
Abstract
D614G is one of the most reported mutations in the spike protein of SARS-COV-2 that has altered some crucial characteristics of coronaviruses, such as rate of infection and binding affinities. The binding affinity of different antiviral drugs was evaluated using rigid molecular docking. The reliability of the docking results was evaluated with the induced-fit docking method, and a better understanding of the drug-protein interactions was performed using molecular dynamics simulation. The results show that the D614G variant could change the binding affinity of antiviral drugs and spike protein remarkably. Although Cytarabine showed an appropriate interaction with the wild spike protein, Ribavirin and PMEG diphosphate exhibited a significant binding affinity to the mutated spike protein. The parameters of the ADME/T analysis showed that these drugs are suitable for further in-vitro and in-vivo investigation. D614G alteration affected the binding affinity of the RBD and its receptor on the cell surface.
Similar content being viewed by others


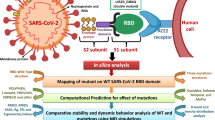
Introduction
COVID-19 is a spreading disease caused by Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [1] and had a worldwide fatality in the last months. SARS-CoV-2 is a member of coronaviruses with a close relation to the bat coronaviruses [2]. Although some approved vaccines have been developed to inhibit the virus infection [3,4,5], there is not any appropriate antiviral drug for this deadly virus to date. This can be assigned to the temporary characteristics of coronaviruses. However, the natural products have beneficial impacts in various aspects [6] such as anti- SARS-CoV-2 impacts, and can be utilized to treat COVID-19 [7]. It is also reported that clinical findings conflict with different groups and ages [8]. According to the nucleotide sequences of SARS-CoV-2, it is a member of Betacoronaviruses, such as the SARS and MERS HCoVs [9]. Seven different strains of human coronaviruses (HCoVs) have been described so far, including the 229E and NL63 strains of HCoVs, and the OC43, HKU1, SARS, MERS, and SARS-CoV-2 HCoVs [10]. Two protein groups characterize HCoVs: structural proteins like Spike marking all coronaviruses; and non-structural proteins, like RdRP [11]. COVID-19 has the capability of rapid genome mutation as it spreads [12]. These mutations are in different parts of virus structure, such as N-protein and S-protein with different stabilities [13]. Such variants have a reputation for changing the characteristics of the virus in various stages. For instance, in 2003/2004, a single amino acid change D480A/G in the receptor-binding domain (RBD) of SARS-COV-1 became a dominant variant. This variant provided the capability for the virus to escape from neutralizing antibody 80R [14]. Although the pace of sequence variant for SARS-COV-2 is not considerably high [15], it could attain more mutation during the world spread [16].
The main target for most vaccines and antibody-based therapeutics is the spike protein, which is also the prominent target for neutralizing antibodies, and the virus uses it for binding to the host cell and entry [17]. The RBD of the virus placed in the spike protein interacts with angiotensin-converting enzyme 2 (ACE2) to trigger the cleavage event. After that, the cleavage N-terminal domain of the spike protein causes the formation of post-fusion conformation, which triggers the virus/host-cell membrane fusion [18]. Accordingly, the monitoring of sequence variants in this protein is crucial, and this is worth noting that the single amino acid changes could be phenotypically pertinent [16]. One of the most reported mutations that have become dominant during the pandemic in numerous regions of the world is the D614G variant [19], and this variant could be solely or with other mutations such as L54F/D614G, D614G/D936Y, and D614G/S939F [13]. Even though the location of D614G is outside of the RDB, this variant is capable of altering RBD-ACE2 interaction [15]. D614G confers some more flexibility to the spike stalk and increases its ability to scan the host-cell surface, which can provide a better interaction with ACE2. The favored interactions are not necessarily more stable in terms of the interaction energy or binding affinity. They could be in terms of the number and type of interactions [20, 21]. Some studies show this variant has increased the viral load in the human body [16]. Korber et al. discussed the hypothesis that G614 causes more infection than D614 [16], and this hypothesis was supported by some other researchers. They investigated the clinical samples and observed a higher level of viral RNA in the infections from G614 [22]. Despite different reports on this mutation, the infection (interactions of SARS-CoV-2 spike protein with ACE2 receptor activating pre-/post-fusion conformational modifications inducing the virus entry into the human cells [23]) or transmissibility (how easy viruses spread and how harmful is a virus to its host [23]) of G614 has not been proved to be more than the viruses containing D614 [19]. Although it has been observed that antibodies generated from viruses containing D614 or G614 could adequately neutralize the virus [16], it does not mean that different drugs would have the same interaction with the wild form and D614G variant of the viruses. Although the impact of this mutation on the anti-viral drugs that inhibit the entrance of the virus to the host cell is unknown [19], our results show that D614G could considerably alter the drug-virus interactions.
