
Abstract
We have characterised the northern Pacific undescribed sponge Haliclona (?gellius) sp. based on rDNA of the sponge and its associated microorganisms. The sponge is closely related to Amphimedon queenslandica from the Great Barrier Reef as the near-complete 18S rDNA sequences of both sponges were identical. The microbial fingerprint of three specimens harvested at different times and of a transplanted specimen was compared to identify stably associated microorganisms. Most bacterial phyla were detected in each sample, but only a few bacterial species were determined to be stably associated with the sponge. A sponge-specific β- and γ-Proteobacterium were abundant clones and both of them were present in three of the four specimens analysed. In addition, a Planctomycete and a Crenarchaea were detected in all sponge individuals. Both were closely related to operational taxonomic units that have been found in other sponges, but not exclusively in sponges. Interestingly, also a number of clones that are closely related to intracellular symbionts from insects and amoeba were detected.
Similar content being viewed by others

Introduction
Marine sponges have proven to be a rich reservoir for bioactive metabolites (e.g. [8, 47]). An early, conservative estimate based upon thousands of assayed sponge species suggested that as many as 11% produce cytotoxic compounds [10]. This percentage is high compared to other organisms and possibly may result from the intimate association between sponges and diverse microbial communities. Marine sponges provide a home to species from most bacterial phyla [15, 54], but in most cases it is unclear whether the cytotoxic compounds are produced by the sponges or by their associated bacteria. The issue is difficult to resolve, as neither sponges nor their associated bacteria can be readily cultured in the laboratory [2, 11, 15, 39] and to produce secondary metabolites from sponges it is necessary to identify their source. In a few instances, the producer of the secondary metabolite has been identified. For example, the sequiterpenoid hydroquinone avarol, isolated from the marine sponge Dysidea avara, was localised inside choanocytes of the sponge [57], while the cytotoxic macrolide swinholide isolated from the sponge Theonella swinhoei was produced by unicellular bacteria inhabiting the sponge [3]. Other important sponge-derived secondary metabolites, such as polyketides, are generally associated with bacterial metabolism [42], but their putative production by bacterial sponge symbionts can only be confirmed if a polyketide-producing strain is found in stable association with the sponge.
The current paradigm is that certain bacterial clades are found only in marine sponges [14] and should be regarded as an integral part of the sponge. Sponges that harbour microorganisms of sponge-specific clades are generally high-microbial-density sponges, whereas low-microbial-density sponges are thought to have less specific associated microorganisms [16]. The existence of sponge-specific clades suggests a certain dependency of these bacteria on the sponge (e.g. providing an appropriate growth environment). It has been documented that sponges provide a habitat that is richer in nutrients than seawater and sediments (reviewed in [29]) and could prevent the inhabitants from drifting away to less favourable environments. While this may be the case, it does not explain species specificity of the symbionts that reside in marine sponges. High levels of specificity suggest that the interaction is more likely to be based on metabolite exchange. Such relationships were predicted by Reiswig [41] and a number of examples support this hypothesis, including the translocation of glucose from symbiotic green algae to their sponge host [65] and translocation of nitrogen from bacteria to the sponge [63]. Of many other bacterial metabolic processes that take place in marine sponges, it is yet unclear whether they are part of a commensal or a mutualistic relationship, such as sulphate reduction [18], methane oxidation [59] and putative ammonia oxidation by symbiotic crenarchaeota [12].
The hypothesis that bacteria from certain clades are only found in marine sponges does not imply that they need to be present in every individual of a certain sponge species. To date, studies of the intraspecific variation of sponge-associated bacteria have precluded generalisation. While the sponge species Chondrilla nucula [56], Ircinia felix, Aplysina cauliformis, Niphates erecta [63] and Cymbastela concentrica [53] have low intraspecific bacterial variation, the sponge species Tethya aurantium [55], Halichondria panicea [1, 64] and Ircinia felix larvae [44] exhibit considerable intraspecific variation.
In the present study, we have analysed the sponge–microbe consortium of Haliclona (?gellius) sp. from western North America, a region that has been characterised as an underrepresented location with regard to 16S rRNA gene libraries from sponges [54]. It is our hypothesis that if certain associated microbes are present in the sponge at different times of the year and if they persist in the sponge when it is translocated to a different environment, they are potentially stables associates of the sponge.
Materials and Methods
Sample Collection
Sponges of the genus Haliclona (Fig. 1a) were collected near the Monterey harbour at a depth of approximately 12 m on April 15, 2006 (Hg1 and Hg9), May 24, 2006 (Hg5), January 23, 2007 (Hg6) and October 15, 2007 (for electron microscopy). The sponges were rinsed three times with sterile artificial seawater (natural sea salt mix, Oceanic Systems, Dallas, TX, USA) before grinding the tissue with a sterilised mortar and pestle. Two tissue volumes of sterile artificial seawater (ASW) were used to obtain a homogeneous cell suspension. The cell suspension was divided in aliquots of 1.2 ml and mixed with 0.6 ml 50% glycerol in ASW. The samples were gently frozen until −20°C before they were stored at −80°C. One specimen (Hg9) was maintained in an aquarium with 0.22 µm filtered natural seawater at 10°C for 1 month before macerating the tissue. The identity of the specimens was confirmed by 18S rRNA gene analysis.

Haliclona (?gellius) sp. in the sea (a) and in the lab (b). Formation of new tissue at the tip of an extension is marked with the white arrow. Amphimedon queenslandica (c) is closely related to Haliclona (?gellius) based on 18S rDNA comparison [A. queenslandica picture courtesy of John Hooper]
DNA Library Construction
Genomic DNA was extracted from cryopreserved cells from four specimens (Hg1, Hg5, Hg6 and Hg9) using the Puregene DNA isolation kit (Gentra, Minneapolis, MN, USA). Cryopreserved cell suspension (400 µl) was used for DNA isolation. The cell suspension was centrifuged at 17,900 ×g and the supernatant was decanted. DNA was isolated according to manufacturer’s instructions.
