
Abstract
Purpose
Assessment of the absorption, metabolism and excretion of [14C]-lenalidomide in healthy male subjects following a single oral dose.
Methods
Six healthy male subjects were administered a single 25 mg oral suspension dose of [14C]-lenalidomide. Blood (plasma), semen and excreta were collected. Mass balance assessments were done by radioactivity measurements. Metabolite profiling and quantitation were accomplished using liquid chromatography with mass spectrometric and radiochemical detection.
Results
[14C]-Lenalidomide was rapidly absorbed (T max 0.77–1.0 h), and the levels declined with a terminal half-life of approximately 3 h, with similar profiles for total blood and plasma radioactivity as well as plasma lenalidomide. The whole blood to plasma radioactivity exposure levels were comparable, suggesting equal distribution between plasma and blood cells. On average, 94% of the administered radioactivity was recovered within 10 days, with >88% recovered within 24 h. Urinary excretion was the primary route of elimination (90% of radioactive dose), with minor amounts excreted in feces (4%). Semen contained a small amount of the radioactive dose (0.0062%). Lenalidomide was the primary radioactive component in plasma (92% of the [14C]-area under the concentration–time curve) and urine (>90% of the radioactivity in urine). The remaining radioactivity was composed of primarily two metabolites: 5-hydroxy-lenalidomide and N-acetyl-lenalidomide, each accounting for less than 5% of the total radioactivity as well as lenalidomide levels in plasma and excreta.
Conclusions
In summary, following oral administration, lenalidomide is highly absorbed and bioavailable, metabolized minimally, and eliminated predominantly via urinary excretion in the unchanged form in humans.
Similar content being viewed by others

Introduction
Lenalidomide (3-(4′aminoisoindoline-1′-one)-1-piperidine-2,6-dione; Revlimid®; Fig. 1) is an oral immunomodulatory drug (IMiD®) [1], with antineoplastic, antiangiogenic and anti-inflammatory properties [2, 3]. It has been used for the treatment of transfusion-dependent anemia due to low- or intermediate-risk myelodysplastic syndromes (MDS) associated with a deletion 5q cytogenetic abnormality with or without additional cytogenetic abnormalities [4, 5], and in combination with dexamethasone for the treatment of multiple myeloma (MM) patients who have received at least one prior therapy [6, 7]. Lenalidomide, as a single agent or in combination with other agents, is also being evaluated for the treatment of newly diagnosed MM, chronic lymphocytic leukemia, non-Hodgkin’s lymphoma and certain solid tumors [8–12].

Structures of lenalidomide and its human metabolites
In both healthy subjects and patients with normal renal function, lenalidomide displays linear pharmacokinetics. The plasma exposure of lenalidomide is proportional to dose up to 400 mg and it does not accumulate with multiple doses [13, 14]. The mean terminal elimination half-life (t1/2) was approximately 3–4 h in clinically relevant dose range (5–50 mg) [13, 14]. Excretion of the unchanged parent in urine is the predominant clearance mechanism of lenalidomide, accounting for approximately 84% of the dose [15].
Incubation of lenalidomide with human liver microsomes, recombinant-CYP isozymes and human hepatocytes did not result in Phase I or Phase II metabolism [16]. Lenalidomide undergoes hydrolysis in human plasma, with an in vitro half-life of approximately 8 h, due to hydrolytic cleavage of glutarimide amide bonds (unpublished data). Although metabolism data on lenalidomide have been collected from in vitro studies, a thorough investigation of the metabolism of lenalidomide has not yet been conducted in vivo in humans.
The purposes of this study were to characterize the disposition of [14C]-lenalidomide in humans and to identify metabolites in circulation and excreta after a single oral suspension dose. The results of this study will help determine whether lenalidomide exposure is subject to alteration in patients with hepatic insufficiency, potential for drug–drug interactions during polypharmacy, and the potential for the exposure of female partner during a heterosexual intercourse when the male partner is on lenalidomide therapy.