In the fight against coronavirus, experts have employed three different developing drug strategies. The first strategy is to utilize the current broad scope of antivirals [24]. Nevertheless, the metabolic characteristics, dosages employed, implied effectiveness, and unfavorable impacts of these medicines are obvious, but they could not defeat coronaviruses in a targeted way. That is because of their extensive spectrum, and their destructive impacts could not be disregarded [25]. The next method is to discover molecules that can have remedial impacts on coronaviruses by selecting molecules given in the current molecular databases [26], such as DrugBank, Center Watch, and DailyMed. As this method uses the High-throughput screening of the compounds, various targets with different functions can be screened. Reclaiming approved drugs attempting to treat unknown illnesses, saving shelved drugs, and expanding patients’ lives make drug repurposing (also comprehended as drug repositioning) an interesting form of drug discovery [27]. In the third method, the classification of the coronaviruses is based on the genomic features and pathological characteristics. This classification will be used for generating different targeted medicines [25]. However, the medicines that would be obtained by this approach are more targeted for coronaviruses, but this method may need a large amount of time [28].
The quickest technique to obtain curative agents versus coronaviruses is to identify the possible therapeutics between established or emerging drugs. For this goal, various computational methods can be applied as profitable approaches [29, 30].
In this study, we have investigated the impact of the D614G variant on the virus-drug interactions of 28 approved or under investigation antiviral drugs. These results could be further used for new vaccines and drug discovery.
Materials and Methods
Sequence Alignment and Modeling
The mutated model of the SASRS-COV-2 spike was built using the Swiss Model web server [31]. As there is not a crystal structure of the D614G mutation with a resolution under 2.8 Å, we used the SASRS-COV-2 spike (PDB: 6ZB5) as the template, and the D614 residue was mutated to G614 in the FSATA sequences. The template with the electron microscopy method (6ZB5) was downloaded from the protein data bank (http://www.rcsb.org/pdb). The model was then aligned to the template for analyzing their identity, and the quality of 3D structures was next evaluated with the Ramachandran plot [32] using the Schrödinger suite.
Protein Preparation
For the preparation of the spike protein and the built model, the protein preparation module from the Schrödinger suite was used to add the hydrogen atoms, remove the waters beyond 5 Å of the binding sites, and optimization of the structure for creating an H-bonds network. Finally, the energy was minimized using the OPLS3e force field with a default setting of 0.30 Å RMSD. The mentioned process was performed at pH 7.0 ± 0.4 to produce correct protonation states.
Receptor Grid Generation
The receptor grid was generated using the Glide (Glide, Schrödinger, generated LLC, New York, NY, 2020). The cubic boxes with the size of 20˟20˟20 Å3 were centroid of site 1 and site 2. Site 1 is the location of D614G, and site 2 is the binding site of the RBD that is determined by the SiteMap module of Schrodinger. The default parameters of van der Waals scaling factor of 1 Å, partial charge cut off at 0.25, and 20 Å docked ligand length was used for the receptor grid generation.
Ligand Preparation
We created a library of twenty-eight approved or under investigation antiviral drugs as test ligand molecules against the built models. The ligands were prepared using the LigPrep module of Schrödinger, and the OPLS3e force field was used for the generation of ionization and tautomeric states at pH 7.0 ± 0.4 using the Epik module of LigPrep. The name and structures of chosen drugs are presented in Table1. The name and structure of chosen drugs are given in Table S1.
Molecular Docking
The molecular docking of selected drugs against the target was performed utilizing the GLIDE from the Schrödinger Suite with default parameters [33]. Standard precision (SP) and extra precision (XP) docking calculations were accomplished. The values of the scaling factor and partial charge cutoff were set at 0.80 and 0.15, respectively. As there could be several protonation states during the ligand preparation, the ligands that have a sufficient association with the receptor would be authorized. In the last step, the pose viewer was used for investigating the interactions of chosen drugs and protein docked systems (Table 2).
Molecular Docking Reproducibility
The reliability of the docking results was further evaluated using induced fit docking. The benefit of this method is the flexibility of the receptor, which produces more reliable poses. For the box option, the centroid of the residues was chosen, and the residues of previous boxes were selected. The energy window of the sample ring conformation was set to 2.5 kcal/mol, and the residues were refined within 5 Å of the ligand poses [34].
