
Abstract
The yeast Kluyveromyces marxianus is a convenient host for industrial synthesis of biomolecules. However, despite its potential, there are few studies reporting the expression of heterologous proteins using this yeast. Here, we report expression of a dengue virus protein in K. marxianus for the first time. The dengue virus type 1 nonstructural protein 1 (NS1) was integrated into the K. marxianus UFV-3 genome at the LAC4 locus using an adapted integrative vector designed for high-level expression of recombinant protein in Kluyveromyces lactis. The NS1 gene sequence was codon-optimized to increase the level of protein expression in yeast. The synthetic gene was cloned in frame with K. lactis α-mating factor signal peptide, and the recombinant plasmid obtained was used to transform K. marxianus UFV-3 by electroporation. The transformed cells, selected in yeast extract peptone dextrose containing 200 μg mL−1 Geneticin, were mitotically stable. Analysis of recombinant strains by RT-PCR and protein detection using blot analysis confirmed both transcription and expression of extracellular NS1 polypeptide. After induction with galactose, the NS1 protein was analyzed by sodium dodecyl sulfate-PAGE and immunogenic detection. Protein production was investigated under two conditions: with galactose and biotin pulses at 24-h intervals during 96 h of induction and without galactose and biotin supplementation. Protease activity was not detected in post-growth medium. Our results indicate that recombinant K. marxianus is a good host for the production of dengue virus NS1 protein, which has potential for diagnostic applications.
Similar content being viewed by others


Introduction
The yeasts Kluyveromyces marxianus and Kluyveromyces lactis are phylogenetically related to the baker’s yeast Saccharomyces cerevisiae, but they have unique advantages for biotechnological applications. Unlike S. cerevisiae, K. marxianus and K. lactis have the ability to assimilate lactose and use it as a sole carbon and energy source. Owing to this feature, these yeasts are common in dairy products such as milk, cheese, and yogurt (Lane and Morrissey 2010), giving Kluyveromyces the status of Generally Recognized as Safe (GRAS) and allowing its use in the pharmaceutical and food industries. Moreover, K. marxianus has been isolated from several other environments, which reflects its high metabolic diversity. As a consequence, several biotechnological applications have been investigated for this yeast, for example the production of aromatic compounds and bioingredients, including cheese whey (Fonseca et al. 2008), ethanol (dos Santos et al. 2013; Silveira et al. 2005), and most recently as a host for heterologous protein synthesis (Rocha et al. 2010; Rocha et al. 2011). In addition, the potential of K. marxianus for industrial use is further supported by its thermotolerance, high growth rate, and broad substrate spectrum (Fonseca et al. 2008). We are interested in the potential of K. marxianus as a host for the synthesis and secretion of recombinant proteins with immunogenic or vaccinal functions.
Dengue is a tropical mosquito-borne viral disease caused by infection with one of four serotypes of dengue virus (Coller et al. 2011), for which there is no preventive vaccine or effective treatment currently available (Swaminathan and Khanna 2009). Dengue virus contains a single positive-stranded RNA genome of about 11 kb that encodes three structural proteins and seven nonstructural proteins (Guzman et al. 2010). Dengue nonstructural protein 1 (NS1) is a glycoprotein of approximately 40–46 kDa, with two N-linked glycosylation sites, that may form homodimers and is detected in serum of infected individuals and also in infected cells in vitro (Noisakran et al. 2007). NS1 is a protective antigen (Athmaram et al. 2012) and an important biomarker of dengue infection, because it can be detected before formation of antibodies (Alcon et al. 2002; Singh et al. 2010). Thus, the use of NS1 antigen has been suggested for early diagnosis of dengue infection after the onset of fever (Chaiyaratana et al. 2009; McBride 2009; Ramirez et al. 2009).
There are several reports on the use of heterologous expression systems (e.g., vaccinia virus, cell lines, Pichia pastoris, and Escherichia coli) for expressing NS1 protein (Athmaram et al. 2012; Noisakran et al. 2007; Zhao et al. 1987; Zhou et al. 2006). However, no systematic studies have been carried out using Kluyveromyces yeasts for protein synthesis from synthetic genes encoding viral proteins. Our goal is to establish a system for large-scale synthesis of recombinant NS1 (rNS1) protein utilizing standard yeast medium, which does not require the explosion-proof fermentation equipment necessary for large-scale growth of methylotrophic yeasts such as P. pastoris, and to drive secretion of the protein into the culture supernatant, facilitating downstream operations. With this background, the current study was focused on the construction of K. marxianus UFV-3 strains to produce rNS1 protein, which has an important application in diagnostic testing.
Materials and methods
Microorganisms and maintenance
The yeast strain used in this work is designated as K. marxianus UFV-3 (collection number CCT 7735). It was isolated from a regional dairy industry in southeast Brazil and taxonomically identified by the Centraalbureau voor Schimmelcultures (Utrecht, The Netherlands) as Candida kefyr (Beijerinck) van Uden & Buckley [non-ascospore-forming state of K. marxianus (Hansen) van der Walt]. The yeast strain was maintained on YPD plates [1 % (w/v) yeast extract, 2 % (w/v) peptone, 2 % (w/v) glucose, 2 % (w/v) agar]. For long-term storage, cells were maintained frozen at −80 °C in 20 % (v/v) glycerol. All K. marxianus UFV-3 strains (wild type and selected mutants) were precultured in YPD medium [1 % (w/v) yeast extract, 2 % (w/v) peptone, 2 % (w/v) glucose]. YPD agar containing 200 μg mL−1 of Geneticin (G418, Sigma®) was used to select transformants. E. coli strain DH5α™ was used for routine recombinant plasmid manipulations. Nontransformed and recombinant E. coli cells were subcultured in Luria-Bertani (LB) medium at 37 °C, supplemented with ampicillin (50 μg mL−1) as appropriate.