PCR was performed with several primer sets (Table 1) according to the following profile: initial denaturation for 10 min at 95°C; ten cycles of denaturation for 30 s at 95°C, annealing for 30 s at 58°C (touch down to 49°C) and elongation for 1 min 45 s at 72°C; 25 cycles of denaturation for 30 s at 95°C, annealing for 30 s at 55°C and elongation for 1 min 45 s at 72°C; and a final extension step for 10 min at 72°C.
PCR products of successful reactions were cloned with a TOPO TA cloning kit (Invitrogen, Carlsbad, CA, USA) in accordance with the manufacturer’s instructions. Of the bacterial 16S products that were obtained with the primer set 8F–1492R, a total of 837 colonies were picked, cultured and plasmids purified (Hg1/276 colonies; Hg5/184; Hg6/193; Hg9/184). A total of 559 clones with an archaeal 16S PCR product (primers 21F–958R) were picked, cultured and plasmids purified (Hg1/192; Hg5/192; Hg6/96; Hg9/79). Twenty clones containing a eukaryotic 18S rRNA gene segment were picked, cultured and plasmids purified. For Hg1 (four clones), Hg5 (four) and Hg9 (four), the EUKF–EUKR primer set was used, while for Hg6 (eight) only the 82FE–690RE primer set was successful.
Plasmids containing a PCR product were sequenced using the primers that were used for the PCR. Sequence chromatograms were manually inspected and assembled using the Contig Express software (Invitrogen). Sequences with similarities >99% were considered as one operational taxonomic unit (OTU) [49]. In addition, they were checked for possible chimeric origins by using the program Bellerophon (version 3) from the Greengenes website [22]. Sequences were deposited in the NCBI Genbank under accession numbers: EU095523 (18S rRNA gene H. (?gellius) sp.), EU236274–EU236437 and EU817104–EU817119 (16S rRNA bacteria) and EU251478–EU251483 (16S rRNA archaea).
Phylogenetic Analysis
The DNA sequences of the OTUs and nearest neighbours (as determined using the Greengenes database http://greengenes.lbl.gov) were imported in the ARB software package [32]. DNA sequences were aligned using the FastAlign function of the alignment editor implemented in the ARB program and refined manually. Ambiguous regions of the alignment were systematically removed using the program Gblocks v.0.91b [4]. The default program parameters were used, exclusive of allowing a minimum block length of 5 and gaps in 50% of positions. Bayesian (MB), maximum likelihood (ML) and maximum parsimony (MP) phylogenetic methods were employed to estimate phylogenetic relationships in the data sets. MP trees were generated with PAUP* [52]. Heuristic searches consisted of 1,000 random step-wise-addition-sequence replicates using TBR branch swapping. ML trees were calculated with the GTR model and estimated proportion of invariable sites and gamma distribution in GARLI v0.951 [66]. Runs were automatically terminated when tree topology did not change for 10,000 generations. Five separate runs were performed and the tree with the best likelihood was selected. Bayesian analyses were performed using MrBayes v3.0b4 [23]. All parameters were treated as unknown variables with uniform prior-probability densities at the beginning of each run and their values were estimated from the data during the analysis [43]. All Bayesian analyses were initiated with random starting trees and were run for 1 × 107 generations.
Rarefaction Analysis
The extent of diversity examined in the clone libraries was determined by rarefaction analysis. Rarefaction curves were obtained using the algorithm described by Hurlbert [24] with the freeware program aRarefactWin (http://www.uga.edu/~strata/software.html). Rarefaction curves were plotted and regressions performed using two different regression equations [55]:
where x is the sample size, y the observed number of OTUs, a the expected number of OTUs with infinite sample size and b and c are regression variables.
Transmission Electron Microscopy
A fresh sponge was cut into fragments of 1 mm3. The fragments were prepared for transmission electron microscopy (TEM) by overnight fixation with 2.5% glutaraldehyde in autoclaved natural seawater. After rinsing the sample 3 × 15 min with autoclaved natural seawater, the sample was postfixed in autoclaved natural seawater containing 1% osmium tetroxide for 2 h. After rinsing 3 × 15 min with autoclaved natural seawater and 3 × 10 min with distilled water the sample was incubated overnight in 0.5% uracyl acetate in the dark at 4°C. After the overnight incubation, the sample was rinsed 3 × 15 min with distilled water and dehydrated in an acetone series (35, 50, 70, 80, 95, 100 v/v). The dehydrated sample was infiltrated with resin (epon–araldite mixture) by 1 h incubation steps with 2:1, 1:1 and 1:2 proportions of acetone:resin. After overnight incubation in pure resin, the resin was polymerised in moulds and the sample was incubated at 60°C for 48 h. The sample was sectioned by using a microtome (RMC MTX, Reichert Ultracut E, RMC MT6000) with a diamond blade and the tissue was observed with a FEI Tecnai 12 transmission electron microscope.
Results
Sponge
Based upon outer appearance, spicule size and shape (oxeas 147–178–198 µm; sigmas 29–36– 41 µm C shaped), colour (beige with purple tinge to rose–lavender) and habitat, the sponge species was identified as Haliclona (?gellius) sp. (personal communication R.W.M. van Soest and W.L. Lee; [7]). The species was first named Sigmadocia sp. [13] and further analysed by Lee et al. [30]. A characteristic that was never observed in the sea, but was observed every time when individuals of H. (?gellius) sp. were transplanted to an aquarium in the laboratory, was the formation of finger-shaped extensions (Fig. 1b) and the formation of new sponge tissue at the tips of some extensions. The process resembles budding although the extensions never had a well-defined shape at the tips.