Molecular Dynamics Simulation for Dynamic Investigation
For a better understanding of the interactions between the drugs and protein, a MD simulation was carried out. For this purpose, the compound with the highest binding affinity for the mutated protein was chosen to be investigated with MD simulation. The complexes of the ligand and protein obtained from the docking study were used for the MD simulation. The MD simulation was carried out employing Desmond Software, Schrödinger. Before the simulation, the ligand was aligned with the protein, and the energy of the whole system was minimized with the minimization step of desmond. An orthorhombic box was used for the system, and the size of the box was then minimized by utilizing a solvent system of transferable intermolecular potential with 3 points (TIP3P) [35]. The system neutralization was performed with a suitable amount of Na + /Cl − ions and a salt concentration of 0.15 M using the system setup of Schrödinger [36]. Finally, the system was taken into the MD simulation for 60 ns with default relaxation protocol and the number of atoms, pressure, and temperature (NPT) ensemble. The Nose–Hoover protocol and isotropic scaling were used to set the pressure and temperature at 310.15 K (37 °C) and 1 atm, respectively [37].
Binding Free Energy Calculations
The MM/GBS method was utilized for the calculation of the binding free energy. The prime module of Schrödinger was used for this purpose, and the parameters were set as default. The VSGB and OPLS-2005 were used for the solvation model and force field, respectively. The formula of the binding free energy between the ligand and protein is as follows:
∆G bind = G complex – (G protein + G ligand),
where ∆G bind indicates the binding free energy, G complex is the binding free energy of the complex. The G protein and G ligand indicate the binding free energy of the protein and ligand, respectively.
ADME/T Analysis for Examination of Compounds’ Druggability
After the virtual screening, the ADME/T (absorption, distribution, metabolism, excretion, and toxicity) analysis was accomplished for antiviral drugs. This analysis will evaluate the potential of these antiviral drugs for being used in further in-vitro and in-vivo studies [38]. The ADME/T was conducted using QikProp of Schrödinger.
Results
SARS-COV-2 Spike D614G Modeling
The model of the mutated SARS-COV-2 spike exhibited a high sequence identity to the template with an identification of 99.9%. A graphical description of the wild and mutated form of SARS-COV-2 spike protein is presented in Fig. S0a. The RMSD obtained by the superimposition of the generated 3D model of D614G and the protein used as the template was calculated. This value was 0.508 Å. The alignment of surface glycoprotein SARS-COV-2 (YP_009724390.1), and spike protein is presented in Fig. S0b. The sequence alignment and the position of mutated residue for the built model and template (6ZB5) are shown in Fig. S0c. For a better comparison, the location of site 1 and site 2 is presented in Fig. S0d. The quality of the 3D structure for the homology model was evaluated via the Ramachandran plot using Schrödinger suites. The Ramachandran plot is presented in Fig. S0e. More than 99.6% of residues are in the allowed regions for the mutated model (98.9% in the favored regions and 0.7% in the allowed regions), which implies the model’s reliability.
Drug Binding to Wild and Mutated SARS-COV-2 Spike Glycoprotein
Site 1
The results of molecular docking against the mutated site (residue 614), as a critical target, are presented in Table 1. As could be seen, some antiviral drugs have experienced a significant alteration in the docking score from the original form of residues to the mutated one. It means that the D614G could cause a remarkable change in the stereochemical aspects of the residues.
The stars indicate the drugs with significant alteration in the docking score. a) The drug with the lowest docking score in the mutated form. b) The drug that experienced the most increase in the docking score. c) The drug with the lowest docking score in the wild form.
The results show that some drugs have experienced a remarkable alteration in docking scores. For instance, the docking score of Ribavirin in the wild protein is not significantly high, but it has the highest score against the mutated model between different antiviral drugs. Figure 1A and 1B represent Ribavirin among the residues, and its interactions with the mutated and not-mutated spike protein, respectively. A more detailed 2D illustration of the interactions with Ligplot is presented in Fig. S1-a and S1-b. As could be seen, G614 has created a hydrophobic interaction with Ribavirin (Fig. S1-b), while D614 has not participated in the interaction (Fig. S1-a).

Docking pose of Ribavirin among the residues of wild (A) and mutated (B) protein. Most of the labled residues have created non-cocalant bonds. The residues with hydrogen bond in the wild form are Thr675 (two hydrogen bonds) and Lys730. The residues with hydrogen bonds in the mutated form are Ile847, Leu855, and Val857. A detailed 2D view of hydrogen bond and hydrophobic contacts are presented in Fig S1a and S1b
The score of Ribavirin on the mutated spike is closely followed by PMEG diphosphate and BVDUTP with the scores of -7.597 and -6.858, respectively. BVDUTP is another drug that is alternated significantly from -3.263 to -6.858. Its docking score has decreased more than twofold from -3.263 to -6.858. Figure 2A and 2B represent BVDUTP among the residues, and its interactions with the mutated and not-mutated spike protein, respectively.