Methods
Materials
The specific activity, chemical purity and radiochemical purity of [14C]-lenalidomide (synthesized by Girindus Solvay Organics for Celgene Corporation) used for the preparation of the dosage formulation in this study were approximately 200.9 μCi/mg (52.43 mCi/mmol), 99.8 and 99.8%, respectively. The study dosage form was manufactured by Girindus Solvay Organics (for Celgene Corporation) in individual 20 ml clear glass scintillation vials containing approximately 25 mg of preweighed powder, comprising a mixture of racemic [14C]-lenalidomide and racemic unlabeled lenalidomide powders. The actual radioactivity contained in each dosage was approximately 85 μCi, with a specific activity of approximately 3.4 μCi/mg.
Study design and subjects
This was an open-label, single-center, single oral dose study in healthy male subjects. The study was conducted at Quintiles Phase I Study Center (Overland Park, Kansas). The study protocol and Informed Consent Form were approved by the Institution Review Board (Lenexa, Kansas) and Radiation Safety Committee at Quintiles Phase I Services prior to study initiation.
Subjects were eligible for the study if they met the following criteria: were male; aged 18–45 years; weighed 60–100 kg, with a body mass index of 19–30 kg/m2; were healthy as determined by medical history, physical examination, 12-lead electrocardiogram (ECG), serum biochemistry, hematology and urinalysis. All subjects must agree to use a reliable method(s) of contraception during study and for at least 28 days after study. Subjects were excluded, if they had the following conditions: positive results to the tests for drugs of abuse, alcohol or cotinine; positive results for hepatitis B surface antigen, hepatitis C antibody or human immunodeficiency virus (HIV) antibody; exposure to excess radiation or participation in a study involving radioisotopes within the previous 12 months; current or a history of vasectomy, vasoligation, an undescended testicle(s) or ejaculation problems.
The subjects entered the study center on the day prior to dosing and remained at the center for up to 10 days following dosing or until the total radioactivity recovered in urine and fecal samples from two consecutive days was ≤1% of the administered radioactive dose. All subjects were asked to abstain from any study-unrelated sexual activities including self-masturbation from 72 h before dosing until after the last semen sample was taken.
Safety assessments
Physical examination, clinical laboratory tests (hematology, serum chemistry and urinalysis) and 12-lead ECG were performed at the selected times before and after the administration of the study drug. All subjects were monitored throughout the study for adverse events and vital signs and for the use of concomitant medications. Adverse events were evaluated by the investigator as to their intensity, seriousness and relationship to study drug.
Treatment
Within 15 min prior to dosing, 15 ml of distilled water was added to the dosage vial, and the vial was sonicated for 5 min to obtain a homogenous suspension of the study drug. Within 10 min of completion of sonication, the subject ingested the drug suspension. The dosing vial was rinsed with 15 ml of distilled water, sonicated for 1 min and the rinse ingested by the subject. This procedure was repeated twice more (i.e., total 3 rinses). Following ingesting the three rinses, the subject drank an additional 180 ml of water. The entire procedure for dosing each subject (from ingestion of the initial drug suspension to ingestion of the final 180 ml water) was completed within no more than 5 min. All dosing vials were saved for the measurement of residual radioactivity. The actually administered mass of dose and radioactivity (μCi) were recorded.
Subjects were fasted for at least 8 h before dosing and for 4 h after dosing. Water was not allowed for 2 h before dosing and for 4 h after dosing. To aid in daily bowel movements, subjects received high-fiber meals and a stool softer (Fibercon in each evening) from check-in until discharge from the study center.
Biologic sample collections
Whole blood samples were collected into EDTA tubes at predose, 0.25, 0.5, 1, 1.5, 2, 4, 6, 8, 10, 12, 24, 48, 72, 96, 120, 144 and 168 h postdose. At each time point, a portion of the blood sample was saved for counting the total radioactivity in the whole blood and the remaining blood was used to prepare plasma. Within 30 min of collection, blood samples were centrifuged at 4°C to obtain plasma, which were stored at −70°C until analysis.