Isolation and manipulation of nucleic acids and dengue NS1 nucleotide sequences
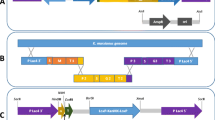
Maps of the plasmids used in this study are shown in Fig. 1. The vector pKLAC2 (New England Biolabs®) was used as a base. The construct pKLJC/GME (Rosa et al. 2013) was used to obtain the LoxP-KanMX-LoxP cassette that confers resistance to the aminoglycoside antibiotic G418 and kanamycin, and the cassette was subcloned into the BsrGI and XmaI sites of pKLAC2, replacing the acetamidase selectable marker (amdS) to yield a new plasmid designated as pKMCL. The construct pTZ57R/T+NS1-Denv1 (see below) was used as the template for PCR amplification of recombinant NSI/Denv1 (rNS1/Denv1) with the primers ns1/denv1-F and ns1/denv1-R (Table 1). The rNS1 product was synthesized using the amplification parameters 30 s at 98 °C, followed by 35 cycles of 10 s at 98 °C, 1 min at 70 °C, and 1 min at 72 °C, and cloned into pGEM®-T Easy Vector (Promega) and further transferred into pKMCL to generate the plasmid pKMCL/NS1-Denv1. All ligation reactions were performed with a T4 DNA Ligase kit from Invitrogen®.

Maps of the plasmid vectors used for rNS1 gene expression: pKLAC2 (a), pKlJC/GME (b), and pKMCL (c). pKLAC2 was derived from pKLAC1. pKMCL was created by inserting the LoxP-KanMX-LoxP cassette of pK1JC/GME between the restriction sites BsrGI and XmaI of pKLAC2 and deleting the amdS gene. The cassette confers resistance to Geneticin
The nucleotide sequence encoding the viral protein was designed from the primary sequence of the NS1/Denv1 peptide, with the codons optimized for expression in yeast (GenBank accession number KJ418180). The two N-linked glycosylation sites were maintained. This 1,056-bp sequence constitutes the open reading frame (ORF) encoding the NS1 protein from dengue virus type 1 and was cloned into the pTZ57 R / T vector. The construct pTZ57R/T+NS1-Denv1 was a gift of Dr. Eduardo Rezende Honda.
Yeast transformation
K. marxianus UFV-3 transformation was carried out according to Sanchez et al. (1993), with some modifications. Fresh K. marxianus UFV-3 cells were plated on YPD agar medium and incubated overnight at 37 °C. An isolated colony was inoculated in 50 mL YED medium [1 % (w/v) yeast extract, 1 % (w/v) glucose] at 30 °C and 200 rpm overnight. With these precultured cells, 50 mL YED was inoculated to start at OD600 0.0025 per milliliter (0.1 OD). When OD600 reached approximately 0.8, the cells were harvested at 3,000 g for 5 min at 4 °C and washed with 20 mL ice-cold sterile distilled water. At 30 °C for 30 min and 100 rpm, 20 mL of pretreatment buffer (YED, 25 mM DTT, and 20 mM HEPES-Tris pH 8.0) was added and further incubated. Cells were collected at 3,000 g for 5 min at 4 °C and resuspended in 0.3 mL electroporation buffer (EB) (10 mM Tris-HCl, pH 7.5, 270 mM sucrose, and 1 mM MgCl2) and divided into 60-μL aliquots of competent cells in Eppendorf tubes on ice. To each aliquot were added 50 μg Salmon Sperm DNA (SS-DNA) and 2 μg transforming DNA, and the tubes were kept on ice for 15 min. The mixture was transferred to a chilled electroporation cuvette (2 mm) on ice and eletroporated at 1 kV, 25 μF, and 400 Ω. Immediately, 1 mL of ice-cold YED medium was added and the mixture was incubated on ice for 15 min and at 37 °C for 1 h with shaking (200 rpm). The cells were plated on YPD agar plates containing 200 μg mL−1 of Geneticin and kept at 37 °C for 2 days (note: Geneticin should be spread on the surface of the plates).
Total DNA extraction and yeast transformant screening
K. marxianus UFV-3 cells were grown in 5 mL YPD containing selective antibiotic at 37 °C to saturation. The cell mass was collected by centrifugation, resuspended in 0.2 mL lysis buffer (2 % Triton X-100, 1 % sodium dodecyl sulfate (SDS), 100 mM NaCl, 10 mM Tris pH 8, 1 mM EDTA), and transferred to 2-mL screw-cap tubes. Next, 0.2 mL of PCI [phenol pH 6.7-chloroform-isoamyl alcohol (25:24:1)] and 0.3 g of glass beads were added. The cells were broken by using a FastPrep homogenizer (speed 6 for 20 s) followed by centrifugation at 10,000 g for 10 min. The supernatant was transferred to a new tube, 0.5 mL ethanol was added, and the mixture was kept at −20 °C for at least 20 min. Total DNA was pelleted by centrifugation (14,000 g, 10 min), washed with 70 % ethanol, and dried at room temperature. The DNA samples were dissolved in 20 μL nuclease-free water and kept at −20 °C. An aliquot of 1 μL was used as template in a 50-μL PCR. The specific primers used to detect the single cassette insertion into the LAC4 promoter locus are shown in Table 1. The amplification parameters were as follows: 5 min at 98 °C; 35 cycles of 10 s at 98 °C, 30 s at 70 °C, and 1 min at 72 °C; and 5 min at 72 °C.
Total RNA extraction from yeast and RT-PCR
The transformed yeast cells were grown in 20 mL YPD medium containing selective antibiotic at 37 °C and 200 rpm overnight. The cells were pelleted by centrifugation at 9,000 g for 5 min at 4 °C, and the supernatant was discarded. Total RNA from recombinant K. marxianus UFV-3 yeast cells was extracted using the hot acid phenol method as described by Collart and Oliviero (2001). cDNA synthesis from total RNA was carried out using the Reverse Transcription System from Promega®. A 2-μL cDNA aliquot from each sample was used in a 50-μL PCR to detect mRNA expression of the rNS1 gene inserted into the K. marxianus UFV-3 genome. The RT-PCR was carried out using the same primers and amplification cycles used for viral gene amplification.