Docking pose of BVDUTP among the residues of wild wild (A) and mutated (B) RBD. The residues with hydrogen bond in the wild RBD are Asn16 Phe592, and Lys851. The residues with hydrogen bond in the mutated RBD are Phe592, Asn616, Phe830, Lys 832, and Lys851 (two hydrogen bonds). A detailed 2D view of hydrogen bond and hydrophobic contacts are presented in Fig S4a and S4b
On the other hand, there are some drugs with an increase in their docking score. For example, HPmpa has experienced remarkable growth from -3.286 to -0.015, which shows the different impacts of this mutation on separate components and their interactions. HPmpa and its interactions with the spike are presented in Fig. S2-a and S2-b. Despite the alteration in the docking score, most of the residues that participated in the interactions are common in wild and mutated models. This alteration could be attributed to the length of the bonds. The number of hydrogen bonds made by Lys730 is changed from two in the wild form to one in the mutated form. That could be considered as another reason for the alteration in the binding affinity of this antiviral drug.
For better insight, the drugs with considerable alternations were investigated cautiously. These drugs are Dolutegravir, BVDUTP, HPmpa, Didanosine, and Ribavirin with alterations of − 3.155, − 3.595, + 3.271, − 2.290 and − 2.742 respectively. Ribavirin, the drug with the lowest score, and HPmpa as the highest one have been analyzed before. This part tends to investigate the interactions of Dolutegravir, BVDUTP, and Didanosine. Fig. S3-a and S3-b show the interactions of Dultegrovir. The number of hydrophobic contacts in the not-mutated model is 6, which has increased to 11 in the mutated form. It could be the reason for the docking score increase.
Fig. S4-a and S4-b present various interactions of BVDUTP, and it can be seen that the number of hydrogen bonds in the mutated form is 5, while this number is 3 in the not-mutated form. It is also clear that G614 could make two hydrophobic interactions with C11 and O3 atoms of the drug, but D614 could make only one hydrophobic contact with the drug. It means that G614 is more active than D614.
Fig. S5 shows the interactions of Didaosine with an increase of 110% from not-mutated to mutated form. As could be seen, G614 has made a hydrophobic contact with the drug, but D614 is not active enough to create a link with Didanosine. Albeit the number of interactions in the mutated and wild model is almost similar, the only common residue that has participated in the interactions of mutated and non-mutated form is Thr856, and other links are different from one model to another. As mentioned, some drugs experienced considerable alteration in the docking score. For a better insight, the Ligplots of two models for these drugs were aligned. The results are presented in Fig. S6 a-e.
Site 2
The docking results of site 2, RBD, as a crucial target for receptor binding inhibition are presented in Table3.
The results of docking against RBD show that the mutation site is more active than RBD, and even some of the antiviral drugs had no confident interaction with the RBD. As could be seen, BVDUTP has the lowest docking score among drugs, and Ribavirirn and Valganciclovir have shown no interaction with the mutated and wild protein, respectively. Figure 2 presents the docking pose of BVDUTP. As could be seen in Fig. S7, the number and the residues that are participated in the interaction are almost the same, but the length of bonds in the mutated RBD is less than bonds in a wild protein. For example, Arg403 has created Hydrogen bonds in both wild and mutated RBD, but the lengths of bonds in wild RBD are 3.03 and 2.81, while these bonds are 2.91and 2.55 in the mutated form. These differences are caused by the alteration in the stereochemical characteristics of RBD from wild form to mutated one.
Reproducibility of Docking Results for Evaluation of the Reliability
Another docking method was employed to evaluate the reliability of the obtained results from the conventional docking study (previous step). For this purpose, the compounds with the lowest docking scores for each site were selected to be evaluated. The induced-fit docking was performed for six antiviral agents in both wild and mutated protein for site 1 and site 2. As regards the information presented in Tables 3, 4, 5, 6, the docking scores have decreased, but their orders have not changed, except for PMEG diphosphate (in the mutated spike protein, site 1). As it is obvious in Table 3, PMEG diphosphate has the lowest docking score with -10.226, and Ribavirin (the previous best component) with a docking score of -9.064 is in the next place. The docking pose of PMEG diphosphate is presented in Fig. 3, and its interactions with wild and mutated SARS-COV-2 are shown in Fig. S8.

Docking pose of PMEG diphosphate in the mutated spike protein. The residues with hydrogen bond are Gln314 (two hydrogen bonds), Gln613, Ile666, Lys730, Asn761, and Arg762. A detailed 2D view of hydrogen bond and hydrophobic contacts are presented in Fig S8
The induced-fit docking process is closer to the natural mechanism of drug-protein interactions. Therefore, PMEG diphosphate was considered the drug with the most binding affinity to the mutated spike protein. For a better understanding, the interactions of this compound were further investigated with MD simulation.
