
Abstract
The mechanism of osmoregulation is crucial for maintaining growth, development, and life activities in teleosts. Takifugu obscurus, the only euryhaline species in the genus Takifugu, is a proper model organism for studying the mechanism of low-salt tolerance and hypoosmotic regulation. In this study, whole-genome sequencing data were obtained from 90 pufferfish representing five species within this genus, T. rubripes, T. obscurus, T. flavidus, T. niphobles, and T. bimaculatus. Using a phylogeny, PCA, and population structure analyses, we observed similar amounts of population genetic differentiation among species. The five species are closely related to each other and have differentiated within a relatively short period, while T. bimaculatus and T. flavidus shared the most similar genetic backgrounds. We further identified hundreds of genes under selection related to hypoosmotic regulation between T. obscurus and other Takifugu species, including 16 representative genes involving ion transporters (atp1a3, atp2a2, atp2a3, slc13a1, slc5a8, slc12a2, slc12a4, slc26a2, scn1b, and kcna2/3/10), genes involved in hormone regulation (fyn, prlr, and grb2), and a gene associated with water absorption (aqp3). Our findings provide preliminary insight into the mechanism of osmoregulation and will facilitate follow-up validation of candidate genes related to osmoregulation in T. obscurus.
Similar content being viewed by others

Introduction
Pufferfish are a group of migratory benthic teleost fishes belonging to the genus Takifugu (Tetraodontiformes, Tetraodontidae). They are naturally distributed in the Western Pacific Ocean, spanning the Bohai Sea, Yellow Sea, East China Sea, and the Sea of Japan. Some pufferfish, e.g., Takifugu rubripes, Takifugu flavidus, are commercially important fish in China, Japan, and Korea due to their high nutritional value and extraordinary flavor (Song et al. 2001). The genus Takifugu includes 25 species, most of which are closely related, that are able to produce fertile hybrids (e.g., T. rubripes × T. niphobles, T. rubripes × T. flavidus) (Hosoya et al. 2013). Hybrids can occur in wild populations or in artificially-crossed breeding. The extensive existence of hybrids is mainly due to the relatively recent evolutionary time scale of Takifugu species diversification. Takifugu species underwent explosive speciation in the marine waters of East Asia within 1.8–5.3 Ma during the Pliocene (Yamanoue et al. 2009). This was consistent with the theoretical result (Bolnick and Near 2005) that more recently diverged species pairs are more likely to produce viable hybrids. It should not go unnoticed that Takifugu species display speciation-related phenotypic variations that could potentially improve our understanding of speciation mechanisms (Noor and Feder 2006; Seehausen et al. 2008).
Pufferfish have speciated and radiated under different salinity levels of the aquatic environment. What is noteworthy is that the habitats of most pufferfish are restricted to salt water, with only one exception that can survive and develop normally in freshwater. T. obscurus, as that exceptional case, is a euryhaline fish that can survive in both freshwater and saline water (Kato et al. 2005). In its natural habitat, T. obscurus is an anadromous fish that migrates to freshwater to spawn, and newly hatched larvae live there for months (Yan et al. 2005). Meanwhile, artificial culture of T. obscurus has developed rapidly in China, and the fish are able to grow and reproduce in freshwater over the entire life cycle (Yang and Chen 2003). Some pufferfish species have the potential to adapt to low salinity to some extent; such species include T. niphobles, which is considered as a peripheral freshwater fish that can survive in freshwater for several days (Kato et al. 2010). Fluctuating salinity, an important environmental factor, directly influences the fluid osmotic pressure of fish, in turn impacting the survival, growth, and reproduction of the fish (Sampaio et al. 2007; Shi et al. 2008).