Urine were collected as voided at predose (spot collection), pooled postdosing over the intervals of 0–4, 4–8, 8–12, 12–24 h on Day 1, and over intervals of 24 h on Days 2–10. The total volume of urine collected over each interval was recorded. Subsequently, an aliquot of urine taken, acidified with an equal volume of Sorenson’s citrate buffer (pH 1.5) and stored at 4°C (for counting radioactivity) or −70°C until assayed.
Feces were collected as individual bowel movements at predose and pooled postdosing over 24 h intervals for up to 10 days. The fecal samples were homogenized to a slurry (using 4 volumes, by weight, of pH 1.5 Sorensen’s buffer), and the homogenized samples were stored at −70°C until assayed.
Semen samples were collected during screening (baseline sample), between 2 and 3 h postdose, and between 72 and 73 h postdose. To avoid contamination with urinary [14C]-lenalidomide/metabolites in the semen sample collected during 2 to 3 h postdose, the subject was asked to empty his bladder immediately prior to dosing and to produce this semen sample before collection of any urine for the first postdose collection interval. Semen was ejaculated, by masturbation, into a container. Within 15 min of collection, the semen was mixed with two volumes, by weight, of pH 1.5 Sorensen’s citrate buffer to give a final semen/buffer ratio of 1:2 [w/w]. The acidified semen samples were stored at −70°C until assayed.
Determination of radioactivity
The radioactivity was measured by liquid scintillation counter (Beckman LS6000SC or LS6500) either for 20 min or until the 2-sigma error is less than or equal to 2%, whichever came first. Duplicate aliquots for plasma and urine samples were directly counted for radioactivity in Ultima Gold scintillation cocktail (Packard). Duplicate aliquots for whole blood, fecal homogenates and semen samples were combusted in a Packard model 307 oxidizer. After combustion, the resulting [14C]-carbon dioxide was collected into CarboSorb and Permafluor (Packard Instrument Co.) and counted. The mean percent recoveries of quality control samples were within 100 ± 15% for all matrices. The amount of radioactivity (dpm) in whole blood, plasma, urine, feces and semen were converted to equivalent concentrations of lenalidomide (ngEq/g, ngEq/ml or μgEq/g) based on the specific activity of the administered compound (3.4 μCi/mg).
Determination of lenalidomide
Plasma samples were analyzed for lenalidomide concentrations by a validated achiral liquid chromatography-tandem mass spectrometry (LC–MS/MS) method. Briefly, plasma samples, with 13C5-lenalidomide as the internal standard, were processed by combining with acidified acetonitrile. The supernatants were processed, and the chromatographic separation was achieved by high-performance liquid chromatography (HPLC) using a Monochrom C18 column (5 μ, 100 × 4.6 mm) operated at ambient temperature. The gradient mobile phase consisted of 0.1% formic acid in water and 0.1% formic acid in acetonitrile at a flow rate of 0.8 ml/min with a run time of 5.5 min. Detection was performed by an AB Sciex API 4000 mass spectrometer configured with electro spray ionization in the positive ion mode. Data were acquired by multiple-reaction monitoring of mass transition pairs at m/z of 260.3 > 149.1 for lenalidomide and 265.3 > 149.1 for the internal standard. The lenalidomide standard curve was linear from 5 to 1,000 ng/ml, with a lower limit of quantitation of 5 ng/ml.
Pharmacokinetic analysis
Total radioactivity excreted in urine, feces and semen were converted to percentage of the actually administered radioactive dose (% dose). The total recovery of radioactivity was computed as the sum of the cumulative excretion (as % dose) in urine, feces and semen.
Standard pharmacokinetic parameters were estimated from the lenalidomide concentration versus time profile or the blood and plasma radioactive concentration equivalents versus time profiles by established noncompartmental methods using WinNonlin Professional Version 4.1b (Pharsight Corporation, Mountain View, CA) or SAS® Version 8.2 (SAS Institute, Cary, NC).
The distribution of radioactivity in red blood cells as percentage of the whole blood radioactivity was estimated by the following equation:
where Cwb and Cp were the radioactive concentration equivalent in whole blood and plasma, respectively, and Ht was the mean value of the hematocrit obtained from Day 0 (baseline) and follow-up.