MD Simulation for Dynamic Investigation
The MD simulation is a powerful and reliable approach for the evaluation of interactions between the ligand and protein in a dynamic condition. It can provide a beneficial understanding of the stabilities in a physiological environment situation. The simulation study was performed on the compound with the most binding affinity, PMEG diphosphate, using Desmond, Schrodinger, and different characteristics such as RMSD and contacts histogram were used to investigate the structural stability and dynamic behavior of the complexes. The overall RMSD of mutated protein was found to be stable throughout the 60 ns simulation with no considerable fluctuations after 20 ns (Fig. S9). As could be seen in Fig. S9, the RMSD of wild spike protein has more fluctuations than mutated protein, which indicated more stability of the mutated protein. The hydrogen bonds and hydrophobic interactions of the protein after MD simulation are presented in Fig. S10A. As it is obvious, G614 has created some interactions with the drug, while Asp614 has no confident interaction with the ligand (Fig. S10B). These results elucidate the more active characteristics of the D614G variant.
Binding Free Energy
The calculations of binding free energy using the MM/GBSA method were conducted for PMEG diphosphate. The results are shown in Table 7. As could be seen in the table, the binding free energy of PMEG diphosphate in the mutated protein is higher than in wild protein. It shows the higher stability of the PMEG diphosphate-mutated protein complex. This table also indicates that Waals energy, H-bond, and lipophilic energy contribute to system stability. However, Van der Waals energy plays a key role in the system as the Van der Waals energy values are more coincided with the previous results.
ADME/T Analysis for Examination of Compounds’ Druggability
For theoretical prediction of the impacts and responses of the antiviral drugs, the ADME/T analysis was conducted. Several parameters were obtained that are presented in Table S2. Since the molecules should have a determined molecular weight, the first parameter in this table is the molecular weight. Another parameter is SASA, the total solvent accessible surface area in square angstroms using a probe with a 1.4 Å radius. One other parameter is QP polarizability, which is the predicted polarizability in cubic angstroms. An essential parameter is QPlog HERG, the numerical amount of the estimated IC50 value for blockage of HERG K + channels. Another parameter is QPPCaco, which is the predicted Caco‐2 cell permeability in nm/sec for passive transport. The next parameter is QPlog BB, which is the Predicted brain/blood partition coefficient of an orally delivered drug. Another parameter is human oral absorption, which is the predicted qualitative human oral absorption. It should be 1, 2, or 3 for low, medium, or high, respectively. Another two parameters which are the most important are the rule of five and the rule of three. The rule of five is Lipinski's fifth rule of Pfizer. The rules are: mol_MW < 500, QPlogPo/w < 5, donorHB ≤ 5, accptHB ≤ 10. The rule of three is the three rules of Jorgensen [39]. These three rules are: QPlogS > -5.7, QP PCaco > 22 nm/s, # Primary Metabolites < 7. These two parameters are among the parameters that must be checked for the molecules to be theoretical drugs. The results of ADME/T analysis for the rest of the drugs are provided in the supplementary information.
Discussion
Although several types of research have focused on the treatment of COVID-19, there is not a specific address for this pandemic. This could be attributed to the transient nature of coronaviruses and also the rate of mutation in their structure. A mutation that has become dominant in many parts of the world through the spread of the virus is the D614G variant [40,41,42]. Some reports indicate the viruses bearing the G614 is more infectious than those with D614 [43]. Even though the impact of this variant on the different interactions of viruses is not thoroughly understood [19], the G614 can affect the interaction of the virus with the ACE2 receptor. The alteration in the manner of the virus is due to the flexibility induced by glycine [20, 44]. This replacement can change the orientation of the site affecting the contact between the virus and receptor [21]. D614G confers some more flexibility to the spike stalk and increases its ability to scan the host-cell surface, which can provide a better interaction with ACE2. The favored interactions are not necessarily more stable in terms of the interaction energy or binding affinity. They could be in terms of the number and type of interactions [20, 21]. This alteration in receptor-virus interactions could happen in the other interactions of the virus, such as drug-virus interactions. According to the difficulties and time-consuming process of clinical trials, computational studies such as molecular docking and molecular dynamics could be used as versatile approaches to address such problems [45, 46]. For this reason, we investigated the impact of this variant on the efficacy of different antiviral drugs with the molecular docking study and MD simulation. The results show that the alternation of D614 to G614 could change the binding affinity of drugs considerably. The D614G affects the behavior of RBD considerably. Therefore, it could be a crucial target for antiviral drugs. Ribavirin as a drug against respiratory syncytial virus-RSV and PMEG diphosphate exhibited the highest binding affinity to the mutated SARS-COV-2 spike protein among different antiviral drugs. MD simulation outcomes confirmed the obtained results of the docking study. The results of ADME/T analysis showed that the selected antiviral drugs have the potential to be further investigated for in-vitro and in-vivo evaluation. Although some of the ADME/T parameters are out of the acceptable range, these results are theoretical and need to be more investigated. There are some antiviral drugs such as HPmpa that have experienced a significant decrease in docking scores during the D614G mutation. The results of HPmpa showed that the length of H-bonds could affect the binding affinity of ligands to the protein. This means that researchers and drug manufacturers need to find a more reliable insight into the alternated characteristics of coronaviruses and their mutations. The validated drugs could be easily transported to the target sites using virosome [47] or bioconjugated nanoparticles [48]. The spike glycoprotein of coronaviruses is the virus key for cell entrance [49], and residue 614 is placed in this protein. Albeit this residue is remote from the RBD [15], considering the folding of the protein, residue 614 is capable of providing communication with RBD. The optimal communication between D614 and RBD (T500) is only found in one chain (chain B), but this optimal communication route between G614 and RBD (T500) is reported to be in two chains (chains B and C) [50]. This finding could lead us to find the reason for changing the binding affinity of the mutated model of the virus compared to the wild one.