Evaluating the genetic stability of the recombinant strains
Recombinant strains were transferred to YPD agar and incubated at 37 °C until the appearance of isolated colonies. Next, five colonies of each transformant were successively transferred to nonselective complete YPD medium in a total of five replatings. Each replating was incubated at 37 °C for 48 h. At the end of the fifth replating, the colonies were transferred to selective medium (YPD agar plates containing 200 μg mL−1 of Geneticin) and incubated at 37 °C for over 48 h. The transformed cells were then subjected to an induction phase test.
Induction of protein expression
The recombinant yeast cells were precultured overnight in 50 mL YPD at 200 rpm and 37 °C to obtain a cell mass. The culture was washed with peptone water 0.01 % (w/v) and used to inoculate 1 L Yeast Nitrogen Base with amino acids and 5 g L−1 ammonium sulfate (YNB Sigma, St. Louis, USA) containing 4 g L−1 of galactose (inducer), and yeast extract 0.5 % (w/v) supplemented with biotin 4 × 10−5 % (w/v). The cells were cultured at 37 °C (optimum temperature determined for growth) and 250 rpm, in a 2.5-L BioFlo® & CelliGen® 310 Fermentor/Bioreactor (New Brunswick); pH was maintained at 5.0 by addition of either 10 % (w/v) HCl or 10 % (w/v) NaOH as required. Samples were collected every 24 h for measurement of specific protease activity, SDS-PAGE, and rNS1 detection. An induction experiment was carried out in parallel, during which every 24 h, a pulse of 4 g L−1 galactose solution and 4 × 10−5 % (w/v) biotin was added to the culture. Foaming was prevented by addition of sterile antifoam solution (Sigma Chemicals) as required. Shake-flask experiments were performed under the same conditions described above for comparison.
Total protein content was measured according to Bradford (1976), using bovine serum albumin (BSA) as the standard. Specific protease activity was measured according to Ray et al. (1992) with some modifications and assayed with cell-free culture supernatants, using azocasein as a substrate at a concentration of 1 % (w/v), pH 7.5. Enzymatic hydrolysis of azocasein releases stable dye-labeled peptides and amino acids into the reaction mixture which can be measured easily. Azocasein protease activity was measured by incubating 100 μL of culture supernatant and 100 μL of 0.5 % (w/v) azocasein for 12 h at 30 °C. The reaction was stopped by adding 1 mL of 10 % (w/v) trichloroacetic acid and allowed to stand for 15 min at room temperature. The mixture was centrifuged at 10,000 g for 10 min to remove a yellow precipitate. The absorbance of the supernatant was measured at 440 nm using a spectrophotometer. The activity of the protease was expressed in arbitrary units, where 1 unit of activity was defined as the absorbance obtained divided by the total protein concentration in milligrams · per liter per time unit.
Fractionation of cultures for rNS1 SDS-PAGE analysis and immunogenic detection using positive human serum for dengue virus
Both cells and culture supernatants were routinely assayed for rNS1 detection. Cultures were fractionated into supernatant, cell wall (retained inside the periplasmic space), and cell-bound protein fractions according to the method of Rouwenhorst et al. (1988). Total protein content was collected during the induction phase and precipitated with trichloroacetic acid and acetone, resuspended in SDS sample buffer [150 mM Tris-HCL pH 7.0, 200 mM DTT, 12 % (w/v) SDS, 30 % glycerol, 0.05 % (w/v) Coomassie Blue G-250, 8 M urea (instead of water)] and stored at −20 °C. Samples were heated to 100 °C for 5 min, and proteins were separated by SDS-PAGE. Electrophoresed proteins were transferred to a nitrocellulose membrane, and positive human serum for dengue virus was used to detect rNS1 by western blot analysis. In brief, for the western blot analysis, the extracted proteins along with prestained protein markers on adjacent lanes were transferred electrophoretically to a nitrocellulose membrane. The membrane was blocked overnight with 5 % (w/v) bovine serum albumin in TBS buffer (10 mM Tris-HCl, pH 7.6, 1.4 mM NaCl) at 4 °C and then washed with TBS buffer plus 0.1 % (w/v) Tween 20 (TBS-T) for 20 min. Serum samples were diluted 1:10,000 with blocking buffer and incubated with membranes for 2 h at room temperature. After incubation, membranes were washed with TBS-T and incubated for 1 h with human anti-IgG and anti-IgM (Sigma Chemicals). The protein bands were visualized by incubating in the substrate 3,3′-diaminobenzidine (Sigma Chemicals). In order to evaluate whether the secreted rNS1 protein was in a monomeric or dimeric form, samples were prepared under nonreducing conditions.
rNS1 isolation
The culture supernatant containing rNS1 protein was rapidly adjusted to pH 8.0 using 5 M NaOH to minimize precipitation of rNS1 (isoelectric point (pI) 5.7) and was freshly diluted with an equal volume of binding buffer, pH 8.0 (20 mM Tris-HCl). The diluted supernatant was clarified by filtration through a 0.45-μm membrane and then submitted to ion chromatography (IC). The clarified supernatant was loaded onto a 1-mL HiTrap Capto Q column, previously equilibrated with binding buffer and attached to an AKTA purifier system (GE Healthcare Life Sciences, Uppsala, Sweden). Ten-column volumes of binding buffer were passed through the column to remove nonspecifically bound proteins prior to elution with 10, 25, 50, and 100 % elution buffer (20 mM Tris-HCl, 500 mM NaCl). Peak fractions were collected and dialyzed against PBS pH 8.0 using centrifugal filtration devices with a 10,000-molecular-weight cutoff (Millipore). Protein concentration was determined with a BCA kit (Pierce Chemical Co., Rockford, USA). For characterization, the purified proteins were subjected to 15 % SDS-PAGE.