18S rDNA data were used to determine the phylogenetic position of H. (?gellius) sp. in the phylogeny of the order Haplosclerida recently published by Redmond and colleagues (2007). The maximum likelihood phylogram obtained after including H. (?gellius) sp. and its nearest neighbour, Amphimedon queenslandica (which was identical to H. (?gellius) sp.), was highly similar to the ML tree published by Redmond et al. [40]. Both species branch deeply in the order Haplosclerida in a clade with Xestospongia muta, Oceanapia sp. and Dasychalina fragilis while they are only distantly related to the other Haliclona spp. (Fig. 2). A slightly different topology from Redmond et al. was obtained due to repositioning of D. fragilis and Oceanapia sp. in a separate clade (posterior probability 97%, maximum parsimony bootstrap 68%) with the two new species in the tree and X. muta. Phylogenetic tree topologies generated with parsimony, maximum likelihood and a Bayesian model were highly similar (data not shown).

Maximum likelihood phylogram of 18S rRNA gene sequences. The numbers above the branches correspond to posterior probability (PP) values of the Bayesian analysis. The numbers shown below the branches are MP bootstrap values. Nodes with PP or bootstrap values of <70% are indicated with an ‘–’. Some PP and bootstrap values were not included due to clarity (only those not relevant to the manuscript). The species in the tree are based on [40] with the addition of Amphimedon queenslandica (EF654521) and Haliclona (?gellius) sp. (EU095523)
Bacteria
Bacteria were determined to reside primarily extracellularly in the mesohyl (Fig. 3) in moderate numbers. A total of 796 high-quality clone sequences were obtained and, based on the rule of thumb of Stackebrandt and Ebers [49] that sequences that share <99% similarity are considered to be derived from different operational taxonomic units (OTUs), these represented 170 OTUs. Rarefaction curves showed that actual bacterial diversity is probably well over 250 different OTUs for H. (?gellius) sp. (Fig. 4). Regression model 2 gave a slightly better fit, which is to be expected when a three-parameter model is compared to a two-parameter model. Coverage of the bacterial diversity is estimated between 37% and 63%, for regression models 2 and 1, respectively (Table 2). Coverage of bacterial diversity of individual specimens was generally higher than for the combined sponge samples. Despite average coverage of the samples of approximately 55%, the high number of analysed clones implies that the part of bacterial diversity that was not detected probably represents a minor quantitative fraction in the sponge. Clone libraries for sponge individuals typically contained 17–36 OTUs, but 101 OTUs were found in Hg9, which accounted for more than 50% of the total diversity that was observed.

Transmission electron micrograph of a section of H. (?gellius) sp. Bacteria (A) are embedded in the mesohyl outside of sponge cells (B). The dark fragment (C) is part of a fragmented spicule. The diameter of cell B is approximately 9 µm

Example of a rarefaction curve plotted for the total bacterial diversity that was discovered in the four Haliclona (?gellius) sp. specimens. The expected number of OTUs as determined by Hurlbert’s algorithm [24] is plotted against the number of analysed 16S clones (black diamonds). Extrapolated regression curves for equations 1 and 2 are depicted as the light and dark solid line, respectively
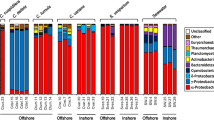
Representatives of most bacterial phyla including α-, β-, γ-, δ- and ε-Proteobacteria, Cyanobacteria, Planctomycetes, Firmicutes, Acidobacteria, Actinobacteria, Bacteroidetes, Chloroflexi, Fusobacteria, Nitrospirae, Verrucomicrobia and candidate division TM6 were present in H. (?gellius) sp. (Fig. 5). γ-Proteobacteria (54.8%) formed the dominant group based on PCR product frequency, followed by Firmicutes (12,1%), Planctomycetes (9.4%), β-Proteobacteria (7.3%), α-Proteobacteria (6.9%) and Bacteroidetes (3.0%). For each sponge individual, the γ-Proteobacteria dominated the cloned 16S PCR products. α-, β-, δ-Proteobacteria and a significant proportion of Planctomycetes were also present in all individuals. Acidobacteria, Actinobacteria, Chloroflexi, ε-Proteobacteria, Fusobacteria, Nitrospirae and Verrucomicrobia-derived sequences were observed in only one of the individuals (this was also the case for Stramenopile and Viridiplantae PCR products). Similarities and differences between individuals on the OTU-level are summarised in Fig. 6. Only OTU 2 was present in all individuals and a BLAST search showed it is closely related to an uncultured Pirellula species (Planctomycetes) from the seawater close to a coral reef at the Palmyra Atoll. OTUs 1, 24 and 26 were observed in three of four individuals and represent different β- and γ-Proteobacteria. Seven OTUs were found in half of the individuals and each individual contained approximately 15–25 singles (OTUs not found in any of the other individuals). Hg9 was an outlier in that respect with 95 singles.

Distribution of 16S PCR products over the bacterial phyla (including Stramenopiles and Viridiplantae) for sponge individuals Hg1 (256 clones), Hg5 (177), Hg6 (188), Hg9 (175) and the total (796). The phyla as listed in the legend are depicted clockwise starting at 12 ‘o clock. The first listed phyla, Acidobacteria and Actinobacteria, are only present in Hg9 and Hg ALL. α-Proteobacteria is the first phylum that is present in all individuals. Hg6 is least diverse and only yielded α-, β-, γ- and δ-Proteobacteria, candidate division TM6 and Planctomycetes related clones

OTUs found in H. (?gellius) sp. individuals. Each number represents an OTU that was observed in more than one individual. Their position on the map shows which sponge individuals harboured the OTUs. OTUs that were found in only one individual are included in the category ‘singles’
In order to obtain more detailed insight in the bacterial populations associated with H. (?gellius) sp., phylogenetic trees of these bacteria and their nearest neighbours were constructed (Figs. 7, 8, 9 and 10). In the α-Proteobacteria tree, two Roseobacter-affiliated OTUs were detected in both Hg5 and Hg9 (OTUHg15 and OTUHg19), while 38 other OTUs were found in only one of the sponge individuals (Fig. 7).

