-Gourfinkel I, Brice A, LeGuern E, Moulard B, Chaigne D, Buresi C, Malafosse A (2000) Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet 24:343–345
Fairfield H, Srivastava A, Ananda G, Liu R, Kircher M, Lakshminarayana A, Harris BS, Karst SY, Dionne LA, Kane CC, Curtain M, Berry ML, Ward-Bailey PF, Greenstein I, Byers C, Czechanski A, Sharp J, Palmer K, Gudis P, Martin W, Tadenev A, Bogdanik L, Pratt CH, Chang B, Schroeder DG, Cox GA, Cliften P, Milbrandt J, Murray S, Burgess R, Bergstrom DE, Donahue LR, Hamamy H, Masri A, Santoni FA, Makrythanasis P, Antonarakis SE, Shendure J, Reinholdt LG (2015) Exome sequencing reveals pathogenic mutations in 91 strains of mice with Mendelian disorders. Genome Res 25:948–957. https://doi.org/10.1101/gr.186882.114
Favero M, Sotuyo NP, Lopez E, Kearney JA, Goldberg EM (2018) A Transient developmental window of fast-spiking interneuron dysfunction in a mouse model of Dravet syndrome. J Neurosci 38:7912–7927. https://doi.org/10.1523/jneurosci.0193-18.2018
Feng YCA, Howrigan DP, Abbott LE, Tashman K, Cerrato F, Singh T, Heyne H, Byrnes A, Churchhouse C, Watts N, Solomonson M, Lal D, Heinzen EL, Dhindsa RS, Stanley KE, Cavalleri GL, Hakonarson H, Helbig I, Krause R, May P, Weckhuysen S, Petrovski S, Kamalakaran S, Sisodiya SM, Cossette P, Cotsapas C, De Jonghe P, Dixon-Salazar T, Guerrini R, Kwan P, Marson AG, Stewart R, Depondt C, Dlugos DJ, Scheffer IE, Striano P, Freyer C, McKenna K, Regan BM, Bellows ST, Leu C, Bennett CA, Johns EMC, Macdonald A, Shilling H, Burgess R, Weckhuysen D, Bahlo M, O’Brien TJ, Todaro M, Stamberger H, Andrade DM, Sadoway TR, Mo K, Krestel H, Gallati S, Papacostas SS, Kousiappa I, Tanteles GA, Štěrbová K, Vlčková M, Sedláčková L, Laššuthová P, Klein KM, Rosenow F, Reif PS, Knake S, Kunz WS, Zsurka G, Elger CE, Bauer J, Rademacher M, Pendziwiat M, Muhle H, Rademacher A, van Baalen A, von Spiczak S, Stephani U, Afawi Z, Korczyn AD, Kanaan M, Canavati C, Kurlemann G, Müller-Schlüter K, Kluger G, Häusler M, Blatt I, Lemke JR, Krey I, Weber YG, Wolking S, Becker F, Hengsbach C, Rau S, Maisch AF, Steinhoff BJ, Schulze-Bonhage A, Schubert-Bast S, Schreiber H, Borggräfe I, Schankin CJ, Mayer T, Korinthenberg R, Brockmann K, Kurlemann G, Dennig D, Madeleyn R, Kälviäinen R, Auvinen P, Saarela A, Linnankivi T, Lehesjoki A-E, Rees MI, Chung S-K, Pickrell WO, Powell R, Schneider N, Balestrini S, Zagaglia S, Braatz V, Johnson MR, Auce P, Sills GJ, Baum LW, Sham PC, Cherny SS, Lui CHT, Barišić N, Delanty N, Doherty CP, Shukralla A, McCormack M, El-Naggar H, Canafoglia L, Franceschetti S, Castellotti B, Granata T, Zara F, Iacomino M, Madia F, Vari MS, Mancardi MM, Salpietro V, Bisulli F, Tinuper P, Licchetta L, Pippucci T, Stipa C, Minardi R, Gambardella A, Labate A, Annesi G, Manna L, Gagliardi M, Parrini E, Mei D, Vetro A, Bianchini C, Montomoli M, Doccini V, Marini C, Suzuki T, Inoue Y, Yamakawa K, Tumiene B, Sadleir LG, King C, Mountier E, Caglayan SH, Arslan M, Yapıcı Z, Yis U, Topaloglu P, Kara B, Turkdogan D, Gundogdu-Eken A, Bebek N, Uğur-İşeri S, Baykan B, Salman B, Haryanyan G, Yücesan E, Kesim Y, Özkara Ç, Poduri A, Shiedley BR, Shain C, Buono RJ, Ferraro TN, Sperling MR, Lo W, Privitera M, French JA, Schachter S, Kuzniecky RI, Devinsky O, Hegde M, Khankhanian P, Helbig KL, Ellis CA, Spalletta G, Piras F, Piras F, Gili T, Ciullo V, Reif A, McQuillin A, Bass N, McIntosh A, Blackwood D, Johnstone M, Palotie A, Pato MT, Pato CN, Bromet EJ, Carvalho CB, Achtyes ED, Azevedo MH, Kotov R, Lehrer DS, Malaspina D, Marder SR, Medeiros H, Morley CP, Perkins DO, Sobell JL, Buckley PF, Macciardi F, Rapaport MH, Knowles JA, Fanous AH, McCarroll SA, Gupta N, Gabriel SB, Daly MJ, Lander ES, Lowenstein DH, Goldstein DB, Lerche H, Berkovic SF, Neale BM (2019) Ultra-rare genetic variation in the epilepsies: a whole-exome sequencing study of 17,606 individuals. Am J Human Genet 105:267–282. https://doi.org/10.1016/j.ajhg.2019.05.020