Results
Construction of an expression vector encoding a Geneticin marker and the dengue virus NS1 gene to function in K. marxianus UFV-3
In order to replace the amdS from pKLAC2, the 1,592-bp LoxP-KanMX-LoxP cassette that confers resistance to Geneticin was obtained from the vector pKLJC/GME by cleavage with BsrGI and XmaI restriction endonucleases. The DNA fragment was recovered from agarose gel and subcloned into pKLAC2 vector, now designated as pKMCL.
After confirming the construction of the pKMCL vector (Fig. 1), the DNA encoding the amino acid sequence of the NS1/Denv1 peptide was obtained by PCR. The amplification resulted in a fragment of about 1,056 bp, consistent with the size expected for the NS1 gene. This DNA fragment was used to construct the expression vector pKMCL-rNS1/Denv1, in which the NS1 region from DENV-1 was cloned in frame with the K. lactis α-mating factor secretion domain (α-MF) to direct the fusion protein to the general secretory pathway, thus allowing secretion of the recombinant protein into the medium. The construct was confirmed by cleavage with EcoRI and NdeI restriction endonucleases, resulting in a DNA fragment with the expected size of approximately 1,056 bp (Fig. 2).

Confirmation of vector construction. a pKMCL-rNS1/Denv1 expression vector map. b Agarose gel electrophoresis, 1.2 % (w/v): the insert was released after cleavage of the construct with EcoRI and NdeI restriction endonucleases (lane 1); 1-kb Plus DNA Ladder (Invitrogen) (lane 2)
Transformation of K. marxianus UFV-3 with pKMCL-rNS1/Denv1 and mitotic stability of recombinants
Before transformation of K. marxianus UFV-3 with pKMCL-rNS1/Denv1, the vector was linearized with SacII enzyme. Cleavage resulted in a cassette containing the sequence of interest flanked by regions 3′PLAC4 and 5′PLAC4, which direct integration of the cassette by homologous recombination into the promoter region of the K. marxianus LAC4 locus. Transformants were selected by growth in the presence of 200 μg mL−1 Geneticin and then tested for the presence of the integrated NS1 gene by direct colony PCR. Transformation efficiency was 504 transformants per microgram of plasmid DNA. To analyze if the expression fragment had inserted at the targeted LAC4 locus, genomic DNA was extracted from the transformants and subjected to PCR amplification with primers 1 and 2 (Table 1), which resulted in amplification of a diagnostic DNA fragment of 2.4 kb (Fig. 3) from a selected clone. No nonspecific amplification band was found. Several of the PCR-positive NS1 gene-harboring clones were tested for mitotic stability. All recombinants analyzed were stable (data not shown).

Agarose gel electrophoresis [1.2 % (w/v)] of the PCR product from wild type (lane 1) and a recombinant strain (lane 2). The fragment inserted into the promoter of the LAC4 locus. Single-copy integration at the LAC4 locus was detected using primers 1 and 2 to amplify a 2.4-kb diagnostic fragment (lane 2) and 1-kb Plus DNA Ladder (Invitrogen) (lane 3)
Selection of a K. marxianus UFV-3 recombinant strain and expression and function of the NS1 protein
To analyze the expression of the rNS1/Denv1 protein, an induction test of the transcript and detection of secreted protein by using dot-blot immunoassay with human serum positive for dengue virus were performed. Several recombinant clones were tested in a small-scale expression assay in induction medium. DNA fragments of approximately 1,056 bp obtained by RT-PCR (data not shown) confirmed transcription of the integrated NS1 gene. Cell-free culture supernatants from recombinant cells were used to perform a dot-blot immunoassay, which revealed that the recombinant strain was able to secrete rNS1/Denv1 (Fig. 4), demonstrating that the signal sequences cloned upstream of the NS1 gene indeed targeted the recombinant protein to be secreted.

Induction test of the transcript and recognition of secreted proteins by using a dot-blot immunoassay. Dot-blot analysis of rNS1 secreted by K. marxianus UFV-3. Lanes 1–4: Culture supernatant (100 μL) of recombinant strains and wild type (C-) were spotted onto a nitrocellulose membrane and immunodetected with human serum positive for dengue virus. All the recombinants analyzed were able to express and secrete the rNS1 peptide
To identify the best expressing clone, logarithmically growing test tube cultures of 10 clones were galactose-induced for 72 h, and supernatants were assayed for rNS1 expression by SDS-PAGE (data not shown). One clone, #1, which expressed higher levels of rNS1 among all the clones tested, was used for further study.
Growth of recombinant K. marxianus UFV-3 and production of rNS1 protein in a shake flask and bioreactor
Because it is our objective to develop a cost-effective means of production of the viral protein, we sought to explore culture conditions that would help maximize rNS1 synthesis and secretion. Recombinant protein expression on a laboratory scale is generally performed using a complex medium in shake-flask cultures. However, for producing proteins in bioreactors, the composition of the medium seems to be a critical factor to ensure both cell growth and rNS1 protein production. To optimize synthesis of rNS1 protein by the K. marxianus UFV-3 recombinant strain, the culturing was carried out in two stages. In the first stage, a cell mass of approximately 10 g L−1, corresponding to OD600 of 20, was obtained. This stage was conducted by growing a starter culture of cells (OD600 of 2) in YPD medium for 16 h. In the second stage, the recombinant cell mass was harvested by centrifugation and resuspended in induction medium [YNB buffered at pH 5.0, containing 4 g L−1 of galactose and 0.5 % (w/v) yeast extract supplemented with 4 × 10−5 % (w/v) biotin]. The experiments were conducted in a BioFlo® & CelliGen® 310 Fermentor/Bioreactor (New Brunswick). Shake-flask experiments were also performed in 250-mL Erlenmeyer flasks containing 50 mL of the same cultivation medium. Samples of supernatants of the induction medium were evaluated for total protein and proteolytic activity. Total protein was analyzed during the induction phase from culture aliquots withdrawn at various time points. It was observed that the recombinant strains secreted a greater amount of total protein when a galactose pulse was applied every 24 h (data not shown). Protein quantitation (according to Bradford) revealed that the concentrations reached a maximum of ∼1.2 mg/mL−1 after 72 h of induction. The accumulation of total protein in the culture medium indicated the stability of the secreted proteins and the absence of proteases, which was confirmed by protease activity assay of the supernatants of both recombinant and wild-type strains cultured in bioreactors and shake flasks (Tables 2, 3, 4, and 5).