Variation in salinity could lead to a series of physiological responses in osmoregulation-related organs and tissues, including kidney, gill, and intestine (Perry 1997; Kato et al. 2005; Grosell 2006). The mechanism of salinity adaptation in teleosts is a complex physiological procedure that cannot be examined using freshwater fish. As an ideal model, the mechanisms of low-salinity tolerance differences between T. obscurus and other Takifugu species have been studied from different aspects. In general, T. obscurus has a solid osmotic adjustment capability and a superior osmoregulatory mechanism. In regard to physiological structure, T. obscurus has a freshwater fish-type nephron with a distal tubule in the kidney, while other Takifugu species have a seawater fish-type nephron without the distal tubule (Kato et al. 2005). Two cotransporters, Na+–K+–Cl– cotransporter 2 (NKCC2) and Na+–Cl– cotransporter (NCC), have been demonstrated to be differentially expressed in the distal nephrons of T. obscurus and other Takifugu fishes during the process of NaCl reabsorption (Kato et al. 2011). The gill is another important osmoregulatory organ for maintaining ion gradients in teleosts (Lee et al. 2005). Na+/K+-ATPase (NKA), as a key enzyme of ion transport maintaining the Na+ and K+ gradient, is mainly distributed in the chloride cells of the gills and has a crucial function in the ion exchange and osmoregulation in T. obscurus (Hwang and Lee 2007). The activity of NKA changes dramatically with the variation of environmental salinity (Li et al. 2014). In newly hatched T. obscurus larvae, NKA activity significantly increased after the exposure to high salinity (Wang et al. 2019). In Tetraodon nigroviridis kidney tubules, the expression of FXYD8 protein has the downregulation effect on NKA activity under salinity pressure (Wang et al. 2017). In hypoosmotic environments, the intestine plays a pivotal osmoregulatory role in absorbing water and balancing ions through the intestinal epithelia (Grosell 2006). Meanwhile, kidneys and gills respond to hypoosmotic conditions by participating in the excretion of hypotonic urine and transport of ions. Slc26 has been demonstrated to be an important gene family for exchange of anions such as HCO3− and SO2− (Mount and Romero 2004). HCO3− and SO2− transporters, which are mainly located in the intestinal epithelial cells and the proximal tubules of the kidney, respectively, are two genes closely related to the adaptation to a seawater environment in T. obscurus (Kurita et al. 2008; Kato et al. 2009). In other marine organisms, transcripts also differentially expressed after low-salinity exposure. Pathways, ontologies and protein complexes relating to DNA damage, cell cycle arrest and protein unfolding significantly up-regulated in response to low-salinity exposure (Maynard et al. 2018).
In brief, the mechanism of osmoregulation plays a vital role in maintaining life activities in teleosts and has been studied from various aspects at the organ and tissue levels as well as at the cellular and ion channel protein levels. There have also been a few studies using genomic and transcriptomic methods to explore the genetic mechanism of fish osmoregulation in recent years. The adaptive divergence of osmoregulatory genes in prickly sculpin has been studied using whole-genome sequencing of pooled DNA samples (Dennenmoser et al. 2017). Several adaptive candidate genes have been characterized, such as the ion transport gene sodium–potassium ATPase and the Na+–Cl– cotransporter (NCC). Meanwhile, in a threespine stickleback salinity tolerance study, an integrated genomic approach, including QTL mapping, whole-genome sequencing and RNA sequencing, has been conducted to identify candidate genes (Kusakabe et al. 2017). A handful of genes identified by transcriptomic analysis or genome sequencing were involved in ATP synthesis and hormonal signaling, which could be functional for the variation of osmoregulation ability. In tilapia, GWAS and QTL-seq were used to identify a major QTL region and a long QTL cluster on chrLG18 controlling the ability of osmoregulation in multiple tilapia populations (Jiang et al. 2019). However, the specific genes and co-regulators within their upstream and downstream pathways regulating body fluid osmotic pressure in teleosts have not been well characterized. Therefore, T. obscurus could serve as an ideal experimental model for investigating the molecular mechanisms of osmoregulation and adaptation to low-salinity environments in teleosts. Considering that the complete genome sequence of T. obscurus is not yet available, given the high genomic similarity among Takifugu species, the genome of its closely-related species T. rubripes could be considered as an alternative. T. rubripes was one of the earliest model species for vertebrates because of its compact genome size (~ 400 Mb) and genetic repertoire in comparison to other fishes (Brenner et al. 1993). A draft sequence of the pufferfish was generated using a whole-genome shotgun strategy for T. rubripes (Aparicio et al. 2002). A comprehensive genetic map of T. rubripes (Kai et al. 2011) has been generated using 1220 microsatellites that facilitated anchoring the assembled scaffolds to the 22 chromosomes of T. rubripes on the basis of preliminary research (Kai et al. 2005). This consolidated genome map covered 86% of the genome assembly, and 72% of the scaffolds have been oriented by integrating with the genetic linkage map (https://www.fugu-sg.org/). This genome has been widely used as a valuable resource for comparative genomic and subsequent genetic analysis (Hosoya et al. 2015; Ieda et al. 2018; Zhang et al. 2018). Furthermore, high-throughput sequencing has been developing rapidly during the last decade. Whole-genome re-sequencing is already widely used as a routine tool for identifying genomic differences of samples and for detecting genetic variation.
In this study, the whole genomes of five closely-related pufferfish species were sequenced using Illumina HiSeq X Ten platform. Using the high-quality genomic sequencing data, we elaborated the population genetic differences among the five species. Then, a selective sweep analysis using salt water versus freshwater species was performed to explore the genetic architecture underlying the adaptive evolution of osmoregulation. This study may assist in delineating the genetic diversity of pufferfish and in identifying candidate genes and pathways involved in the regulation networks of low-salinity tolerance.
Materials and methods
Statement of human and animal rights
This study was carried out in accordance with the recommendations of the care and use of animals for scientific purposes set up by the Animal Care and Use Committee of the Chinese Academy of Fishery Sciences (ACUC-CAFS). The sample collection and protocols were approved by the ACUC-CAFS.