Metabolite profiling
A total of six pooled inter-subject plasma samples were prepared before the extraction by combining equal volume (2 ml) of plasma from the six subjects by time points. The prepared pooled plasma samples were acidified and subjected to solid phase extraction using C-18 Strata SEP Pak column; the eluates were dried under a nitrogen stream, and the residue was reconstituted with 0.1% aqueous acetic acid for further analysis. Urine samples were centrifuged to sediment particulate matter, and the supernatants were analyzed without further processing. Six individual semen samples collected at 2–3 h postdose (one from each subject) were analyzed after processing in a manner similar to plasma samples described above. Based on the radioactivity in the fecal homogenates, selected fecal samples from each subject were used for metabolic profiling. Aliquots of fecal homogenates were combined with 0.1% acetic acid in acetone, vortexed and centrifuged at 4,500–5,000 rpm and 4°C for 10 min. The supernatant was transferred to a fresh tube, and the extraction procedure was repeated twice, and the pooled supernatants were concentrated under nitrogen stream to a final volume of 0.5–2 ml.
Liquid chromatography with radiochemical detection was performed for quantitative analysis of [14C]-lenalidomide and its metabolites. Separations were achieved using a Varian Monochrom 5 μ, C18, 4.6 × 100 mm, with a gradient mobile phase of A (0.1% acetic acid in water) and B (0.1% acetic acid in acetonitrile) at a flow rate of 1 ml/min and a run length of 55 min.
Metabolite identification was accomplished by mass spectrometry with API 4000 Q-trap spectrometer (AB Sciex, Foster City, CA) run in two modes, ESI (electro spray ionization) or APCI (atmospheric pressure chemical ionization) in positive ionization mode. Accurate mass measurements were performed using LTQ OrbiTrap (Thermo, San Jose, CA). The definitive structural assignments were accomplished by the comparison of chromatographic retention times, the molecular masses and the fragmentation patterns of parent and metabolites compared to their respective synthetic standards.
Results
Subjects and dosing
The 6 healthy male subjects who participated in the study had a mean age of 29.7 years (range: 20–42 years), a mean height of 178.92 cm (range: 173.4–181.5 cm), a mean weight of 83.95 kg (range: 77.7–91.2 kg) and a mean BMI of 26.22 kg/m2 (range: 24.1–27.8 kg/m2). Three subjects (50%) were Black and 3 subjects (50%) were White. The mean (±SD) dose administered was 84.6 ± 0.47 μCi of the [14C]-radioactivity and 24.9 ± 0.12 mg of lenalidomide.
Safety
There were no deaths, serious adverse events or discontinuations from the study due to adverse events. Overall, 5 adverse events were reported by 4 subjects (66.7%) after dosing. Gastrointestinal disorders (hard feces, gastro-esophageal reflux and stomach discomfort), reported by 3 (50%) of the 6 subjects, were the most common AE in this study. Of the 5 adverse events, only the event of hard feces was assessed by the investigator as having a suspected relationship to study medication. All the adverse events were assessed as mild in intensity by the investigator and all resolved prior to study conclusion. There were no drug-related changes in clinical laboratory results, vital signs, ECG values and physical examinations.
Mass balance
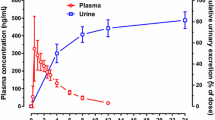
The total recovery of the radioactivity over 10 days averaged 94.3 ± 2.9% (range: 90.5–97.2%) of the administered dose, with majority (88.5%) of the radioactivity recovered in 24 h postdose (Fig. 2, Table 1). A total of 90.3 ± 3.0% (range: 87.0–94.6%) of dose was recovered in urine, which indicates the oral absorption of lenalidomide was high. Furthermore, urinary excretion of the radioactivity was rapid, with approximately 84% of the administered dose recovered in urine during the first 12 h and 88.5% recovered during 24 h (Fig. 2). In contrast, mean total fecal elimination of radioactivity accounted for only 3.95 ± 1.94% of the dose. Most fecal excretion was completed by Day 4 (96 h). Only a very small amount of the radioactivity (0.0062% of the dose; 1.479 μg equivalent of [14C]-lenalidomide) was recovered from semen.