Detection and isolation of rNS1 in supernatant culture
To analyze the distribution of the rNS1 protein, the culture was fractionated into supernatant, cell-bound, and cell-wall protein fractions; separated by SDS-PAGE; and analyzed by western blot (Fig. 5). The results confirmed that rNS1 is secreted (Fig. 5a) and that it appears in all three fractions examined (Fig. 5b, c). The concentration of rNS1 released by the cell-wall and cell-bound fractions was lower than the total protein secreted into the supernatant. In particular, the secretion efficiency of rNS1 using the pKMCL expression vector adapted from pKLAC2 was confirmed by western blot analysis using a dengue-infected human serum sample (Fig. 6). No similar band appeared in the negative control. To evaluate whether the secreted rNS1 protein was in a monomeric or dimeric form, samples were prepared under nonreducing conditions (data not shown); the results indicate that NS1 protein was secreted mainly in a monomeric form of approximately 45 kDa.

Coomassie blue-stained 12 % (w/v) SDS-polyacrylamide gel (a, b) and western blot (c) for the analysis of rNS1 expressed in K. marxianus UFV-3. a K. marxianus supernatant: prestained protein ladder (Thermo Scientific) (1), wild type (2), and recombinant strain producing rNS1 (3). b Fractionation of cultures for rNS1 detection in K. marxianus: prestained protein ladder (Thermo Scientific) (1), negative control (2), K. marxianus cell-bound rNS1 (3, 4), and K. marxianus cell wall-associated rNS1 (5–8). The arrow and the arrowheads identify rNS1. c Fractionation of cultures for rNS1 detection in K. marxianus: negative control (1), K. marxianus cell-bound rNS1 (2), K. marxianus cell wall-associated rNS1 (3), and prestained molecular weight standard (Thermo Scientific)

Expression of dengue virus type 1 rNS1 in K. marxianus UFV-3. Prestained molecular weight standard (Thermo Scientific) (1). Western blot analysis of rNS1 protein using human serum positive for dengue virus (2)
The 96-h cell-free culture supernatant from bioreactor cultures was diluted with an equal volume of binding buffer, pH 8.0 (20 mM sodium phosphate, 500 mM NaCl), clarified by filtration through a 0.45-μm membrane, and then examined by ion chromatography, which yielded four distinct peaks (Fig. 7a). The recombinant protein emerged in peak 2, with other proteins found in the supernatant (Fig. 7b).

Ion exchange chromatography profile of 10 mL K. marxianus UFV-3 bioreactor supernatant containing rNS1. a Chromatogram of the IC. Four distinct peaks are shown. Peaks 1 to 4 were eluted with 10, 25, 50, and 100 % elution buffer, respectively. b SDS-PAGE analysis of the peak fractions collected. Prestained protein ladder (Thermo Scientific) (1), peaks 1 to 4, respectively (2–5), and nonspecifically bound proteins (6). The arrowhead indicates the position of the rNS1 protein
Discussion
Successful production of dengue virus protein has been reported in several vector systems (Batra et al. 2010; Blaney et al. 2005; Block et al. 2010; Jaiswal et al. 2004; Saxena et al. 2008; Zhou et al. 2006). Although prokaryotic expression systems such as those in E. coli are facile systems, it is difficult to purify recombinant protein from inclusion bodies in bacteria (Wei et al. 2003). Furthermore, they lack the modification mechanisms (e.g., posttranslational modifications) that many eukaryotic proteins require for optimal biological activity and stability (Coller et al. 2011). One should be aware however that protein glycosylation is a major posttranslational modification process in the yeast secretory pathway and confers an advantage in the secretory production of heterologous proteins that require glycosylation for proper folding and biological activity (Idiris et al. 2010). One drawback to the use of yeast and fungal expression systems for the production of therapeutics is that secreted proteins that become glycosylated will bear a nonhuman glycosylation pattern. This can adversely affect the half-life, tissue distribution, and immunogenicity of a therapeutic protein (Van Ooyen et al. 2006). Therefore, more suitable hosts for heterologous expression of eukaryotic proteins are required. In the present study, we investigated the suitability of K. marxianus UFV-3 yeast to produce dengue-1 NS1 protein, because of its ease of molecular genetic manipulation, its ability to express foreign proteins either intra- or extracellularly, its high growth rate and capacity to produce cell mass, and its status of being GRAS for the production of food- and pharmaceutical-related proteins.
The biggest disadvantage in utilizing K. marxianus as a host strain for recombinant protein production is the absence of a commercial cloning and expression system. We first tested a system designed for high-level expression of recombinant protein in K. lactis for the selection of recombinant K. marxianus UFV-3, but the nontransformed strains were able to utilize acetamide, the selective agent, as the sole nitrogen source. We tested several concentrations of acetamide (5–40 mM), and at all concentrations, we observed growth of the K. marxianus strains. In order to use this vector in K. marxianus UFV-3 cells, it was necessary to replace the amdS from pKLAC2 with a LoxP-KanMX-LoxP cassette that confers resistance to Geneticin. To transform the K. marxianus UFV-3 cells, several protocols were tested but only the protocol described in “Materials and methods” was successful.