Conclusions
The D614G mutation in the SARS-COV-2 spike protein has changed some crucial characteristics of coronaviruses, such as the rate of infection and binding affinity. This alteration could effectively change the behavior of coronaviruses in the presence of antiviral drugs. It can also change the interactions between the virus and the ACE2 receptor of the host cell. D614G confers some more flexibility to the spike stalk and increases its ability to scan the host-cell surface, which can provide a better interaction with ACE2. The favored interactions are not necessarily more stable in terms of the interaction energy or binding affinity. They could be in terms of the number and type of interactions. Therefore, in addition to the RBD, the mutation D614G could also be considered a crucial target for antiviral drugs. Ribavirin and PMEG diphosphate have shown a remarkable binding affinity to the mutated form of the spike protein. These antiviral drugs could be considered an effective inhibitor for the mutated form of coronaviruses.
Availability of Data and Material
Not applicable.
References:
Wang Q, Wu J, Wang H et al (2020) Structural basis for RNA replication by the SARS-CoV-2 polymerase. Cell 182:417-428.e13. https://doi.org/10.1016/j.cell.2020.05.034
Chen Y, Liu Q, Guo D (2020) Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol 92:418–423
Baden LR, El Sahly HM, Essink B et al (2021) Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N Engl J Med 384:403–416. https://doi.org/10.1056/nejmoa2035389
Falsey AR, Sobieszczyk ME, Hirsch I et al (2021) Phase 3 safety and efficacy of AZD1222 (ChAdOx1 nCoV-19) Covid-19 vaccine. N Engl J Med 385:2348–2360. https://doi.org/10.1056/nejmoa2105290
Heath PT, Galiza EP, Baxter DN et al (2021) Safety and efficacy of NVX-CoV2373 Covid-19 vaccine. N Engl J Med 385:1172–1183. https://doi.org/10.1056/nejmoa2107659
Mehrabani M, Raeiszadeh M, Najafipour H et al (2020) Evaluation of the cytotoxicity, antibacterial, antioxidant, and anti-inflammatory effects of different extracts of Punica granatum var. Pleniflora. J Kerman Univ Med Sci 27:414–425. https://doi.org/10.22062/JKMU.2020.91474
Singla RK, He X, Chopra H et al (2021) Natural products for the prevention and control of the COVID-19 pandemic: sustainable bioresources. Front Pharmacol 12:3215
Vakili S, Savardashtaki A, Jamalnia S et al (2020) Laboratory findings of COVID-19 infection are conflicting in different age groups and pregnant women: a literature review. Arch Med Res 51:603–607. https://doi.org/10.1016/j.arcmed.2020.06.007
Ibrahim IM, Abdelmalek DH, Elshahat ME, Elfiky AA (2020) COVID-19 spike-host cell receptor GRP78 binding site prediction. J Infect 80:554–562. https://doi.org/10.1016/j.jinf.2020.02.026
Hui DS, Azhar EI, Madani TA et al (2020) The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health—the latest 2019 novel coronavirus outbreak in Wuhan, China. Int J Infect Dis 91:264–266
Elfiky AA (2020) Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): a molecular docking study. Life Sci 253:117592. https://doi.org/10.1016/j.lfs.2020.117592
Lu R, Zhao X, Li J et al (2020) Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395:565–574. https://doi.org/10.1016/S0140-6736(20)30251-8
Laha S, Chakraborty J, Das S et al (2020) Characterizations of SARS-CoV-2 mutational profile, spike protein stability and viral transmission. Infect Genet Evol 85:104445. https://doi.org/10.1016/j.meegid.2020.104445
Sui J, Aird DR, Tamin A et al (2008) Broadening of neutralization activity to directly block a dominant antibody-driven SARS-coronavirus evolution pathway. PLoS Pathog 4:e1000197. https://doi.org/10.1371/journal.ppat.1000197
Yurkovetskiy L, Wang X, Pascal KE et al (2020) Structural and functional analysis of the D614G SARS-CoV-2 spike protein variant. Cell 183:739-751.e8. https://doi.org/10.1016/j.cell.2020.09.032
Korber B, Fischer WM, Gnanakaran S et al (2020) Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 182:812-827.e19. https://doi.org/10.1016/j.cell.2020.06.043
Yuan M, Wu NC, Zhu X et al (2020) A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science 368:630–633. https://doi.org/10.1126/science.abb7269