Bayesian phylogram of α-Proteobacteria 16S rRNA gene sequences from H. (?gellius) sp. bacteria and their nearest neighbours (Greengenes database May 31, 2007). The numbers above or below the branches correspond to posterior probability (PP) values of the Bayesian analysis. OTUs that were found in more than one individual start with the letters ‘OTU’, OTUs that were found in only one individual are encoded as a combination of the name of the individual (e.g. Hg9) + the clone name. The grey boxes indicate OTUs with invertebrate-derived neighbours. Thermocrinis sp. and Sulfurihydrogenibium azorense were used as outgroup. If H. (?gellius) sp. OTUs were more closely related to each other than to any other sequence, only one of the OTUs was used to construct the tree


a, b Bayesian phylogram of β- and γ-Proteobacteria 16S rRNA gene sequences from H. (?gellius) sp. bacteria and their nearest neighbours (for details, see Fig. 7)

Bayesian phylogram of Bacteroidetes 16S rRNA gene sequences from H. (?gellius) sp. bacteria and their nearest neighbours (for details, see Fig. 7)


a, b Bayesian phylogram of 16S rRNA gene sequences from H. (?gellius) sp. and their nearest neighbours belonging to other phyla (for details, see Fig. 7)
Two β-Proteobacteria-affiliated OTUs, OTUHg1 and OTUHg24, were found in three (of four) individuals. Including the singles, a total of ten β-Proteobacteria OTUs were detected in H. (?gellius) specimens (Fig. 8b). The nearest neighbour of OTUHg24 is a clone from the marine sponge Tethya aurantium, but it is only distantly related (identity = 83%). OTUHg1 was found in Hg1, Hg5 and Hg9. The nearest neighbours are a Cupriavidus species isolated from cystic fibrosis patients and a DNA fragment detected on old Antarctic ice.
One γ-Proteobacteria OTU, OTU26, was found in three individuals and represented the most abundant sequence in the clone library with more than 30% of the clones. The nearest neighbours are bacteria from other marine sponges: Axinella verrucosa, Chondrilla nucula and Tethya aurantium (Fig. 8a). Three other γ-Proteobacteria-related OTUs were found in two individuals, while 43 OTUs were singles.
Twenty-three OTUs grouped within the Bacteroidetes (Fig. 9). All of them were singles detected in Hg9, except for one Bacteroidetes-affiliated clone in Hg1 and in Hg5.
In the other phyla, three OTUs were found in more than one individual (Fig. 10). In addition to the Planctomycetes species that was found in all four individuals (OTUHg2), another Planctomycete species (OTUHg29) was found in Hg5 and Hg6. Moreover, 16 single Planctomycete-related OTUs were found (Fig. 10b). The Cyanobacteria-affiliated OTUHg9 was detected in both Hg1 and Hg5 and has a Synechococcus species isolated from Californian seawater as its nearest neighbour (Fig. 10a). All OTUs that were discovered in other phyla were singles. Seven δ-Proteobacteria OTUs were discovered and each sponge individual harboured different δ-Proteobacteria. In most individuals (not in Hg1), members of candidate division TM6 were found. All but one Firmicutes OTU was found in Hg1. They represented a large proportion of the clones from this individual (37%) but were negligible in the other individuals. A similar situation was observed for Nitrospirae-related clones in Hg9, although not to the same extent as they represented 4% of the Hg9 clones. Only one or two clones belonging to Verrucomicrobia, Actinobacteria, Chloroflexi, ε-Proteobacteria and Fusobacteria were detected.
Archaea
Five archaeal OTUs were found in H. (?gellius) sp. (Fig. 11). One of them was found in all individuals (OTUHgAr2) and belongs to the C1a-α group. Its closest relative is an archaeal clone from the marine sponge Astrosclera willeyana. The other OTUs, also belonging to the C1a-α group, were only detected in Hg5 or Hg6.

Bayesian phylogram of archaea 16S rRNA gene sequences from H. (?gellius) sp. and their near neighbours. The goldmine clones SAGMA-A and SAGMA-8 were used as an outgroup (for details, see Fig. 7)
Discussion
Sponge
Haliclona (?gellius) sp. has been called an uncommon species by Hartman [13], but that may be because it generally resides in cold water under rocks and is not easily detectable. In addition, the species has not been studied in subsequent scientific publications except as part of systematic surveys [7, 30].
This is a sharp contrast to Amphimedon queenslandica, which is being developed as a model experimental system and is subject of a genome project (Joint Genome Institute). A. queenslandica is only found in the warm waters around the Great Barrier Reef [21]. Despite these differences and their superficially different appearance (compare Fig. 1a to 1c), the A. queenslandica 18S rRNA gene sequence is identical to the near-complete sequence of H. (?gellius) sp. This is of particular interest, because of its similarity on the 18S level to A. queenslandica, H. (?gellius) sp. may present easily accessible genetic targets. Some preliminary results suggest that EST sequences from H. (?gellius) sp. of randomly chosen protein coding sequences share high identity at the nucleotide level with A. queenslandica (unpublished data).