A versatile feature of K. marxianus (and K. lactis) as a host system is that it can be transformed with vectors that replicate extrachromosomally (episomal vectors) or that integrate into the genome (integrative vectors). Both vector systems have been used to direct expression of recombinant proteins in K. marxianus, as described in Van Ooyen et al. (2006) and Rocha et al. (2010). Episomal vectors provide a higher copy number to the expression constructs but can be unstable in the absence of selection. This instability leads to expression problems of some heterologous proteins, especially in large industrial applications where cells are often subjected to prolonged growth in the absence of selection. Rocha et al. (2010) employed different episomal and integrative constructs and demonstrated that, based on plasmid stability studies, both K. marxianus and K. lactis cells lost nearly all their episomal expression plasmids after 48 h. In another study, after growth of a K. lactis strain secreting human lysozyme expressed from a pKD1 vector, only 17.3 % of cells retained the episomal vector in contrast with >91.5 % of cells that retained an integrated expression vector producing the same enzyme under the same growth conditions (Iwata et al. 2004). The integration cassette used in the present work has regions of homology with the LAC4 promoter, which reduces but does not eliminate the possibility of ectopic integration. To verify the correct genomic integration of the linear expression cassette into the promoter region of the K. marxianus LAC4 locus, we used specific primers designed to K. lactis LAC4. Our results demonstrate that these primers can also be used in K. marxianus, because nonspecific bands were not amplified. Analysis of K. marxianus UFV-3 recombinant strains by RT-PCR and protein detection using positive human serum for dengue virus confirmed the transcription and secretion of rNS1. A major benefit of using an integrative expression vector is the increased genetic stability of the recombinant strains, confirmed in our study by a mitotic stability test, because the use of mitotically stable recombinant lines reduces the difficulties of maintaining selective pressure in the culture medium in large-scale industrial applications.
We evaluated different culture conditions in an effort to maximize rNS1 production. Early experiments in our laboratory showed that, among the different media tested, highest yields were obtained using YNB containing 4 g L−1 of galactose and 0.5 % (w/v) yeast extract supplemented with biotin. In the present work, we evaluated a variety of induction conditions, including different pH levels and galactose and biotin pulses, to enhance the cell mass and level of rNS1 induction in YNB, rather than in YPGal [1 % (w/v) yeast extract, 2 % (w/v) peptone, 2 % (w/v) galactose]. We observed that the induction performed in YNB [containing 5 g L−1 ammonium, 4 g L−1 of galactose, and 0.5 % (w/v) yeast extract supplemented with biotin 4 × 10−5 %] buffered at pH 5.0 increased the cell mass and rNS1 levels approximately 50-fold compared to YPGal medium. Periodic monitoring of the cell density during the induction phase revealed that the cells grew best at pH 5.0. Since at this pH cell viability increased and no proteolysis was detected, there was a higher accumulation of recombinant protein in K. marxianus UFV-3 cultured in a bioreactor. When recombinant cells were cultured in shake flasks, a maximum OD600 of 13 was reached during 96 h of induction in buffered medium. In contrast, when the recombinant cells were grown in a bioreactor under the same conditions described above, a maximum OD600 of 24 was reached. These results suggest that the use of bioreactors for recombinant protein production is recommended, since the production of recombinant protein is directly related to the cell mass in the medium. We also evaluated the effect of pH on rNS1 production. As the pI of NS1 is predicted to be ∼5.7, we used YNB at pH 3, 4, and 5, to preclude isoelectric precipitation of the recombinant protein. YNB medium buffered at pH 5.0 was selected. The effect of galactose concentration on the induction of gene expression and cell mass was also evaluated. Several studies used 2 % (w/v) galactose or lactose as the inducer for heterologous protein production (Bartkeviciute and Sasnauskas 2003; Donnini et al. 2004; Ganatra et al. 2011; Kooistra et al. 2004; Li et al. 2011), but we determined that the use of 4 % (w/v) galactose increased the concentration of cell mass and hence the recombinant protein in the bioreactor.
rNS1 synthesis by K. marxianus UFV-3 was performed in two stages and conducted in both a bioreactor and shake flasks containing the same cultivation medium. In the first stage, glucose was used as the carbon source for generating cell mass by allowing high rates of K. marxianus growth; maximizing cell mass is crucial for improving rNS1 yields. However, this culture medium can compromise the recovery of recombinant peptides due to the presence of self-peptides in the culture medium. In the second step, induction of rNS1synthesis in 4 g L−1 of galactose showed that induction parameters are also important and can directly affect the yield of the recombinant protein. Two induction conditions were tested: with and without pulses of galactose and biotin every 24 h during 96 h of induction, since biotin is an essential vitamin for the growth of many microorganisms (Jungo et al. 2007). The concentration of extracellular proteins in those cultures pulsed with galactose and biotin was higher compared to that in cultures not receiving galactose and biotin supplementation. Therefore, it can be inferred that the inducer is consumed over time and the addition of galactose maintains the synthesis and secretion of proteins. In this way, the galactose and biotin pulse must be considered as a strategy for increasing rNS1 yields. One of the major problems that hinders effective secretion and purification of recombinant proteins from yeast is proteolytic degradation in the culture medium of the recombinant gene products by extracellular proteases, cell-bound proteases, and/or by intracellular proteases from lysed cells. These proteases are present in relatively high levels in yeast and are induced readily by environmental stresses, especially during high-density fermentation processes (Idiris et al. 2010). Thus, two approaches have been attempted as a solution: controlling the cultivation conditions (culture pH) and changing the composition of the medium. The accumulation of total protein in the supernatant revealed the stability of secreted proteins and the absence of proteases, which was confirmed by protease activity assay. This result is important because the absence of detectable proteases in the supernatant reduces the cost and improves the efficiency of obtaining the desired recombinant proteins. Sugrue et al. (1997) showed that expression of the dengue protein in P. pastoris was accompanied by extensive proteolytic degradation, making downstream operations difficult.
Currently, most P. pastoris fed-batch cultivations for the production of recombinant proteins, using the AOX1 expression system, consist of three or four distinct phases, which include glycerol batch, glycerol fed batch, starvation, and methanol fed batch. However, we have recently demonstrated a simple two-step strategy, which only includes a glucose batch phase, to generate sufficient cell mass, directly followed by the induction phase with galactose for recombinant protein synthesis. In the present work, we successfully employed this two-step strategy for the secretion of rNS1. Recombinant NS1 protein was obtained in the first 24 h of cultivation. SDS-PAGE and western blot analysis showed that larger quantities of secreted rNS1 were present in the supernatant than were retained inside the periplasmic space or cell-bound. The cell-free culture supernatant obtained was loaded onto an ion chromatography column for rNS1 isolation, but other proteins were also isolated in the same fractions.