Mercurio I, Tragni V, Busto F et al (2021) Protein structure analysis of the interactions between SARS-CoV-2 spike protein and the human ACE2 receptor: from conformational changes to novel neutralizing antibodies. Cell Mol Life Sci 78:1501–1522. https://doi.org/10.1007/s00018-020-03580-1
Grubaugh ND, Hanage WP, Rasmussen AL (2020) Making sense of mutation: what D614G means for the COVID-19 pandemic remains unclear. Cell 182:794–795. https://doi.org/10.1016/j.cell.2020.06.040
Pierri CL (2020) SARS-CoV-2 spike protein: flexibility as a new target for fighting infection. Signal Transduct Target Ther 5:1–3. https://doi.org/10.1038/s41392-020-00369-3
Turoňová B, Sikora M, Schürmann C et al (2020) In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 370:203–208. https://doi.org/10.1126/science.abd5223
A Unique Clade of SARS-CoV-2 Viruses is Associated with Lower Viral Loads in Patient Upper Airways | medRxiv. https://pubmed.ncbi.nlm.nih.gov/32511558/. Accessed 2 Oct 2020
Tragni V, Preziusi F, Laera L et al (2022) Modeling SARS-CoV-2 spike/ACE2 protein–protein interactions for predicting the binding affinity of new spike variants for ACE2, and novel ACE2 structurally related human protein targets, for COVID-19 handling in the 3PM context. EPMA J 13:149–175. https://doi.org/10.1007/s13167-021-00267-w
Chan JFW, Chan KH, Kao RYT et al (2013) Broad-spectrum antivirals for the emerging Middle East respiratory syndrome coronavirus. J Infect 67:606–616. https://doi.org/10.1016/j.jinf.2013.09.029
Wu C, Liu Y, Yang Y et al (2020) Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B 10:766–788. https://doi.org/10.1016/j.apsb.2020.02.008
Dyall J, Coleman CM, Hart BJ et al (2014) Repurposing of clinically developed drugs for treatment of Middle East respiratory syndrome coronavirus infection. Antimicrob Agents Chemother 58:4885–4893. https://doi.org/10.1128/AAC.03036-14
Dotolo S, Marabotti A, Facchiano A, Tagliaferri R (2021) A review on drug repurposing applicable to COVID-19. Brief Bioinform 22:726–741
Omrani AS, Saad MM, Baig K et al (2014) Ribavirin and interferon alfa-2a for severe Middle East respiratory syndrome coronavirus infection: a retrospective cohort study. Lancet Infect Dis 14:1090–1095. https://doi.org/10.1016/S1473-3099(14)70920-X
Kumar Y, Singh H, Patel CN (2020) In silico prediction of potential inhibitors for the main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. J Infect Public Health 13:1210–1223. https://doi.org/10.1016/j.jiph.2020.06.016
Poustforoosh A, Hashemipour H, Pardakhty A, Kalantari Pour M (2021) Preparation of nano-micelles of meloxicam for transdermal drug delivery and simulation of drug release: a computational supported experimental study. Can J Chem Eng. https://doi.org/10.1002/cjce.24339
Biasini M, Bienert S, Waterhouse A et al (2014) SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42:252–258. https://doi.org/10.1093/nar/gku340
Yadav M (2019) Homology modeling and molecular dynamics dimulation study of β carbonic anhydrase of Ascaris lumbricoides. Bioinformation 15:572–578. https://doi.org/10.6026/97320630015572
Poustforoosh A, Faramarz S, Nematollahi MH et al (2022) In silico SELEX screening and statistical analysis of newly designed 5mer peptide-aptamers as Bcl-xl inhibitors using the Taguchi method. Comput Biol Med 146:105632. https://doi.org/10.1016/J.COMPBIOMED.2022.105632
Poustforoosh A, Hashemipour H, Tüzün B et al (2021) Evaluation of potential anti-RNA-dependent RNA polymerase (RdRP) drugs against the newly emerged model of COVID-19 RdRP using computational methods. Biophys Chem 272:106564. https://doi.org/10.1016/j.bpc.2021.106564
Poustforoosh A, Faramarz S, Nematollahi MH et al (2022) 3D-QSAR, molecular docking, molecular dynamics, and ADME/T analysis of marketed and newly designed flavonoids as inhibitors of Bcl-2 family proteins for targeting U-87 glioblastoma. J Cell Biochem 123:390–405. https://doi.org/10.1002/jcb.30178
Sirin S, Pearlman DA, Sherman W (2014) Physics-based enzyme design: predicting binding affinity and catalytic activity. Protien Struct Funct Bioinform 82:3397–3409. https://doi.org/10.1002/prot.24694