Sample collection
A total of 90 samples from five populations were collected, with 29 T. rubripes, 30 T. obscurus, 8 T. flavidus, 8 T. bimaculatus, and 15 T. niphobles. The samples of T. rubripes, T. flavidus, and T. bimaculatus were from the Tanghai breeding base, Beidaihe Central Experiment Station, Chinese Academy of Fishery Sciences (119°31′32′′ E, 39°48′58′′ N). A natural population of T. niphobles was obtained from the open seas of Bohai Bay located near Beidaihe, Qinhuangdao, Hebei in China (119°32′55′′ E, 39°48′56′′ N). T. obscurus were sampled from the Zhongyang pufferfish manor in Hai’an, Jiangsu, China (120°53′58′′ E, 32°36′44′′ N). The salinity of the corresponding water environment was measured using a salinometer. The water salinity for T. obscurus was 6 ppt, while the water salinity for the other Takifugu species was equivalent to the natural salinity of seawater, 30 ppt.
Whole-genome sequencing and SNP discovery
The collected fish were euthanized in MS222 solution (30 mg/L) for three minutes before tissue collection. Genomic DNA of 90 fish was extracted from the caudal fin using DNeasy 96 Blood & Tissue Kits (Qiagen, Shanghai, China) in accordance with the manufacturer’s instructions. The integrity of DNA was checked via electrophoresis on 1.0% agarose gels. Genomic DNA was quantified by Nanodrop-1000 (Thermo Scientific, Wilmington, DE, USA), qualified by the fluorometer Qubit 4.0 (Thermo Scientific) and diluted to 50 ng/μL for sequencing. The total amount of DNA for whole-genome sequencing was no less than 2 μg per sample. After DNA quality verification, all individuals belonging to T. rubripes (N = 29), T. obscurus (N = 30), T. flavidus (N = 8), T. bimaculatus (N = 8), and T. niphobles (N = 15) were used for genome re-sequencing. DNA libraries were constructed and barcoded using the DNA Library Prep Reference Guide (Illumina). Sequencing was performed on the Illumina HiSeq X Ten platform with 150 bp paired-end reads. The average sequencing depth was 10× coverage per sample. The quality of raw reads was assessed using FastQC (https://www.bioinfor-matics.babraham.ac.uk/projects/fastqc/). The sequences were filtered by the following steps: (1) potential remnants of adapter sequences were removed; (2) low-quality paired reads with higher than 10% unrecognized bases in a single-end read were eliminated; and (3) low-quality bases with a Phred quality score lower than 20 were trimmed. After filtering, clean sequencing data were used for SNP calling. The sequencing data from each individual were aligned to the genome assembly of T. rubripes FUGU5 (Kai et al. 2011) using the Burrows-Wheeler Aligner (BWA) (Li and Durbin 2009). SNP calling was conducted using SAMTOOLS and BCFTOOLS with default parameters (Li et al. 2009).
Phylogeny, principle component analysis, and genetic structure
To understand the evolutionary relationships of pufferfish species, maximum-likelihood (ML) analysis was performed to construct a phylogenetic tree using RAxML version 8 (Stamatakis 2014). The bootstrap analysis was conducted by the best-scoring topology with 1000 replicates. The iTOL software (https://itol.embl.de/upload.cgi) was used to display the phylogenetic tree. PCA was conducted using PLINK 1.9 (Chang et al. 2015) and Genome-wide Complex Trait Analysis (GCTA) software (Yang et al. 2011). The population structure was investigated by the Bayesian clustering program STRUCTURE 2.3.1 (Falush et al. 2007) using all of the SNPs, with 2000 iterations. The number of genetic clusters K was predefined from two to five. The exported structure matrix was visualized by the StructurePlot 2.0 software (Ramasamy et al. 2014).
LD decay and haplotype construction
LD decay for five populations, saline populations, and all samples was estimated within a range of 5 kb using PLINK 1.9 (Chang et al. 2015). The average R2 value per kb region was calculated and plotted against the physical distances of SNPs in units of kb. Haplotype blocks that showed strong linkages were constructed using the PLINK “-blocks” parameter for each population.
Identification of selective signatures between populations
Whole-genome scanning between Saline vs Ob (Saline: T. rubripes, T. flavidus, T. niphobles and T. bimaculatus, n = 60; Ob: T. obscurus,n = 30), as well as between each seawater species vs Ob one-by-one, was conducted using three methods: π, Fst, and Tajima’s D tests. The π distribution was calculated using a sliding-window method in VCFTOOLS (Danecek et al. 2011). The genome was divided into 100 kb sliding windows, and the stepwise distance was 10 kb. The π ratios of Saline/Ob as well as from T. obscurus and the other four species were calculated, and the ratios were sorted. Fst and Tajima’s D test were conducted using VCFTOOLS “-weir-fst-pop” and “-TajimaD” parameters, respectively. The regions with the highest 5% π ratio and Fst values were identified and considered as candidate regions under selective sweeps. The scatter plots of π ratios and Fst values were visualized using the R ggplot2 package (https://cran.r-project.org/package=ggplot2).