Fernandes HB, Riordan S, Nomura T, Remmers CL, Kraniotis S, Marshall JJ, Kukreja L, Vassar R, Contractor A (2015) Epac2 mediates cAMP-dependent potentiation of neurotransmission in the hippocampus. J Neurosci 35:6544–6553. https://doi.org/10.1523/jneurosci.0314-14.2015
Gardella E, Becker F, Møller RS, Schubert J, Lemke JR, Larsen LHG, Eiberg H, Nothnagel M, Thiele H, Altmüller J, Syrbe S, Merkenschlager A, Bast T, Steinhoff B, Nürnberg P, Mang Y, Bakke Møller L, Gellert P, Heron SE, Dibbens LM, Weckhuysen S, Dahl HA, Biskup S, Tommerup N, Hjalgrim H, Lerche H, Beniczky S, Weber YG (2016) Benign infantile seizures and paroxysmal dyskinesia caused by an SCN8A mutation. Ann Neurol 79:428–436. https://doi.org/10.1002/ana.24580
Goldberg-Stern H, Aharoni S, Afawi Z, Bennett O, Appenzeller S, Pendziwiat M, Kuhlenbäumer G, Basel-Vanagaite L, Shuper A, Korczyn AD, Helbig I (2014) Broad phenotypic heterogeneity due to a novel SCN1A mutation in a family with genetic epilepsy with febrile seizures plus. J Child Neurol 29:221–226. https://doi.org/10.1177/0883073813509016
GTEx Consortium (2015) Human genomics the genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348:648–660. https://doi.org/10.1126/science.1262110
Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB, Rubenstein JL, Scheuer T, de la Iglesia HO, Catterall WA (2012) Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission. Nature 489:385–390
Hartshorne RP, Catterall WA (1984) The sodium channel from rat brain. Purification and subunit composition. J Biol Chem 259:1667–1675
Hawkins NA, Anderson LL, Gertler TS, Laux L, George AL Jr, Kearney JA (2017a) Screening of conventional anticonvulsants in a genetic mouse model of epilepsy. Annals Clin Translational Neurol 4:326–339. https://doi.org/10.1002/acn3.413
Hawkins NA, Calhoun JD, Huffman AM, Kearney JA (2019) Gene expression profiling in a mouse model of Dravet syndrome. Exp Neurol 311:247–256. https://doi.org/10.1016/j.expneurol.2018.10.010
Hawkins NA, Kearney JA (2012) Confirmation of an epilepsy modifier locus on mouse chromosome 11 and candidate gene analysis by RNA-Seq. Genes Brain Behav 11:452–460
Hawkins NA, Kearney JA (2016) Hlf is a genetic modifier of epilepsy caused by voltage-gated sodium channel mutations. Epilepsy Res 119:20–23. https://doi.org/10.1016/j.eplepsyres.2015.11.016
Hawkins NA, Lewis M, Hammond RS, Doherty JJ, Kearney JA (2017b) The synthetic neuroactive steroid SGE-516 reduces seizure burden and improves survival in a Dravet syndrome mouse model. Sci Rep 7:15327. https://doi.org/10.1038/s41598-017-15609-w
Hawkins NA, Martin MS, Frankel WN, Kearney JA, Escayg A (2011) Neuronal voltage-gated ion channels are genetic modifiers of generalized epilepsy with febrile seizures plus. Neurobiol Dis 41:655–660
Hawkins NA, Zachwieja NJ, Miller AR, Anderson LL, Kearney JA (2016) Fine mapping of a Dravet syndrome modifier locus on mouse chromosome 5 and candidate gene analysis by RNA-Seq. PLoS Genet 12:e1006398. https://doi.org/10.1371/journal.pgen.1006398
Helbig I, Tayoun AA (2016) Understanding genotypes and phenotypes in epileptic encephalopathies. Mol Syndromol 7:172–181. https://doi.org/10.1159/000448530