In summary, a novel approach to generate recombinant NS1 protein was developed using K. marxianus UFV-3. Our rNS1 protein exhibited the same molecular size as an authentic dengue virus protein. To our knowledge, this is the first study concerning the production of a viral protein with immunogenic potential of pharmaceutical interest using the yeast K. marxianus. The rNS1 produced has potential applications in the detection of anti-dengue IgM and IgG antibodies as well as in further studies in vaccine development for dengue virus infection. By using this strategy, other recombinant viral proteins could also be produced. Further studies of the structure and functions of the rNS1 protein will be performed.
References
Alcon S, Talarmin A, Debruyne M, Falconar A, Deubel V, Flamand M (2002) Enzyme-linked immunosorbent assay specific to dengue virus type 1 nonstructural protein NS1 reveals circulation of the antigen in the blood during the acute phase of disease in patients experiencing primary or secondary infections. J Clin Microbiol 40:376–381. doi:10.1128/JCM.40.2.376
Athmaram TN, Saraswat S, Misra P, Shrivastava S, Singh AK, Verma SK, Gopalan N, Behara PK, Rao PVL (2012) Optimization of dengue-3 recombinant NS1 protein expression in E. coli and in vitro refolding for diagnostic applications. Virus Genes. doi:10.1007/s11262-012-0851-5
Bartkeviciute D, Sasnauskas K (2003) Studies of yeast Kluyveromyces lactis mutations conferring super-secretion of recombinant proteins. Yeast 20:1–11. doi:10.1002/yea.935
Batra G, Gurramkonda C, Nemani SK, Jain SK, Swaminathan S, Khanna N (2010) Optimization of conditions for secretion of dengue virus type 2 envelope domain III using Pichia pastoris. J Biosci Bioeng 110:408–414. doi:10.1016/j.jbiosc.2010.05.001
Blaney JE, Matro JM, Murphy BR, Whitehead SS (2005) Recombinant, live-attenuated tetravalent dengue virus vaccine formulations induce a balanced, broad, and protective neutralizing antibody response against each of the four serotypes in rhesus monkeys. J Virol 79:5516–5528. doi:10.1128/JVI.79.9.5516
Block OKT, Rodrigo WWSI, Quinn M, Jin X, Rose RC, Schlesinger JJ (2010) A tetravalent recombinant dengue domain III protein vaccine stimulates neutralizing and enhancing antibodies in mice. Vaccine 28:8085–8094. doi:10.1016/j.vaccine.2010.10.004
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–54
Chaiyaratana W, Chuansumrit A, Pongthanapisith V, Tangnararatchakit K, Lertwongrath S, Yoksan S (2009) Evaluation of dengue nonstructural protein 1 antigen strip for the rapid diagnosis of patients with dengue infection. Diagn Microbiol Infect Dis 64:83–84. doi:10.1016/j.diagmicrobio.2009.01.004
Collart MA, Oliviero S (2001) Preparation of yeast RNA. Curr Protoc Mol Biol Chapter 13:Unit13.12. doi:10.1002/0471142727.mb1312s23
Coller B-AG, Clements DE, Bett AJ, Sagar SL, Ter Meulen JH (2011) The development of recombinant subunit envelope-based vaccines to protect against dengue virus induced disease. Vaccine 29:7267–7275. doi:10.1016/j.vaccine.2011.07.021
Donnini C, Farina F, Neglia B, Compagno MC, Uccelletti D, Goffrini P, Palleschi C (2004) Improved production of heterologous proteins by a glucose repression-defective mutant of Kluyveromyces lactis. Society 70:2632–2638. doi:10.1128/AEM.70.5.2632
Dos Santos VC, Bragança CRS, Passos FJV, Passos FML (2013) Kinetics of growth and ethanol formation from a mix of glucose/xylose substrate by Kluyveromyces marxianus UFV-3. Antonie Van Leeuwenhoek 103:153–161. doi:10.1007/s10482-012-9794-z
Fonseca GG, Heinzle E, Wittmann C, Gombert AK (2008) The yeast Kluyveromyces marxianus and its biotechnological potential. Appl Microbiol Biotechnol 79:339–354. doi:10.1007/s00253-008-1458-6
Ganatra MB, Vainauskas S, Hong JM, Taylor TE, Denson J-PM, Esposito D, Read JD, Schmeisser H, Zoon KC, Hartley JL, Taron CH (2011) A set of aspartyl protease-deficient strains for improved expression of heterologous proteins in Kluyveromyces lactis. FEMS Yeast Res 11:168–178. doi:10.1111/j.1567-1364.2010.00703.x
Guzman MG, Halstead SB, Artsob H, Buchy P, Farrar J, Gubler DJ, Hunsperger E, Kroeger A, Margolis HS, Martínez E, Nathan MB, Pelegrino JL, Simmons C, Yoksan S, Peeling RW (2010) Dengue: a continuing global threat. Nat Rev Microbiol 8:S7–S16. doi:10.1038/nrmicro2460
Idiris A, Tohda H, Kumagai H, Takegawa K (2010) Engineering of protein secretion in yeast: strategies and impact on protein production. Appl Microbiol Biotechnol 86:403–417. doi:10.1007/s00253-010-2447-0
Iwata T, Tanaka R, Suetsugu M, Ishibashi M, Tokunaga H, Kikuchi M, Tokunaga M (2004) Efficient secretion of human lysozyme from the yeast, Kluyveromyces lactis. Biotechnol Lett 26:1803–1808. doi:10.1007/s10529-004-4614-9
Jaiswal S, Khanna N, Swaminathan S (2004) High-level expression and one-step purification of recombinant dengue virus type 2 envelope domain III protein in Escherichia coli. Protein Expr Purif 33:80–91. doi:10.1016/j.pep.2003.09.009