Enrichment analysis
Genes involved in the regions of selective sweeps were chosen for functional annotation. GO and KEGG pathway analyses were performed using DAVID 6.8 (Huang et al. 2009) between T. obscurus and four saline water species, as well as between Saline and Ob.
Results
Sample collection and genotyping
A total of five Takifugu populations comprising 90 individuals were collected from three sites in China (Fig. 1, Supplementary Table 1). After quality control of extracted DNA, all of the individuals were re-sequenced to explore the population structure and the selective signals under adaptive evolution. In total, 438.67 Gb of sequencing data were generated, while for each individual, 4.87 Gb sequences with an average of ~ 12.43-fold depth per sample were obtained. From 2.92 G raw reads, 2.57 G reads were mapped to the reference genome for SNP calling with an average mapping ratio of 87.87%. After the sequence alignment and filtering, a total of 11,344,115 candidate SNPs were identified for further analysis (Supplementary Table 2).

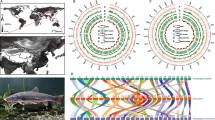
Geographic distribution of five Takifugu populations
Population structure
To investigate the genetic relationships of different Takifugu species, a phylogenetic tree was constructed using all of the informative SNPs (Fig. 2a). Each population formed a distinct clade that reflected the evolutionary differences among species as expected. Similar results were observed in the plot of the PCA (principle components analysis) analysis. Overall, the populations of T. rubripes, T. niphobles, and T. obscurus clustered separately and tightly, while the individuals of T. flavidus and T. bimaculatus were closely clustered together and overlapped. This was consistent with the results of phylogenetic analysis in that the clades of T. flavidus and T. bimaculatus shared one main branch point and were adjacent to each other. Four clusters from the PCA were far apart from each other with no confusing admixture. Population structure of the five species was evaluated using the Bayesian clustering program STRUCTURE. As the model K = 4 obtained the highest log likelihood value, the population structure for K = 4 is shown in Fig. 2c. Unsurprisingly, similar results were observed; the five populations were separated into three subgroups. Outside the T. rubripes and T. obscurus subgroups, T. flavidus, T. niphobles, and T. bimaculatus clustered and shared a common ancestry. T. rubripes, T. niphobles, and T. obscurus showed a simplex inheritance background, while T. flavidus and T. bimaculatus reflected a more admixed genetic structure. T. flavidus and T. bimaculatus have a very similar genetic structure and shared some ancestral sequences with T. obscurus. When K = 2, 3, or 5, only two subgroups existed, T. rubripes and the others (Fig. S1). This information, which was consistent with the phylogenetic analysis, also reflected the relatively earlier differentiation time of T. rubripes and the close genetic relationships and relatively recent species differentiation of the other four species.

Phylogeny, PCA, and population structure of five Takifugu populations. a The maximum-likelihood phylogenetic tree. The tree was constructed from 11,344,115 SNPs of 90 pufferfish from five populations (red, T. rubripes; orange, T. flavidus; yellow, T. bimaculatus; green, T. niphobles and purple, T. obscurus). b The principal components analysis (PCA) of five Takifugu populations. c Genetic structure of the pufferfish inferred by STRUCTURE analysis. The number of ancestral populations K = 4 is shown. Each individual has been indicated as a colored segment representing the proportion of its ancestral population
LD decay and pattern of haplotype blocks
LD (Linkage Disequilibrium) decay was investigated for five populations, and R2 values were calculated by PLINK and sorted by distance ranges. T. flavidus showed the highest R2 value, while the other four populations obtained significantly lower R2 values (Fig. 3a). When the distance was greater than 1 kb, the mean R2 values were 0.55 in T. flavidus, 0.40–0.45 in T. rubripes, T. niphobles, and T. obscurus, and 0.38 in T. bimaculatus (Supplementary Table 3). LD decay of whole samples and saline samples showed higher R2 values than most single populations except for T. flavidus. Because of the relatively small sample sizes for T. flavidus (N = 8) and T. bimaculatus (N = 8), haplotypes were constructed only for the populations of T. rubripes, T. niphobles, and T. obscurus using the PLINK “-block” parameter. The distribution of haplotype block lengths was calculated using the R software ggplot2 package (Fig. 3b). The block patterns were similar in three populations, while all samples and saline samples showed higher densities in smaller blocks. The maximum block lengths were 146.67 kb, 36.51 kb, and 34.22 kb in T. rubripes, T. niphobles, and T. obscurus, respectively, while the corresponding mean block sizes were 3.81 kb, 2.17 kb, and 2.42 kb (Supplementary Table 4).