Bacteria
The bacterial diversity in H. (?gellius) is larger than what has been observed for most other sponge species investigated. One hundred and seventy OTUs (within 796 clone sequences) were found, while in many other 16S rRNA clone-based studies, typically 20–40 OTUs are discerned (Table 3). However, this difference may be attributed to the number of clones analysed in most studies, because if more clones were analysed, as in the cases for Ircinia felix and a Corticium sp., approximately 100 OTUs were obtained [44, 46]. We cannot compare coverage of bacterial diversity between the sponges, because not for all species rarefaction data have been published, but the number of OTUs/clone can be used as an indicator for sampling density. A high number of OTUs/clone implies that the number of OTUs that will be discovered by increasing the sample size will be relatively high. It is shown that, except for K. varialosa and C. australiensis, which have comparative values to H. (?gellius) sp., all other sponge species have a considerable lower sampling density than H. (?gellius) sp. At the same time, we have reasons to believe that the numbers of detected and expected OTUs are overestimated for H. (?gellius) sp. The 99% shared identity standard to distinguish between OTUs [49] requires completely unambiguous sequences, which is not the case for most DNA fragments in our analysis. This will lead to a conservative estimation of overlap between sponge specimens while overestimating diversity. For the expected number of OTUs, as determined by rarefaction analysis, it has been noted that if many OTUs of low abundance are present in the data set (as is the case for H. (?gellius) sp.) the rarefaction curve tends to converge to a line without an apparent limit [55], which limits the usefulness of rarefaction analysis to obtain a reliable estimate of expected diversity. The latter issue is especially true for Hg9 (and therefore also Hg all) with 95 clone sequences that were obtained only once at a total of 175 clones. The experimental difference between Hg9 and the other specimens was that Hg9 was kept in an aquarium for 1 month before DNA was extracted and microorganisms present in the aquarium may have invaded the sponge.
A part of the bacterial diversity detected can probably be attributed to the environment rather than to the sponge due to the presence of bacteria that serve as food, sediment particles and ambient seawater in the sponge tissue. Although tissue with visible particles was discarded before DNA extraction and sponge tissue was rinsed three times with artificial seawater, it is impossible to remove all environmental DNA. When sponge-derived clones are compared to their nearest neighbours in a phylogeny (Figs. 7, 8, 9 and 10), three categories of neighbours can be discerned: invertebrate-derived clones, seawater clones and sediment clones. Twenty-five percent of the OTUs from Hg9 were related to sediment clones, while for the other individuals only 9.5 ± 0.8% (data not shown) had sediment-derived nearest neighbours. It is possible that the water circulation in the aquarium whirled the sediment in the aquarium and thus Hg9 was colonised with a large number of sediment bacteria (the sample looked ‘dirty’ in the aquarium). This could explain a large part of the differences found between Hg9 and the other individuals. In addition, we compared the sponge-derived sequences with 94 16S rDNA sequences that were obtained from Monterey Bay bacterioplankton samples in other studies (data not shown; e.g. [50, 51]). This analysis revealed that three H. (?gellius) sp.-derived OTUs (Hg92F12, Hg92F3 and Hg5a2D8) were closely related to Monterey Bay bacterioplankton samples (Figs. 7, 9 and 10a, respectively).
Most OTUs had high sequence similarity with bacterial clones from different environments and do not belong to sponge-specific clades that were proposed by Hentschel et al. [14]. However, this does not exclude the possibility that they are stably associated with the sponge. For example, many clones of OTUHg2 were derived from all H. (?gellius) sp. individuals and was found in bacterioplankton, but also in the marine sponge Tethya aurantium (AM259827). Nevertheless, based on our results, we cannot make a firm conclusion for OTUs with neighbours from different groups, because we cannot distinguish horizontally acquired associated bacteria from ‘contaminants’ from the seawater. In addition, other sponge-derived sequences, such as the one from T. aurantium, may also have been the result of a seawater ‘contamination’. However, the fact that OTUHg2 persisted in Hg9 when it was kept in an aquarium with filtered seawater for 1 month suggests it could be a stable associate of the sponge.
A number of α-, β- and γ-Proteobacteria OTUs (Figs. 7 and 8) have only invertebrate-derived neighbours (checked with BLAST search; Jan 27, 2008). The α-Proteobacteria from H. (?gellius) sp. with invertebrate neighbours will not be considered, because they are all singles and it is therefore unlikely that they represent important partners for the sponge. A few clones may be worth mentioning because they have intracellular symbionts as neighbours. The α-Proteobacterium Hg5a2C10 and γ-Proteobacteria Hg1bE8 and Hg5a2E8 have intracellular symbionts from one amoeba sp. and a variety of insects as nearest neighbours. Intracellular bacteria have been observed for a number of sponges [25, 58, 60]), but there are currently no 16S rRNA gene sequences marked as ‘derived from intracellular bacteria’ in the NCBI database.
The γ-Proteobacterium OTUHg26 and β-Proteobacterium OTUHg24 that were both detected in three of the four H. (?gellius) sp. specimens belong to sponge-specific clades. Nearest neighbours of OTUHg26, the most abundant clone sequence in the library, were all derived from the Mediterranean and Adriatic sponges Axinella verrucosa, Chondrilla nucula and Tethya aurantium and belong to the sponge-specific SAB-Gamma-III cluster [56]. Maximum identity is only 92%, which indicates that it is only a distantly related species. BLAST search revealed that OTUHg26 has 90% identity with Thiohalomonas denitrificans (EF117911.1), which is a halophilic chemolithoautotrophic bacterium that utilises sulphide and thiosulphate as electron donor for the reduction of nitrate to N2 [48]. A sponge-specific symbiotic sulphur cycle with sulphate reducers and -oxidisers was hypothesised to exist in the marine sponge Polymastia cf. corticata [35] and thus OTUHg26 could be responsible for the oxidation of sulphide in the sponge tissue. The chimera-check earmarked OTUHg26 (and also Hg62F6 and Hg63D7) as putative chimeras, but OTUHg26 was obtained from different samples, which suggests that the sequences are not likely to be chimeras.