Hernandez CC, Klassen TL, Jackson LG, Gurba K, Hu N, Noebels JL, Macdonald RL (2016) Deleterious rare variants reveal risk for loss of GABAA receptor function in patients with genetic epilepsy and in the general population. PLoS ONE 11:e0162883. https://doi.org/10.1371/journal.pone.0162883
Ishimura R, Nagy G, Dotu I, Zhou H, Yang XL, Schimmel P, Senju S, Nishimura Y, Chuang JH, Ackerman SL (2014) RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science 345:455–459. https://doi.org/10.1126/science.1249749
Ito S, Ogiwara I, Yamada K, Miyamoto H, Hensch TK, Osawa M, Yamakawa K (2013) Mouse with Nav1.1 haploinsufficiency, a model for Dravet syndrome, exhibits lowered sociability and learning impairment. Neurobiol Dis 49:29–40. https://doi.org/10.1016/j.nbd.2012.08.003
Jurgensen S, Castillo PE (2015) Selective dysregulation of hippocampal inhibition in the mouse lacking autism candidate gene CNTNAP2. J Neurosci 35:14681–14687. https://doi.org/10.1523/jneurosci.1666-15.2015
Kalume F, Westenbroek RE, Cheah CS, Yu FH, Oakley JC, Scheuer T, Catterall WA (2013) Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Investig 123:1798–1808. https://doi.org/10.1172/JCI66220
Kang SK, Hawkins NA, Kearney JA (2018) C57BL/6J and C57BL/6N substrains differentially influence phenotype severity in the Scn1a (+/-) mouse model of Dravet syndrome. Epilepsia Open 4:164–169. https://doi.org/10.1002/epi4.12287
Kang SK, Hawkins NA, Kearney JA (2019) C57BL/6J and C57BL/6N substrains differentially influence phenotype severity in the Scn1a+/− mouse model of Dravet syndrome. Epilepsia Open 4:164–169. https://doi.org/10.1002/epi4.12287
Kapur M, Ganguly A, Nagy G, Adamson SI, Chuang JH, Frankel WN, Ackerman SL (2020) Expression of the neuronal tRNA n-Tr20 regulates synaptic transmission and seizure susceptibility. Neuron 108:193-208.e199. https://doi.org/10.1016/j.neuron.2020.07.023
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, Neale BM, Daly MJ, MacArthur DG (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581:434–443. https://doi.org/10.1038/s41586-020-2308-7
Kearney JA, Yang Y, Beyer B, Bergren SK, Claes L, Dejonghe P, Frankel WN (2006) Severe epilepsy resulting from genetic interaction between Scn2a and Kcnq2. Hum Mol Genet 15:1043–1048
Korostynski M, Kaminska-Chowaniec D, Piechota M, Przewlocki R (2006) Gene expression profiling in the striatum of inbred mouse strains with distinct opioid-related phenotypes. BMC Genomics 7:146. https://doi.org/10.1186/1471-2164-7-146
Miller AR, Hawkins NA, McCollom CE, Kearney JA (2014) Mapping genetic modifiers of survival in a mouse model of Dravet syndrome. Genes Brain Behav 13:163–172. https://doi.org/10.1111/gbb.12099
Mistry AM, Thompson CH, Miller AR, Vanoye CG, George AL Jr, Kearney JA (2014) Strain- and age-dependent hippocampal neuron sodium currents correlate with epilepsy severity in Dravet syndrome mice. Neurobiol Dis 65:1–11. https://doi.org/10.1016/j.nbd.2014.01.006
Møller RS, Heron SE, Larsen LHG, Lim CX, Ricos MG, Bayly MA, van Kempen MJA, Klinkenberg S, Andrews I, Kelley K, Ronen GM, Callen D, McMahon JM, Yendle SC, Carvill GL, Mefford HC, Nabbout R, Poduri A, Striano P, Baglietto MG, Zara F, Smith NJ, Pridmore C, Gardella E, Nikanorova M, Dahl HA, Gellert P, Scheffer IE, Gunning B, Kragh-Olsen B, Dibbens LM (2015) Mutations in KCNT1 cause a spectrum of focal epilepsies. Epilepsia 56:e114–e120. https://doi.org/10.1111/epi.13071
Mulligan MK, Abreo T, Neuner SM, Parks C, Watkins CE, Houseal MT, Shapaker TM, Hook M, Tan H, Wang X, Ingels J, Peng J, Lu L, Kaczorowski CC, Bryant CD, Homanics GE, Williams RW (2019) Identification of a functional non-coding variant in the GABAA receptor α2 subunit of the C57BL/6J mouse reference genome: major implications for neuroscience research. Frontiers Genet 10:188. https://doi.org/10.3389/fgene.2019.00188