LD decay and haplo-block size distributions. a LD patterns of five Takifugu populations, saline populations, and all individuals. b Distribution of haplotype block length of T. niphobles, T. obscurus, T. rubripes populations, saline populations, and all individuals
Genome-wide selective sweep analysis
The genetic diversity in certain regions of the genome might be dramatically decreased as a result of natural selection or domestication. Among the five species in this study, the T. obscurus population (Ob group) is more strongly euryhaline compared with the other four species (the Saline group), which is probably due to its unique genetic background. To identify genome regions under selection in the T. obscurus genome, we scanned the genome-wide variation and allele frequency spectra based on the approximately 13.69 million SNPs. The π ratios (πSaline/Obscurus) were calculated using a 100 kb sliding-window approach with 10 kb steps. In comparison to the Saline group, the Ob group had a significantly lower level of diversity (median πSaline/Obscurus = 2.860), reflecting the fewer recombination events in the Ob population. We identified a total of 1,548 outlier windows (top 5%, empirical π ratios ≥ 5.856) with median πSaline/Obscurus = 8.230 that included 554 candidate genes (Supplementary Table 5) based on the π ratio analysis. To further validate the genome regions under strong selective pressure in the Ob population, the genome regions with Fst greater than 0.232 (top 5%) were also identified, corresponding to 497 candidate genes (Supplementary Table 5). A total of 379 candidate genes shared by both the π ratio and Fst analyses were recognized as potential genes under selection (Fig. 4a). Among these selected genes, many are involved in ion transportation and ATPase related pathways; those that have been reported in previous studies include atp1a3 (sodium/potassium-transporting ATPase), slc13a1 (sodium/sulfate symporter), kcna2/3/10 (potassium voltage-gated channel subfamily A member 2/3/10), slc5a8 (sodium-coupled monocarboxylate transporter), and scnb1 (sodium channel subunit beta-1) (Fig. 4b). The diversity in the neighboring regions of these selected genes showed significant decrease in the Ob population compared with the Saline group, indicating possible selective sweeps adapting to the long-term changing osmotic environment. For example, in the kcna gene family, kcna2, kcna3, and kcna10 in the same sweep region (> 500 kb) were identified, while another gene, slc5a8, was also found in this region. To better understand these candidate genes and their potential functions, further GO (Gene Ontology) and KEGG (Kyoto Encyclopedia of Genes and Genomes) enrichment analyses also identified several key genes related to osmotic regulation. Two genes, fyn and atp2a3, were enriched in the GO term “ATP binding,” and grb2 was enriched in the “Gap junction” pathway (Fig. 4b, Fig. S2, and Supplementary Table 6). The results based on Tajima’s D analysis also provided supporting evidence identifying strong selective sweep signals (Fig. 5). The results suggested that the genomes of the Ob population have significantly evolved adaptations to the euryhaline environment.

Pi, Fst, and selected genes from selective sweep analysis between the saline populations and T. obscurus. a Distribution of π ratios and Fst values. Red data points were identified as selective sweeps that passed the thresholds of π ratio (the 5% right tail of the empirical π ratio distribution, π ratio ≥ 5.856) and Fst (the 5% right tail of the empirical Fst distribution, Fst ≥ 0.232). b Representative genes with strong selective sweep signals in the T. obscurus population. Genome annotations are shown at the bottom, and red bars represent coding sequences

Distribution of π ratios, Fst, and Tajima’s D values for selective sweep regions between the saline populations and T. obscurus. a Distribution of π ratios and the absolute differences of Tajima’s D values for genomic regions under selective sweeps in comparison with the whole genome. Blue dots denote the selected regions, while black dots denote the unselected regions. b Distribution of Fst and the absolute differences of Tajima’s D values for genomic regions under selective sweeps in comparison with the whole genome. Blue dots denote the selected regions, while black dots denote the unselected regions. c Box plot of the absolute differences of Tajima’s D values for genomic regions under selective sweeps in comparison with the whole genome. Boxes denote the values between the 25th and 75th percentiles, and the black transverse line inside the box denotes the median. Black vertical lines denote the values within 1.5 times the quartile values. Outliers are shown as black dots