Cumulative radioactivity recovery in urine, feces and all excreta over 10 days following a single oral 25 mg dose of [14C]-lenalidomide in healthy male subjects. Shown are mean ± SD (N = 6)
Pharmacokinetics
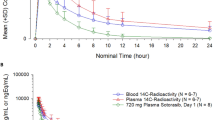
The pharmacokinetic parameters are summarized in Table 2. The mean concentrations for the total radioactivity in plasma and whole blood were comparable to that for the parent compound in plasma. For all the three measures, namely total radioactivity in blood and plasma, as well as intact lenalidomide in plasma, the maximum concentrations were achieved between 0.5 and 1 h postdose followed by similar rates of elimination, with the levels below the limits of quantitation by 24 h postdose. The distribution of radioactivity into erythrocytes averaged from 44 to 36% of the whole blood radioactivity from 0.25 to 12 h postdose. These values were comparable to the mean hematocrit (42–43%) for these subjects. The pharmacokinetic parameters for the total radioactivity in both plasma and whole blood are comparable to that for the parent. Based on the ratio of AUC∞ for lenalidomide and total radioactivity in plasma, lenalidomide comprised 98% of C max and 92% of AUC for the total plasma radioactivity. The mean renal clearance was estimated to be 261 ± 30 mL/min based on the amount of radioactive equivalents recovered in urine and the radioactive plasma AUC from 0 to 12 h postdose.
The mean radioactive concentration in semen collected between 2 and 3 h postdose was 492 ± 20.7 ngEq/g and similar to the radioactive C max equivalent observed in plasma. The semen concentration of radioactivity at 72 h time point was 31.7 ± 5.6 ngEq/g.
Metabolite identification
The plasma and urinary radioactivity were composed of a predominant peak, two small peaks (Metabolites A and B) and a few very minor peaks. A representative chromatogram is shown in Fig. 3.

Representative radio-chromatogram of an 0–4 h urine sample following a single oral 25 mg dose of [14C]-lenalidomide in a healthy male subject
Identification of the predominant component
The HPLC retention time of the predominant peak in both urine and plasma corresponded with that of lenalidomide standard. The protonated molecular ion with an m/z 260 [M + H]+ and the product ions of m/z 187, m/z 149, m/z 106 and m/z 84 were observed with both lenalidomide standard and the predominant peak. Further, neutral losses 73 (corresponding to urea) and 111 (corresponding to glutarimide ring) were also similar. The results of these experiments confirm the identification of the predominant peak of plasma and urinary radioactivity as unchanged lenalidomide.
Identification of metabolite A
The protonated molecular ion of Metabolite A of both plasma and urine was m/z 276. The results of the neutral loss experiment showed m/z 111 amu (corresponding to unchanged glutarimide moiety) and 73 amu (corresponding to urea moiety as observed in parent), indicating that the glutarimide moiety is intact in Metabolite A. A product ion scan for m/z 276 yielded m/z 165, which is indicative of hydroxylation of the iso-indolinone portion of the molecule (Fig. 4). The retention time and fragmentation pattern of this metabolite were similar to that of the synthetic standard of 5-hydroxy-lenalidomide (Fig. 4). Metabolite A had an accurate mass of m/z 276.09765, which suggests the addition of an oxygen atom (16 amu) to the parent molecular ion (lenalidomide m/z 260.10263). Metabolite A and synthetic standard 5-OH-lenalidomide were combined for concurrent analysis to further confirm the consistency of the retention time. Metabolite A and 5-OH-lenalidomide standard eluted as one peak with an m/z 276.09741 confirming the structure of Metabolite A as 5-OH-lenalidomide (Fig. 1).