The β-Proteobacterium OTUHg24 has no near relatives, but it is most similar to clones obtained from Tethya aurantium (AM259836.1, 83% identity), which are part of a sponge-specific cluster with sequences from globally distributed sponges [55]. The lack of similarity to other sequences makes it difficult to speculate about the role of the bacteria in the sponge tissue.
The lack of overlap of Bacteroidetes-affiliated OTUs between H. (?gellius) sp. specimens and the absence of sponge-specific OTUs confirms the idea that Bacteroidetes are not specific partners for sponges [54]. Bacteroidetes were found to represent a large part of the necrotic tissue of the sponge Rhopaloeides odorabile after it was exposed for 24 h to elevated temperatures, while they were nearly absent in control specimens [62]. If we had based our analysis on DGGE bands, we would probably not have detected Bacteroidetes because they would have remained below the detection limit. The presence of low numbers of Bacteroidetes in the tissue of the sponge could be the inoculum for rapid proliferation when conditions change.
Cytotoxicity tests of a tissue extract of H. (?gellius) sp. showed that it was void of cytotoxic metabolites. No activity of a methanol extract was found against cancer cells and only a slight antimicrobial activity at 64 µg/ml, the highest concentration tested (unpublished data, W. Fenical). This finding corresponds with the absence of consistent populations of Actinobacteria and Chloroflexi (only two and one clones, respectively, and only in Hg9) and the low frequency of Cyanobacteria (two OTUs and only present in Hg1 and Hg5) that are generally associated with the production of bioactive compounds [26, 38]. It could be speculated that the preferred habitat of the sponge (i.e. under rocks) is shielded from predators and direct light, and therefore it does not rely on chemical protection. When the bacterial composition of H. (?gellius) sp. is compared to the symbionts of the multi-cytotoxin-producing high-bacterial-density sponge Aplysina aerophoba, it can be noted that the compositions are remarkably different. In A. aerophoba, the Acidobacteria, Actinobacteria and Chloroflexi together make up more than 50% of the 16S rRNA gene sequences [16], while for H. (?gellius) sp. they represented less than 1% of the clone sequences. Despite the presence of regions with increased concentrations of bacteria, H. (?gellius) sp. also has regions that are virtually bacteria-free. This is a sharp contrast to the high-bacterial-density sponge A. aerophoba and H. (?gellius) sp. is more likely to belong to the low-bacterial-density sponges based on TEM micrographs. This corresponds with the theory that in general low-bacterial-density sponges harbour fewer sponge-specific associated bacteria.
Archaea
The only sponge-specific archaea clade, C1a-Porifera, comprises sequences derived from the genus Axinella [33]. All other sponge-derived archaea belong to the C1a-α group, but members are not restricted to sponges [20] and the archaea-affiliated sequences from H. (?gellius) sp. make part of this group. The nearest neighbour of OTUHgAr2, which was obtained from all four individuals, is a sequence derived from the marine sponge Astrosclera willeyana. This OTU is potentially a symbiotic archaea of H. (?gellius) sp., but sequence differences on the 16S level are small for archaea [45], which is confirmed by lower posterior probability values when compared to the bacterial phylograms. Therefore, conclusions solely based on 16S phylogeny should be regarded carefully.
Conclusion
The cold Pacific sponge Haliclona (?gellius) sp. from Northern California is closely related to the Great Barrier Reef sponge Amphimedon queenslandica based on 18S rDNA. It harbours bacteria from many bacterial phyla, but only a few are likely to have a stable association with the sponge. Sponge-specific sequences of a β- and a γ-Proteobacterium were abundant and most abundant clones in the clone library, respectively. Both of them were present in three of the four specimens analysed. This could imply that H. (?gellius) sp. is the restricted environment for growth of these bacteria, but that the sponge is able to proliferate without them. In addition, two OTUs were detected in all specimens: a Planctomycete- and a Crenarchaea-affiliated microbe. Both were closely related to OTUs that have been found in other sponges, but not exclusively in sponges. The fact that these microbes were also present in Hg9, which was maintained in an aquarium with filtered seawater for 1 month, gives rise to the inclination to include them with the stably associated microorganisms of H. (?gellius) sp. Based on TEM micrographs, the sponge is most likely a low-bacterial-density sponge.