Jungo C, Urfer J, Zocchi A, Marison I, von Stockar U (2007) Optimisation of culture conditions with respect to biotin requirement for the production of recombinant avidin in Pichia pastoris. J Biotechnol 127:703–715. doi:10.1016/j.jbiotec.2006.08.001
Kooistra R, Hooykaas PJJ, Steensma HY (2004) Efficient gene targeting in Kluyveromyces lactis. Yeast 21:781–792. doi:10.1002/yea.1131
Lane MM, Morrissey JP (2010) Kluyveromyces marxianus: a yeast emerging from its sister’s shadow. Fungal Biol Rev 24:17–26. doi:10.1016/j.fbr.2010.01.001
Li S, Shen W, Chen X, Shi G, Wang Z (2011) Secretory expression of Rhizopus oryzae -amylase in Kluyveromyces lactis. J Biotechnol 10:4190–4196
McBride WJH (2009) Evaluation of dengue NS1 test kits for the diagnosis of dengue fever. Diagn Microbiol Infect Dis 64:31–36. doi:10.1016/j.diagmicrobio.2009.01.002
Noisakran S, Dechtawewat T, Rinkaewkan P, Puttikhunt C, Kanjanahaluethai A, Kasinrerk W, Sittisombut N, Malasit P (2007) Characterization of dengue virus NS1 stably expressed in 293 T cell lines. J Virol Methods 142:67–80. doi:10.1016/j.jviromet.2007.01.008
Ramirez AH, Moros Z, Comach G, Zambrano J, Bravo L, Pinto B, Vielma S, Cardier J, Liprandi F (2009) Evaluation of dengue NS1 antigen detection tests with acute sera from patients infected with dengue virus in Venezuela. Diagn Microbiol Infect Dis 65:247–253
Ray MK, Devi KU, Kumar GS, Shivaji S (1992) Extracellular protease from the antarctic yeast Candida humicola. Appl Environ Microbiol 58:1918–1923
Rocha SN, Abrahão-Neto J, Cerdán ME, González-Siso MI, Gombert AK (2010) Heterologous expression of glucose oxidase in the yeast Kluyveromyces marxianus. Microb Cell Factories 9:4. doi:10.1186/1475-2859-9-4
Rocha SN, Abrahão-Neto J, Cerdán ME, Gombert AK, González-Siso MI (2011) Heterologous expression of a thermophilic esterase in Kluyveromyces yeasts. Appl Microbiol Biotechnol 89:375–385. doi:10.1007/s00253-010-2869-8
Rosa JC, Colombo LT, Alvim MC, Avonce N, Van Dijck P, Passos FM (2013) Metabolic engineering of Kluyveromyces lactis for L-ascorbic acid (vitamin C) biosynthesis. Microb Cell Factories 12:59. doi:10.1186/1475-2859-12-59
Rouwenhorst RJ, Visser LE, Van Der Baan AA, Scheffers WA, Van Dijken JP (1988) Production, distribution, and kinetic properties of inulinase in continuous cultures of Kluyveromyces marxianus CBS 6556. Appl Environ Microbiol 54:1131–1137
Sanchez M, Iglesias FJ, Santamaria C, Dominguezl A (1993) Transformation of Kluyveromyces lactis by electroporation. Appl Environ Microbiol 59:2087–2092
Saxena P, Tripathi NK, Rao PVL, Jana AM (2008) Heterologous expression of envelope protein (domain III) of dengue virus type 2 for serodiagnosis. 16–20
Silveira WB, Passos FJV, Mantovani HC, Passos FML (2005) Ethanol production from cheese whey permeate by Kluyveromyces marxianus UFV-3: a flux analysis of oxido-reductive metabolism as a function of lactose concentration and oxygen levels. Enzym Microb Technol 36:930–936. doi:10.1016/j.enzmictec.2005.01.018
Singh MP, Goyal K, Ratho RK (2010) Nonstructural protein NS1: giving a new structure to dengue diagnosis. J Clin Microbiol 48:4688. doi:10.1128/JCM.01668-10, author reply 4688–9
Sugrue RJ, Cui T, Xu Q, Fu J, Chan YC (1997) The production of recombinant dengue virus E protein using Escherichia coli and Pichia pastoris. J Virol Methods 69:159–169
Swaminathan S, Khanna N (2009) Dengue: recent advances in biology and current status of translational research. Curr Mol Med 9:152–173
Van Ooyen A, Dekker P, Huang M (2006) Heterologous protein production in the yeast Kluyveromyces lactis. FEMS Yeast 6:381–392. doi:10.1111/j.1567-1364.2006.00049.x
Wei H, Jiang L, Xue Y, Fang D, Guo H (2003) Secreted expression of dengue virus type 2 full-length envelope glycoprotein in Pichia pastoris. J Virol Methods 109:17–23. doi:10.1016/S0166-0934(03)00039-9
Zhao BT, Prince G, Horswood R, Eckels K, Summers P, Chanock R, Lai CJ (1987) Expression of dengue virus structural proteins and nonstructural protein NS1 by a recombinant vaccinia virus. J Virol 61:4019–4022
Zhou J, Tang Y, Fang D, Zhou J, Liang Y, Guo H, Jiang L (2006) Secreted expression and purification of dengue 2 virus full-length nonstructural glycoprotein NS1 in Pichia pastoris. Virus Genes 33:27–32. doi:10.1007/s11262-005-0036-6
Acknowledgments
We thank the Brazilian Agency CNPq (National Science and Technology Development Council) for the scholarship and CAPES (Coordination for the Improvement of Higher Education Personnel) and FAPEMIG (Foundation for Research Support of the State of Minas Gerais) for the financial support.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Bragança, C.R.S., Colombo, L.T., Roberti, A.S. et al. Construction of recombinant Kluyveromyces marxianus UFV-3 to express dengue virus type 1 nonstructural protein 1 (NS1). Appl Microbiol Biotechnol 99, 1191–1203 (2015). https://doi.org/10.1007/s00253-014-5963-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-5963-5