As the Saline group contains four species, the genetic divergence among species could well reduce the common background noise when used as a whole. The comparison would then be focused on the species’ shared lower osmoregulation ability in contrast to the Ob population. Still, it might be useful to identify more potential genes under selection by one-to-one comparisons between the Ob population and the other four populations (shown as Bi, Fl, Ni, Ru). The π ratios and Fst values for the four comparisons are shown in Fig. 6a, and genes within selected regions are summarized in Supplementary Tables 7–10. The most abundant selected genes (211 genes) were found in the comparison Fl vs Ob, while 41 genes, 132 genes, and 92 genes were identified in Bi vs Ob, Ni vs Ob, and Ru vs Ob, respectively. Despite the result that similar genes were found in all four comparisons of Saline vs Ob (e.g., atp1a3, atp2a3, fyn, and scnb1), other important ion transporter genes were also identified (Fig. 6b), including slc12a4 (potassium/chloride transporter) from the Bi vs Ob group and slc12a2 (sodium/potassium/chloride transporter) and slc26a2 (anion exchanger) from the Ru vs Ob group, indicating their vital roles in regulating osmotic balance. Additionally, atp2b2 (calcium-transporting ATPase 2) and aqp3 (aquaporin 3) were also discovered in selective sweep regions from three comparisons (Bi vs Ob, Fl vs Ob, and Ru vs Ob). The prlr (prolactin receptor), which was reported to be involved in salinity tolerance of Nile tilapia (Yamaguchi et al. 2018), was also identified in Bi vs Ob and Fl vs Ob comparisons. We have screened out 132 genes in the comparison of Ni and Ob, however, very few genes (e.g. sodium/potassium-transporting ATPase subunit alpha-3) were closely related to osmoregulation. According to a previous study on T. obscurus osmoregulation (Kato et al. 2005), survival rates of several species were tested for transition from seawater to freshwater. T. obscurus showed 100% survival rate all the time, while T. niphobles showed longest tolerance time among other Takifugu species. Those results indicate the relatively strong low-salinity tolerance in T. niphobles, and we presume Ni and Ob could have a higher similarity in the mechanism of osmoregulation compared to other seawater populations. GO and KEGG enrichment analyses identified crucial pathways including “neuromast development” (contains slc26a2), “integral component of plasma membrane” (contains aqp3), and “neuroactive ligand-receptor interaction” (contains prlr), suggesting involvement of core functions of the neural system in the regulation of salinity (Supplementary Table 11). Further analyses based on Tajima’s D differences also provided evidence for previous selective sweep signals (Fig. S3). These results suggested that in pairwise comparisons between the Ob population and the other four populations, the genome of the Ob population displays significant selection signatures indicating enhancement of the species’ euryhaline ability.

Pi, Fst, and candidate genes identified under selective sweep analysis between T. obscurus and four other Takifugu populations. a Distribution of π ratios and Fst values. Red data points that passed the thresholds of π ratio and Fst (the 5% right tail of the empirical π ratio and Fst distributions) located to the upper right were identified as selected regions for the T. rubripes/T. obscurus, T. flavidus/T. obscurus, T. bimaculatus/T. obscurus, and T. niphobles/T. obscurus comparisons. b Candidate genes with strong selective sweep signals in the T. obscurus population. The π ratios were plotted using 10 kb sliding windows. Genome annotations are shown at the bottom, and red bars represent coding sequences
The most important genes under selection are summarized in Supplementary Table 12. Ion transporters comprise a large proportion of the candidate genes, while growth and neural development-related genes might play upstream roles in the regulation networks.
Discussion
Previous phylogenetic analysis of 15 Takifugu species (Yamanoue et al. 2009) indicated that the main lineage has undergone explosive speciation within 2.4–4.7 Ma, meaning that all species are closely related. From the combined results of the phylogeny, PCA and population structure analysis in our study, four main clades were identified: T. rubripes, T. obscurus, T. niphobles and the group of T. bimaculatus and T. flavidus. This result is consistent with the ML analysis of 24 Takifugu species using cyt b, 12 s rRNA gene sequences and mitogenomic data (Zhang and He 2008; Yamanoue et al. 2009) showing that T. bimaculatus and T. flavidus were clustered together in one branch, while T. rubripes, T. obscurus, and T. niphobles were assigned to different branches.
The genes discovered in selective sweep regions included ion transporters, neural regulators, and growth-related genes, most of which have been previously reported. In a recent physiological and molecular combined study of steelhead trout, candidate genes related to salinity tolerance were mainly involved in the function of ion transportation, metabolites, energy metabolism, and immune responses (Xiong et al. 2020). In the liver transcriptome analysis of yellow croaker, the differentially expressed genes (DEGs) were assigned to some similar pathways, such as material metabolism, growth, energy metabolism, and signal transduction (Lu et al. 2020). The metabolic pathways of salinity stress effectors in oysters were mainly ion transportation, oxidation–reduction, and signal transduction, which were identified by DEG analysis and weighted gene correlation network analysis (WGCNA) (Liu et al. 2019). As expected, the results of the above studies showed some overlap