Identification of metabolite A: a Mass spectrum of 5-hydroxy-lenalidomide synthetic standard. b Mass spectrum of metabolite A isolated from urine. c Radio-chromatogram of metabolite A isolated from urine. d Mass fragments of 5-hydroxy-lenalidomide synthetic standard. e Mass fragments of metabolite A isolated from urine. f Proposed fragmentation pattern of metabolite A
Identification of metabolite B
The protonated molecular ion of Metabolite B from both plasma and urine was observed at m/z 302, which is 42 amu greater than the parent, and two major fragments with m/z 229.1 and m/z 191.3. Metabolite B resulted in neutral losses of 73 and 111 (corresponding to urea and glutarimide ring), suggesting unchanged glutarimide moiety in Metabolite B. A synthetic standard N-acetyl-lenalidomide produced a similar fragmentation pattern as for Metabolite B and had an identical HPLC retention time, confirming Metabolite B as N-acetyl-lenalidomide (Fig. 1).
Plasma metabolite profiling
Lenalidomide was the predominant circulating component of the radioactivity in the pooled plasma samples (Fig. 5). In addition, two metabolites in plasma were 5-OH-lenlidomide and N-acetyl-lenalidomide, representing 2.9 and 4.2% of lenalidomide levels, respectively. Both metabolites appeared to have an elimination rate similar to lenalidomide (Fig. 5).

Concentrations or equivalents versus time profile for radioactivity, lenalidomide and its metabolites in inter-subject pooled plasma samples
Urinary metabolite profiling
Unchanged lenalidomide accounted for a predominant portion of the radioactivity in urine, with minor amounts of 5-OH-lenalidomide and N-acetyl-lenalidomide. The total radioactivity, lenalidomide, 5-OH-lenalidomide and N-acetyl-lenalidomide in urine up to 24 h were 88.5 ± 3.60, 81.74 ± 3.61, 4.03 ± 0.92 and 1.24 ± 0.92% of the administered dose, respectively. Lenalidomide represented 92% of the total radioactivity in the profiled urine samples. 5-OH-lenalidomide and N-acetyl-lenalidomide were approximately 4.9 and 1.5% of the lenalidomide level.
Fecal metabolite profiling
Radioactivity in fecal samples composed of eight peaks, including unchanged lenalidomide, 5-OH-lenalidomide, N-acetyl-lenalidomide and a few other minor unidentified metabolites. None of the peaks, including parent, represented a predominant portion of the fecal radioactivity or accounted for greater than 1% of the total dose. The total radioactivity, lenalidomide, 5-OH-lenalidomide, and N-acetyl-lenalidomide in the profiled fecal samples were 3.89 ± 1.95, 0.45 ± 0.24, 0.56 ± 0.21 and 0.59 ± 0.46% of the administered dose, respectively. Overall, fecal route of elimination is not an important clearance mechanism for lenalidomide in humans.
Semen radioactivity profiling
The 2–3 h semen samples, which represented 0.0059% of the radioactive dose, was composed of lenalidomide (representing approximately half of the total sample radioactivity) and multiple peaks including 5-hydroxy-lenalidomide, N-acetyl-lenalidomide and some very minor unidentified metabolites. The 72 h semen sample had very low radioactivity levels to allow for profiling.
Discussion
Based on the extent of urinary recovery of radioactivity (90% of dose), it can be concluded that the absorption of lenalidomide (at least 90% of the dose absorbed) is very high in humans following oral administration. Further, based on the 82% of the dose excreted in intact form in urine within 24 h postdose, the oral bioavailability of lenalidomide is at least 82% in humans. Both lenalidomide and the [14C]-radioactivity were rapidly absorbed into the circulation, with the median T max ranging from 0.77 to 1 h postdose. The absorption rate for the suspension is similar to that for the capsule formulation (T max = 0.75–0.88 h) [17]. A comparison of the exposure parameters for lenalidomide and total radioactivity in plasma indicate that unchanged lenalidomide is the predominant circulating component.
The whole blood-to-plasma radioactivity exposure levels were comparable, and the percent radioactivity distributed into red blood cells (44–36%) approximated the mean hematocrit value (41–43%). These data suggest that lenalidomide, as the primary radioactive component in circulation, is distributed nearly equally between cellular and plasma components of whole blood. This observation, along with similar pharmacokinetic profiles for whole blood and plasma radioactivity, demonstrates that plasma is a suitable matrix for the representation of lenalidomide concentrations and exposure in whole blood.