References
Althoff K, Schütt C, Steffen R, Batel R, Müller WEG (1998) Evidence for a symbiosis between bacteria of the genus Rhodobacter and the marine sponge Halichondria panicea: harbour also for putatively toxic bacteria? Mar Biol 130:529–536
Becerro MA (2008) Quantitative trends in sponge ecology research. Mar Ecol 29:167–177
Bewley CA, Holland ND, Faulkner DJ (1996) Two classes of metabolites from Theonella swinhoei are localized in distinct populations of bacterial symbionts. Experientia 52:716–722
Castresana J (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17:540–552
Dawson SC, Pace NR (2002) Novel kingdom-level eukaryotic diversity in anoxic environments. Proc Natl Acad Sci 99:8324–8329
DeLong E (1992) Archaea in coastal marine environments. Proc Natl Acad Sci 89:5685–5689
De Weerdt WH (2002) Family Chalinidae Gray, 1867. In: Hooper JNA, Van Soest RWM (eds) Systema Porifera. A guide to the classification of sponges. Kluwer/Pendulum, New York, pp 859–862
Faulkner DJ (2002) Marine natural products. Nat Prod Rep 19:1–48
Felske A, Rheims H, Woltherink A, Stackebrandt E, Akkermans ADL (1997) Ribosome analysis reveals prominent activity of an uncultured member of the class Actinobacteria in grassland soils. Microbiol 143:2983–2989
Garson MJ (1994) The biosynthesis of sponge secondary metabolites: why it is important. In: Van Soest RWM, Van Kempen TMG, Braekman JC (eds) Sponges in time and space. Balkema, Rotterdam, pp 427–440
Grozdanov L, Hentschel U (2007) An environmental genomics perspective on the diversity and function of marine sponge-associated microbiota. Curr Opin Microbiol 10:215–220
Hallam SJ, Mincer TJ, Schleper C, Preston CM, Roberts K, Richardson PM, DeLong EF (2006) Pathways of carbon assimilation and ammonia oxidation suggested by environmental genomic analysis of marine Crenarchaeota. PLoS Biol 4:520–536
Hartman WD (1975) Phylum Porifera. In: Smith RI, Carlton JT (eds) Light’s manual: intertidal invertebrates of the Central California coast. University of California Press, Berkeley, pp 32–64
Hentschel U, Hopke J, Horn M, Friedrich AB, Wagner M, Hacker J, Moore BS (2002) Molecular evidence for a uniform microbial community in sponges from different oceans. Appl Environ Microbiol 68:4431–4440
Hentschel U, Fieseler L, Wehrl M, Gernert C, Steinert M, Hacker J, Horn M (2003) Microbial diversity of marine sponges. In: Müller WEG (ed) Marine Molecular Biotechnology. Springer, Berlin, pp 59–88
Hentschel U, Usher KM, Taylor MW (2006) Marine sponges as microbial fermentors. FEMS Microbiol Ecol 55:167–177
Hill M, Hill A, Lopez N, Harriott O (2006) Sponge-specific bacterial symbionts in the Caribbean sponge, Chondrila nucula (Demospongia, Chondrosida). Mar Biol 148:1221–1230
Hoffmann F, Larsen O, Thiel V, Rapp HT, Pape T, Michaelis W, Reitner J (2005) An anaerobic world in sponges. Geomicrobiol J 22:1–10
Holmes BM (2006) Marine sponges: systematics, symbiosis and cell culture. PhD thesis, University of California Berkeley
Holmes B, Blanch H (2007) Genus-specific associations of marine sponges with group I crenarchaeotes. Mar Biol 150:759–772
Hooper JNA, Van Soest RWM (2006) A new species of Amphimedon (Porifera, Demospongiae, Haplosclerida, Niphatidae) from the Capricorn–Bunker Group of islands, Great Barrier Reef, Australia; target species for the ‘sponge genome project’. Zootaxa 1314:31–39
Huber T, Faulkner G, Hugenholtz P (2004) Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics 20:2317–2319
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Hurlbert S (1971) The nonconcept of species diversity: a critique and alternative parameters. Ecology 52:577–586
Ilan M, Abelson A (1995) The life of a sponge in a sandy lagoon. Biol Bull 189:363–369
Jensen PR, Mincer TJ, Williams PG, Fenical W (2005) Marine actinomycete diversity and natural product discovery. Antonie van Leeuwenhoek 87:43–48
Kennedy J, Codling CE, Jones BV, Dobson ADW, Marchesi JR (2008) Diversity of microbes associated with the marine sponge, Haliclona simulans, isolated from Irish waters and identification of polyketide synthase genes from the sponge metagenome. Environ Microbiol 10:1888–1902
Lane DJ (1991) 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellows M (eds) Nucleic acid techniques in bacterial systematics. Wiley, New York, pp 115–175
Lee YK, Lee JH, Lee HK (2001) Microbial symbiosis in marine sponges. J Microbiol 39:254–264
Lee WL, Elvin DW, Reiswig HM (2007) The Sponges of California. A guide and key to the marine sponges of California, Monterey Bay Sanctuary Foundation, p. 111
Li ZY, Liu Y (2006) Marine sponge Craniella australiensis-associated bacterial diversity revelation based on 16S rDNA library and biologically active Actinomycetes screening, phylogenetic analysis. Lett Appl Microbiol 43:410–416
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar BA, Lai T, Steppi S, Jobb G, Forster W, Brettske I, Gerber S, Ginhart AW, Gross O, Grumann S, Hermann S, Jost R, Konig A, Liss T, Lussmann R, May M, Nonhoff B, Reichel B, Strehlow R, Stamatakis A, Stuckmann N, Vilbig A, Lenke M, Ludwig T, Bode A, Schleifer KH (2004) ARB: a software environment for sequence data. Nucleic Acids Res 32:1363–1371
Margot H, Acebal C, Toril E, Amils R, Puentes JLF (2002) Consistent association of crenarchaeal Archaea with sponges of the genus Axinella. Mar Biol 140:739–745
Medlin L, Elwood HJ, Stickel S, Sogin ML (1988) The characterization of enzymatically amplified eukaryotic 16S-like rRNA coding regions. Gene 71:491–499
Meyer B, Kuever J (2008) Phylogenetic diversity and spatial distribution of the microbial community associated with the Caribbean deep-water sponge Polymastia cf corticata by 16S rRNA, aprA, and amoA gene analysis. Microb Ecol 56:306–321