Mulligan MK, Wang X, Adler AL, Mozhui K, Lu L, Williams RW (2012) Complex control of GABA(A) receptor subunit mRNA expression: variation, covariation, and genetic regulation. PLoS ONE 7:e34586. https://doi.org/10.1371/journal.pone.0034586
Nakamura K, Kato M, Osaka H, Yamashita S, Nakagawa E, Haginoya K, Tohyama J, Okuda M, Wada T, Shimakawa S, Imai K, Takeshita S, Ishiwata H, Lev D, Lerman-Sagie T, Cervantes-Barragan DE, Villarroel CE, Ohfu M, Writzl K, Gnidovec Strazisar B, Hirabayashi S, Chitayat D, Myles Reid D, Nishiyama K, Kodera H, Nakashima M, Tsurusaki Y, Miyake N, Hayasaka K, Matsumoto N, Saitsu H (2013) Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology 81:992–998. https://doi.org/10.1212/WNL.0b013e3182a43e57
Nomura T, Hawkins NA, Kearney JA, George AL Jr, Contractor A (2019) Potentiating α2 subunit containing perisomatic GABAA receptors protects against seizures in a mouse model of Dravet syndrome. J Physiol 597:4293–4307. https://doi.org/10.1113/jp277651
Oakley JC, Kalume F, Yu FH, Scheuer T, Catterall WA (2009) Temperature- and age-dependent seizures in a mouse model of severe myoclonic epilepsy in infancy. Proc Natl Acad Sci USA 106:3994–3999. https://doi.org/10.1073/pnas.0813330106
Ogiwara I, Iwasato T, Miyamoto H, Iwata R, Yamagata T, Mazaki E, Yanagawa Y, Tamamaki N, Hensch TK, Itohara S, Yamakawa K (2013) Nav1.1 haploinsufficiency in excitatory neurons ameliorates seizure-associated sudden death in a mouse model of Dravet syndrome. Hum Mol Genet 22:4784–4804. https://doi.org/10.1093/hmg/ddt331
Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, Takeuchi T, Itohara S, Yanagawa Y, Obata K, Furuichi T, Hensch TK, Yamakawa K (2007) Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci 27:5903–5914
Panzanelli P, Gunn BG, Schlatter MC, Benke D, Tyagarajan SK, Scheiffele P, Belelli D, Lambert JJ, Rudolph U, Fritschy JM (2011) Distinct mechanisms regulate GABAA receptor and gephyrin clustering at perisomatic and axo-axonic synapses on CA1 pyramidal cells. J Physiol 589:4959–4980. https://doi.org/10.1113/jphysiol.2011.216028
Patino GA, Claes LRF, Lopez-Santiago LF, Slat EA, Dondeti RSR, Chen C, O’Malley HA, Gray CBB, Miyazaki H, Nukina N, Oyama F, De Jonghe P, Isom LL (2009) A functional null mutation of SCN1B in a patient with Dravet syndrome. J Neurosci 29:10764–10778. https://doi.org/10.1523/jneurosci.2475-09.2009
Prenosil GA, Gasser EMS, Rudolph U, Keist R, Fritschy J-M, Vogt KE (2006) Specific subtypes of GABAA receptors mediate phasic and tonic forms of inhibition in hippocampal pyramidal neurons. J Neurophysiol 96:846–857. https://doi.org/10.1152/jn.01199.2005
Rubinstein M, Han S, Tai C, Westenbroek RE, Hunker A, Scheuer T, Catterall WA (2015) Dissecting the phenotypes of Dravet syndrome by gene deletion. Brain 138:2219–2233. https://doi.org/10.1093/brain/awv142
Salgueiro-Pereira AR, Duprat F, Pousinha PA, Loucif A, Douchamps V, Regondi C, Ayrault M, Eugie M, Stunault MI, Escayg A, Goutagny R, Gnatkovsky V, Frassoni C, Marie H, Bethus I, Mantegazza M (2019) A two-hit story: seizures and genetic mutation interaction sets phenotype severity in SCN1A epilepsies. Neurobiol Dis 125:31–44. https://doi.org/10.1016/j.nbd.2019.01.006
Sarsani VK, Raghupathy N, Fiddes IT, Armstrong J, Thibaud Nissen F, Zinder O, Bolisetty M, Howe K, Hinerfeld D, Ruan X, Rowe L, Barter M, Ananda G, Paten B, Weinstock GM, Churchill GA, Wiles MV, Schneider VA, Srivastava A, Reinholdt LG (2019) The genome of C57BL/6J eve the mother of the laboratory mouse genome reference strain. G3 Genes Genomes Genetics 9:1795–1805. https://doi.org/10.1534/g3.119.400071