The present study suggests that lenalidomide and its metabolites were distributed into human semen. However, the total radioactivity found in semen accounted only for a minuscule percentage of the administered dose (0.0062%; 1.5 μgEq of lenalidomide). The extent of excretion of lenalidomide in semen was confirmed in another clinical study following multiple oral doses of lenalidomide to healthy men; <0.01% of dose excreted in semen at steady state [17]. Lenalidomide is an analog of thalidomide and has demonstrated teratogenic effects in monkeys [18]. The possible transfer of lenalidomide to a female partner via semen during a heterosexual intercourse with a male on lenalidomide therapy is a concern. Results of both studies suggest that the exposure of lenalidomide to an embryo or fetus in case of nonprotected sexual intercourse between male patient and female partner would be four orders of magnitude lower than the plasma exposure in the treated male subjects.
Total recovery of the [14C]-radioactivity averaged 94.3% of the administered dose, with mean contributions of 90.3, 3.95 and 0.0062% from urine, feces and semen, respectively. Further, the elimination of [14C]-radioactivity was rapid as demonstrated by a relatively short half-life (approximately 3 h) in circulation and a majority (88%) of the radioactivity was recovered within the first 24 h after dosing. The estimation for urinary excretion (82% of the dose) of the unchanged lenalidomide following a radioactive suspension dose is consistent with the previous findings with the capsule formulation (84% of the dose) [15]. In patients with decreased renal function, renal lenalidomide clearance decreased substantially, prolonging its half-life by approximately 6–12 h and increasing plasma exposure by two- to four-fold [15]. Therefore, dose modifications for patients with renal impairment have been implemented [13]. With careful monitoring of renal function and adverse events, and with appropriate dose adjustments, lenalidomide is well tolerated in MM patients with renal impairment [19].
Mean renal clearance (260 ml/min) of the plasma radioactivity was nearly two-fold greater than the normal glomerular filtration rate (125 ml/min), suggesting that renal elimination likely includes both passive and active processes. An in vitro study demonstrated that lenalidomide is a weak substrate of P-glycoprotein, but not a substrate of human organic anionic transporters OAT1/OAT3, human cationic transporter OCT1 and human organic anion transporting polypeptide OATP2 [20]. If there is an active secretion component in the renal excretion of lenalidomide, it is not known which renal transporter(s) may be responsible.
Unchanged lenalidomide is the predominant component of both circulating and excreted radioactivity in humans. Two metabolites, resulting from hydroxylation at the 5 position of the amino-iso-indolinone moiety and N-acetylation, each represented less than 5% of both total radioactivity and unchanged lenalidomide levels in circulation as well as excreta. Based on the results of in vitro pharmacological assays, both of these metabolites are not expected to contribute to the therapeutic activity of lenalidomide (Personal Communication, Dr. P. Schafer, Celgene). Overall, metabolism contributes to a very minor extent to the clearance of lenalidomide in humans. These in vivo results are consistent with in vitro studies that demonstrated minimal ability of cytochrome P450 to metabolize lenalidomide [16]. The data generated in this study demonstrate that lenalidomide is not likely to be subject to drug–drug interactions when coadministered with cytochrome P450 inhibitors during polypharmacy.
In summary, following oral administration, lenalidomide is rapidly absorbed and highly bioavailable, metabolized minimally and eliminated predominantly via urinary excretion in the unchanged form in humans.