Mohamed NM, Enticknap JJ, Lohr JE, McIntosh SM, Hill RT (2008) Changes in bacterial communities of the marine sponge Mycale laxissima on transfer into aquaculture. Appl Environ Microbiol 74:1209–1222
Mohamed NM, Rao V, Hamann MT, Kelly M, Hill RT (2008) Monitoring bacterial diversity of the marine sponge Ircinia strobilina upon transfer into aquaculture. Appl Environ Microbiol 74:4133–4143
Nett M, König GM (2007) The chemistry of gliding bacteria. Nat Prod Rep 24:1245–1261
Osinga R, Tramper J, Wijffels RH (1999) Cultivation of marine sponges. Mar Biotechnol 1:509–532
Redmond NE, Van Soest RWM, Kelly M, Raleigh J, Travers SAA, McCormack GP (2007) Reassessment of the classification of the order Haplosclerida (class Demospongiae, phylum Porifera) using 18S rRNA gene sequence data. Mol Phylogen Evol 43:344–352
Reiswig HM (1974) Water transport, respiration and energetics of three tropical marine sponges. J Exp Mar Biol Ecol 14:231–249
Ridley CP, Lee HY, Khosla C (2008) Evolution of polyketide synthases in bacteria. Proc Natl Acad Sci U S A 105:4595–4600
Ronquist F, Huelsenbeck JP, Van der Mark O (2005) MrBayes 3.1 manual. School of computational science, Florida State University, USA
Schmitt S, Weisz JB, Lindquist N, Hentschel U (2007) Vertical transmission of a phylogenetically complex microbial consortium in the viviparous sponge Ircinia felix. Appl Environ Microbiol 73:2067–2078
Schleper C, DeLong EF, Preston CM, Feldman RA, Wu KY, Swanson RV (1998) Genomic analysis reveals chromosomal variation in natural populations of the uncultured psychrophilic archaeon Cenarchaeum symbiosum. J Bacteriol 180:5003–5009
Sharp KH, Eam B, Faulkner DJ, Haygood MG (2007) Vertical transmission of diverse microbes in the tropical sponge Corticium sp. Appl Environ Microbiol 73:622–629
Sipkema D, Franssen MCR, Osinga R, Tramper J, Wijffels RH (2005) Marine sponges as pharmacy. Mar Biotechnol 7:142–162
Sorokin DY, Tourova TP, Braker G, Muyzer G (2007) Thiohalomonas denitrificans gen. nov., sp. nov. and Thiohalomonas nitratireducens sp. nov., novel obligately chemolithoautotrophic, moderately halophilic, thiodenitrifying Gammaproteobacteria from hypersaline habitats. Int J Syst Evol Microbiol 57:1585–1589
Stackebrandt E, Ebers J (2006) Taxonomic parameters revisited: tarnished gold standards. Microbiol Today, Nov: 152–155
Suzuki MT, Béjà O, Taylor LT, DeLong EF (2001) Phylogenetic analysis of ribosomal RNA operons from uncultivated coastal marine bacterioplankton. Environ Microbiol 3:323–331
Suzuki MT, Preston CM, Béjà O, De la Torre JR, Steward GF, DeLong EF (2004) Phylogenetic screening of ribosomal RNA gene-containing clones in bacterial artificial chromosome (BAC) libraries from different depths in Monterey Bay. Microb Ecol 48:473–488
Swofford DL (2003) Phylogenetic analysis using parsimony (*and other methods). Version 4. Sinauer Associates, Sunderland
Taylor MW, Schupp PJ, Dahllöf I, Kjelleberg S, Steinberg PD (2004) Host specificity in marine sponge-associated bacteria and potential implications for marine microbial diversity. Environ Microbiol 6:121–130
Taylor MW, Radax R, Steger D, Wagner M (2007) Sponge-associated microorganisms: evolution, ecology and biotechnological potential. Microbiol Mol Biol Rev 71:295–347
Thiel V, Neulinger SC, Staufenberger T, Schmaljohann R, Imhoff JF (2007) Spatial distribution of sponge-associated bacteria in the Mediterranean sponge Tethya aurantium. FEMS Microbiol Ecol 59:47–63
Thiel V, Leininger S, Schmaljohann R, Brümmer F, Imhoff J (2007) Sponge-specific bacterial associations of the Mediterranean sponge Chondrilla nucula (Demospongiae, tetractinomorpha). Microb Ecol 54:101–111
Uriz MJ, Turon X, Galera J, Tur JM (1996) New light on the cell location of avarol within the sponge Dysidea avara (Dendroceratida). Cell Tissue Res 286:519–527
Vacelet J, Donadey C (1977) Electron microscopy study of the association between some sponges and bacteria. J Exp Mar Biol Ecol 30:301–314
Vacelet J, Fiala-Medioni A, Fisher CR, Boury-Esnault N (1996) Symbiosis between methane-oxidizing bacteria and a deep-sea carnivorous cladorhizid sponge. Mar Ecol Prog Ser 145:77–85
Webster NS, Wilson KJ, Blackall LL, Hill RT (2001) Phylogenetic diversity of bacteria associated with the marine sponge Rhopaloeides odorabile. Appl Env Microbiol 67:434–444
Webster NS, Negri AP, Munro MHG, Battershill CN (2004) Diverse microbial communities inhabit Antarctic sponges. Environ Microbiol 6:288–300
Webster NS, Cobb RE, Negri AP (2008) Temperature thresholds for bacterial symbiosis with a sponge. ISME J 2:830–842
Weisz JB, Hentschel U, Lindquist N, Martens CS (2007) Linking abundance and diversity of sponge-associated microbial communities to metabolic differences in host sponges. Mar Biol 152:475–483
Wichels A, Würtz S, Döpke H, Schütt C, Gerdts G (2006) Bacterial diversity in the breadcrumb sponge Halichondria panicea (Pallas). FEMS Microbiol Ecol 56:102–118
Wilkinson CR (1980) Nutrient translocation from green algal symbionts to the freshwater sponge Ephidatia fluviatilis. Hydrobiologia 75:241–250
Zwickl DJ (2006) Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. PhD thesis UT Austin, USA
Acknowledgements
The authors would like to thank Rob van Soest and Welton Lee for their help to identify the sponge species. William Fenical kindly tested the bioactivity of the sponge and we are grateful to Wouter Maalcke and Kent McDonald for their help with the TEM images. This work was supported by a Marie Curie grant from the European Union (MOIF-CT-2005-022164).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sipkema, D., Holmes, B., Nichols, S.A. et al. Biological Characterisation of Haliclona (?gellius) sp.: Sponge and Associated Microorganisms. Microb Ecol 58, 903–920 (2009). https://doi.org/10.1007/s00248-009-9534-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-009-9534-8