Panwar U, Singh SK (2021) Atom-based 3D-QSAR, molecular docking, DFT, and simulation studies of acylhydrazone, hydrazine, and diazene derivatives as IN-LEDGF/p75 inhibitors. Struct Chem 32:337–352. https://doi.org/10.1007/s11224-020-01628-3
Taslimi P, Erden Y, Mamedov S et al (2021) The biological activities, molecular docking studies, and anticancer effects of 1-arylsuphonylpyrazole derivatives. J Biomol Struct Dyn 39:3336–3346. https://doi.org/10.1080/07391102.2020.1763838
Jorgensen WL, Duffy EM (2002) Prediction of drug solubility from structure. Adv Drug Deliv Rev 54:355–366. https://doi.org/10.1016/S0169-409X(02)00008-X
Plante JA, Liu Y, Liu J et al (2021) Spike mutation D614G alters SARS-CoV-2 fitness. Nature 592:116–121. https://doi.org/10.1038/s41586-020-2895-3
Tang L, Schulkins A, Chen CN, et al (2020) The SARS-CoV-2 Spike Protein D614G Mutation Shows Increasing Dominance and May Confer a Structural Advantage to the Furin Cleavage Domain. Preprints 1–14. https://doi.org/10.20944/preprints202005.0407.v1
Butowt R, Bilinska K, Von Bartheld CS (2020) Chemosensory dysfunction in COVID-19: integration of genetic and epidemiological data points to D614G spike protein variant as a contributing factor. ACS Chem Neurosci 11:3180–3184. https://doi.org/10.1021/acschemneuro.0c00596
Hu J, He CL, Gao QZ, et al (2020) D614G mutation of SARS-CoV-2 spike protein enhances viral infectivity. bioRxiv 1–37. https://doi.org/10.1101/2020.06.20.161323
Isabel S, Graña-Miraglia L, Gutierrez JM et al (2020) Evolutionary and structural analyses of SARS-CoV-2 D614G spike protein mutation now documented worldwide. Sci Rep 10:14031. https://doi.org/10.1038/s41598-020-70827-z
Suárez D, Díaz N (2020) SARS-CoV-2 main protease: a molecular dynamics study. J Chem Inf Model 60:5815–5831. https://doi.org/10.1021/acs.jcim.0c00575
Das S, Sarmah S, Lyndem S, Singha Roy A (2021) An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. J Biomol Struct Dyn 39:3347–3357. https://doi.org/10.1080/07391102.2020.1763201
Asadikaram G, Poustforoosh A, Pardakhty A et al (2020) Niosomal virosome derived by vesicular stomatitis virus glycoprotein as a new gene carrier. Biochem Biophys Res Commun 534:980–987. https://doi.org/10.1016/j.bbrc.2020.10.054
Poustforoosh A, Nematollahi MH, Hashemipour H, Pardakhty A (2022) Recent advances in Bio-conjugated nanocarriers for crossing the Blood-Brain Barrier in (pre-)clinical studies with an emphasis on vesicles. J Control Release 343:777–797. https://doi.org/10.1016/j.jconrel.2022.02.015
Tsai JC, Zelus BD, Holmes KV, Weiss SR (2003) The N-terminal domain of the murine coronavirus spike glycoprotein determines the CEACAM1 receptor specificity of the virus strain. J Virol 77:841–850. https://doi.org/10.1128/jvi.77.2.841-850.2003
Omotuyi IO, Nash O, Ajiboye OB et al (2020) Atomistic simulation reveals structural mechanisms underlying D614G spike glycoprotein-enhanced fitness in SARS-COV-2. J Comput Chem 41:2158–2161. https://doi.org/10.1002/jcc.26383
Acknowledgements
This study was supported by Kerman University of Medical Sciences, Kerman, Iran by project No. 99000971, and the Scientific Research Project Fund of Sivas Cumhuriyet University, Sivas, Turkey under project number RGD-020.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or nonprofit sectors.
Author information
Authors and Affiliations
Contributions
AP: investigation, methodology, writing the original draft. Hassan Hashemipour: project administration. BT: validation. MA: investigation, methodology. SF: methodology. AP: supervision. MM: writing original draft. MHN: investigation, supervision.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Informed Consent on Studies with Human and Animal Subjects
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Poustforoosh, A., Hashemipour, H., Tüzün, B. et al. The Impact of D614G Mutation of SARS-COV-2 on the Efficacy of Anti-viral Drugs: A Comparative Molecular Docking and Molecular Dynamics Study. Curr Microbiol 79, 241 (2022). https://doi.org/10.1007/s00284-022-02921-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00284-022-02921-6