with our findings. But still these studies have not explained a lot into specific candidate genes and their potential functions in depth. The T. obscurus population possesses stronger low-salt tolerance than the other four species, and thus the comparisons were made by grouping the four salt-water species together and searching for potential genes under selection in T. obscurus. As various characteristic differences may be revealed by selection analysis, including growth, body shape, body color, and low-salinity tolerance, the genetic divergence within the saline populations could well attenuate the genetic background noise of other traits, allowing us to focus on their common features such as weakened low-salinity tolerance. Similar strategies have been reported in a previous study by Feng Cheng, who identified selective sweeps for leaf-heading trait in the two species Brassica rapa and Brassica oleracea (Cheng et al. 2016). Chinese cabbage has a unique heading-trait accession in B. rapa, while cabbage is the only accession with the heading trait in B. oleracea. The non-heading accessions in each species were grouped together to compare with the Chinese cabbage and cabbage. Thus, genetic divergence of other traits among the non-heading accessions would be balanced and would probably not appear in selective sweep analyses. In our study, pairwise comparisons between T. obscurus and the other four species were made as a supplementary analysis to the group comparisons to identify additional candidate genes. We identified a number of slc genes in the Ob population that were embedded in selected regions. These slc genes encode various transmembrane transporters of anions and cations (e.g., slc13a1, slc5a8, slc12a2, slc12a4, slc26a2). The apical membrane Na+-sulfate cotransporter encoded by slc13a1 mediates sulfate reabsorption across the renal proximal tubule and intestinal epithelia, promoting osmoregulation in low-salinity waters (Markovich et al. 2008; Markovich 2014). Similarly, a considerable number of genes were identified encoding energy-dependent ATP-binding transporters (atp1a3, atp2a2, atp2a3) and voltage-gated ion channels (scn1b, kcna2/3/10). Among these genes, atp1a3 that codes Na+/K+-ATPase plays a key role in the Na+ and K+ gradient balance, and this gene has been reported as being differentially expressed in salinity-varying environments in T. obscurus (Li et al. 2014). The voltage-gated ion channel genes (scn1b and kcna family) have been discovered by differential gene expression during spawning (Cui et al. 2015) and selective sweeps adapting freshwater and alkaline waters in Amur Ide (Xu et al. 2013, 2017). Therefore, the genes encoding transcellular ion transporters and channel proteins are reshaped by natural selection from the variable euryhaline environment. Adapting to lower-salinity environments can lead to varied secretion of hormones (e.g., prlr, ghr, igf) that modulate permeability of cells and prevent ion loss and water absorption (Sakamoto and McCormick 2006; Sinha et al. 2012). The genes fyn and grb2 were embedded in our enriched pathways of “Focal adhesion,” while prlr was enriched in “Neuroactive ligand-receptor interaction.” The aqp3 gene was included in the “integral component of plasma membrane” in our selective sweep analysis; this gene has been reported to be involved in regulating water absorption in renal tubules of Atlantic salmon (Engelund and Madsen 2015).
Using whole-genome sequencing data, we analyzed the population relationships of five Takifugu species. The selective signatures of the T. obscurus population were screened for effects on low-salt tolerance and hypoosmotic pressure adjustment capacity. Phylogenetic analysis, PCA, and population structure analysis were conducted, and interspecific genetic relationships among five species within the genus Takifugu were established. In addition, a number of genes within selected regions have indicated potential function in osmotic regulation. Among the results, 16 genes were identified as potential candidate genes that could possibly be associated with hypoosmotic pressure regulation in T. obscurus. The main categories of osmoregulation genes have been covered in our study; however, the detailed mechanisms of gene interaction and regulation of gene expression are not yet fully understood. Further experimental evidence in view of differential expression by RNA-seq or methylation in more populations from different habitats would also be necessary in the future to support our potential candidate genes related to osmoregulation in T. obscurus. Overall, our findings provide an empirical foundation for further insights into the genetic mechanism of osmoregulation in T. obscurus. This study will also facilitate further research into the biological processes underlying physiological adaptations to low-salt tolerance in marine fishes. Furthermore, these results take the first steps towards verifying the genomic basis of adaptive divergence underlying low-salinity tolerance and the difference of osmoregulation ability.
Availability of data
The datasets generated during the current study are available in the NCBI SRA repository (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA522329/).