References
Knight R (2005) IMiDs: a novel class of immunomodulators. Semin Oncol 32(suppl 5):S24–S30
Kotla V, Goel S, Nischal S et al (2009) Mechanism of action of lenalidomide in hematological malignancies. J Hematol Oncol 2:36
Quach H, Ritchie D, Stewart AK et al (2010) Mechanism of action of immunomodulatory drugs (IMiDs) in multiple myeloma. Leukemia 24:22–32
List A, Kurtin S, Roe DJ et al (2005) Efficacy of lenalidomide in myelodysplastic syndromes. N Engl J Med 352:549–557
Melchert M, Kale V, List A (2007) The role of lenalidomide in the treatment of patients with chromosome 5q deletion and other myelodysplastic syndromes. Curr Opin Hematol 14:123–129
Richardson PG, Blood E, Mitsiades CS et al (2006) A randomized phase 2 study of lenalidomide therapy for patients with relapsed or relapsed and refractory multiple myeloma. Blood 108:3458–3464
Wang M, Dimopoulos MA, Chen C et al (2008) Lenalidomide plus dexamethasone is more effective than dexamethasone alone in patients with relapsed or refractory multiple myeloma regardless of prior thalidomide exposure. Blood 112:4445–4451
Rajkumar SV, Hayman SR, Lacy MQ et al (2005) Combination therapy with lenalidomide plus dexamethasone (Rev/Dex) for newly diagnosed myeloma. Blood 106:4050–4053
Awan FT, Johnson AJ, Lapalombella R et al (2010) Thalidomide and lenalidomide as new therapeutics for the treatment of chronic lymphocytic leukemia. Leuk Lymphoma 51:27–38
Witzig TE, Wiernik PH, Moore T et al (2009) Lenalidomide oral monotherapy produces durable responses in relapsed or refractory indolent non-Hodgkin’s Lymphoma. J Clin Oncol 27:5404–5409
Habermann TM, Lossos IS, Justice G et al (2009) Lenalidomide oral monotherapy produces a high response rate in patients with relapsed or refractory mantle cell lymphoma. Br J Haematol 145:344–349
Dalgleish A, Galustian C (2010) The potential of immunomodulatory drugs in the treatment of solid tumors. Future Oncol 6:1479–1484
Revlimid® package insert. Celgene Corporation. Revlimid® (lenalidomide)
Richardson PG, Schlossman RL, Weller E et al (2002) Immunomodulatory drug CC-5013 overcomes drug resistance and is well tolerated in patients with relapsed multiple myeloma. Blood 100:3063–3067
Chen N, Lau H, Kong L et al (2007) Pharmacokinetics of lenalidomide in subjects with various degrees of renal impairment and in subjects on hemodialysis. J Clin Pharmacol 47:1466–1475
Kumar G, Lau H, Laskin O (2009) Lenalidomide: in vitro evaluation of the metabolism and assessment of cytochrome P450 inhibition and induction. Cancer Chemother Pharmacol 63:1171–1175
Chen N, Lau H, Choudhury S, Wang X, Assaf M, Laskin OL (2010) Distribution of lenalidomide into semen of healthy men after multiple doses. J Clin Pharmacol 50:767–774
Amin RP, Fuchs A, Christian MS et al (2009) An embryo-fetal developmental toxicity study of lenalidomide in cynomolgus monkeys. Birth Defects Res A Clin Mol Teratol 85:435
Dimopoulos M, Alegre A, Stadtmauer ET et al (2010) The efficacy and safety of lenalidomide plus dexamethasone in relapsed and/or refractory multiple myeloma patients with impaired renal function. Cancer 116:3807–3814
Kumar G, Surapeneni S, Lau H, Laskin O, Fox L (2008) Interaction of lenalidomide with human drug transporters in vitro. Drug Metab Rev S3:282–283
Acknowledgments
Authors would like to acknowledge the contributions of the following in the conduct of this study: Dr. Mohit Kothare and Dr. Anthony Frank for the synthesis of metabolite standards, Ms. Emily Stropp for coordinating the synthesis of radiolabeled dosage form, Ms. Iris Su, Dr. Hong Li (ABC Laboratories) and Dr. Gaylin Nickell (ABC Laboratories) for sample analysis.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Nianhang Chen, Henry Lau, Sekhar Surapaneni and Gondi Kumar are employees of Celgene Corporation and own company stock.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Chen, N., Wen, L., Lau, H. et al. Pharmacokinetics, metabolism and excretion of [14C]-lenalidomide following oral administration in healthy male subjects. Cancer Chemother Pharmacol 69, 789–797 (2012). https://doi.org/10.1007/s00280-011-1760-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-011-1760-3