Syrbe S, Zhorov BS, Bertsche A, Bernhard MK, Hornemann F, Mütze U, Hoffmann J, Hörtnagel K, Kiess W, Hirsch FW, Lemke JR, Merkenschlager A (2016) Phenotypic variability from benign infantile epilepsy to ohtahara syndrome associated with a novel mutation in SCN2A. Molecular Syndromology 7:182–188. https://doi.org/10.1159/000447526
Teran FA, Kim Y, Crotts MS, Bravo E, Emaus KJ, Richerson GB (2019) Time of day and a ketogenic diet influence susceptibility to SUDEP in Scn1a (R1407X/+) mice. Front Neurol 10:278. https://doi.org/10.3389/fneur.2019.00278
Weckhuysen S, Mandelstam S, Suls A, Audenaert D, Deconinck T, Claes LRF, Deprez L, Smets K, Hristova D, Yordanova I, Jordanova A, Ceulemans B, Jansen A, Hasaerts D, Roelens F, Lagae L, Yendle S, Stanley T, Heron SE, Mulley JC, Berkovic SF, Scheffer IE, Jonghe Pd (2012) KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann Neurol 71:15–25. https://doi.org/10.1002/ana.22644
Yeo S, Hodgkinson CA, Zhou Z, Jung J, Leung M, Yuan Q, Goldman D (2016) The abundance of cis-acting loci leading to differential allele expression in F1 mice and their relationship to loci harboring genes affecting complex traits. BMC Genomics 17:620. https://doi.org/10.1186/s12864-016-2922-9
Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, Catterall WA (2006) Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci 9:1142–1149
Yu W, Hill SF, Xenakis JG, Pardo-Manuel de Villena F, Wagnon JL, Meisler MH (2020) Gabra2 is a genetic modifier of Scn8a encephalopathy in the mouse. Epilepsia 61:2847–2856. https://doi.org/10.1111/epi.16741
Acknowledgements
We thank Tyler Thaxton, Alexandra Huffman and Esha Raut for technical support.
Funding
This work was supported by NIH/NINDS grant NS084959 (JAK); NIH/NIMH Grant MH099114 (AC); NIH/NIAAA Grants U01AA013499 and U01AA016662 (RW and MM); and AA010422 and AA020889 (GH). The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for Fig. 4 in this manuscript were obtained from: the GTEx Portal on 07/16/2020 and dbGaP accession number phs000424.v8.p2 on 07/16/2020.
Author information
Authors and Affiliations
Contributions
Conceptualization: RWW, GEH, NAH, AC, JAK; Formal analysis: NAH, TN, JAK; Investigation: NAH, SD, LB, TN; Methodology: NAH, T, AC, JAK; Project administration: NAH, JA; Supervision: JAK; Visualization: NAH, TN, Anis Contractor, JK; Writing—original draft: NAH, TN, JAK; Writing—review & editing: NH, TN, SD, LB, RWW, GEH, MK, AC, JAK; Funding acquisition: JAK.
Corresponding author
Ethics declarations
Conflict of interest
JAK serves as a consultant to Encoded Genomics, Praxis Precision Medicines and NeuroCycle Therapeutics, and serves on the Scientific Advisory Boards of the Dravet Syndrome Foundation and FamilieSCN2A Foundation. NAH serves as a consultant to NeuroCycle Therapeutics. All other authors have declared that no competing interests exist.
Ethical approval
All animal care and experimental procedures were approved by the Northwestern University Animal Care and Use Committee in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A version of this manuscript has appeared as a preprint on the bioRxiv preprint server- (bioRxiv 2020.04.19.048546; https://doi.org/10.1101/2020.04.19.048546).
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hawkins, N.A., Nomura, T., Duarte, S. et al. Gabra2 is a genetic modifier of Dravet syndrome in mice. Mamm Genome 32, 350–363 (2021). https://doi.org/10.1007/s00335-021-09877-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-021-09877-1