Abbreviations
- SNP:
-
Single nucleotide polymorphism
- PCA:
-
Principal component analysis
- NKCC2:
-
Na+–K+–Cl− cotransporter 2
- NCC:
-
Na+–Cl− cotransporter
- NKA:
-
Na+/K+-ATPase
- LD:
-
Linkage disequilibrium
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
References
Aparicio S, Chapman J et al (2002) Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science 297(5585):1301–1310
Bolnick DI, Near TJ (2005) Tempo of hybrid inviability in centrarchid fishes (Teleostei: Centrarchidae). Evolution 59(8):1754–1767
Brenner S, Elgar G et al (1993) Characterization of the pufferfish (Fugu) genome as a compact model vertebrate genome. Nature 366(6452):265–268
Chang CC, Chow CC et al (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4:7
Cheng F, Sun RF et al (2016) Subgenome parallel selection is associated with morphotype diversification and convergent crop domestication in Brassica rapa and Brassica oleracea. Nat Genet 48(10):1218–1224
Cui J, Xu J et al (2015) Transcriptional profiling reveals differential gene expression of amur ide (Leuciscus waleckii) during spawning migration. Int J Mol Sci 16(6):13959–13972
da Huang W, Sherman BT et al (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37(1):1–13
Danecek P, Auton A et al (2011) The variant call format and VCFtools. Bioinformatics 27(15):2156–2158
Dennenmoser S, Vamosi SM et al (2017) Adaptive genomic divergence under high gene flow between freshwater and brackish-water ecotypes of prickly sculpin (Cottus asper) revealed by Pool-Seq. Mol Ecol 26:25–42
Engelund MB, Madsen SS (2015) Tubular localization and expressional dynamics of aquaporins in the kidney of seawater-challenged Atlantic salmon. J Comparative Physiol B Biochem Syst Environ Physiol 185(2):207–223
Falush D, Stephens M et al (2007) Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes 7(4):574–578
Grosell M (2006) Intestinal anion exchange in marine fish osmoregulation. J Exp Biol 209(Pt 15):2813–2827
Hosoya S, Kai W et al (2013) The genetic architecture of growth rate in juvenile Takifugu species. Evolution 67(2):590–598
Hosoya S, Suetake H et al (2015) Genetic basis underlying behavioral correlation between Fugu Takifugu rubripes and a closely related species, Takifugu niphobles. Behav Genet 45(5):560–572
Hwang PP, Lee TH (2007) New insights into fish ion regulation and mitochondrion-rich cells. Comparative Biochem Physiol A Mol Integr Physiol 148(3):479–497
Ieda R, Hosoya S et al (2018) Identification of the sex-determining locus in grass puffer (Takifugu niphobles) provides evidence for sex-chromosome turnover in a subset of Takifugu species. PLoS ONE 13:1
Jiang DL, Gu XH et al (2019) Identifying a long QTL cluster across chrLG18 associated with salt tolerance in tilapia using GWAS and QTL-seq. Mar Biotechnol 21:250–261
Kai W, Kikuchi K et al (2005) A genetic linkage map for the tiger pufferfish, Takifugu rubripes. Genetics 171(1):227–238
Kai W, Kikuchi K et al (2011) Integration of the genetic map and genome assembly of fugu facilitates insights into distinct features of genome evolution in teleosts and mammals. Genome Biol Evol 3:424–442
Kato A, Doi H et al (2005) Takifugu obscurus is a euryhaline fugu species very close to Takifugu rubripes and suitable for studying osmoregulation. BMC Physiol 5:18
Kato A, Chang MH et al (2009) Identification of renal transporters involved in sulfate excretion in marine teleost fish. Am J Physiol Regul Integr Compar Physiol 297(6):R1647–R1659
Kato A, Maeno Y et al (2010) Brief migration of the grass puffer, Takifugu niphobles, to fresh water from salt water. Ichthyol Res 57(3):298–304
Kato A, Muro T et al (2011) Differential expression of Na+-Cl- cotransporter and Na+-K+-Cl- cotransporter 2 in the distal nephrons of euryhaline and seawater pufferfishes. Am J Physiol Regul Integr Comp Physiol 300(2):R284–297
Kurita Y, Nakada T et al (2008) Identification of intestinal bicarbonate transporters involved in formation of carbonate precipitates to stimulate water absorption in marine teleost fish. Am J Physiol Regul Integr Compar Physiol 294(4):R1402–R1412
Kusakabe M, Ishikawa A et al (2017) Genetic basis for variation in salinity tolerance between stickleback ecotypes. Mol Ecol 26:304–319
Lee KM, Kaneko T et al (2005) Low-salinity tolerance of juvenile fugu Takifugu rubripes. Fish Sci 71(6):1324–1331
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–1760
Li H, Handsaker B et al (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25(16):2078–2079
Li JJ, Wang J et al (2014) Changes in plasma osmolality and Na+/K+ ATPase activity of juvenile obscure puffer Takifugu obscurus following salinity challenge. Biochem Syst Ecol 56:111–117
Liu X, Li L et al (2019) Transcriptome and gene coexpression network analyses of two wild populations provides insight into the high-salinity adaptation mechanisms of Crassostrea ariakensis. Mar Biothechnol 21:596–612
Lu Z, Huang W et al (2020) Liver transcriptome analysis reveals the molecular responses to low-salinity in large yellow croaker Larimichthys crocea. Aquaculture 517:734827
Markovich D (2014) Na+-sulfate cotransporter SLC13A1. Pflugers Archiv Eur J Physiol 466(1):131–137
Markovich D, Romano A et al (2008) Functional and structural characterization of the zebrafish Na+-sulfate cotransporter 1 (NaS1) cDNA and gene (slc13a1). Physiol Genomics 34(3):256–264
Maynard A, Bible JM et al (2018) Transcriptomic responses to extreme low salinity among locally adapted populations of Olympia oyster (Ostrea lurida). Mol Ecol 27(21):4225–4240
Mount DB, Romero MF (2004) The SLC26 gene family of multifunctional anion exchangers. Pflugers Archiv Eur J Physiol 447(5):710–721
Noor MA, Feder JL (2006) Speciation genetics: evolving approaches. Nat Rev Genet 7(11):851–861
Perry SF (1997) The chloride cell: structure and function in the gills of freshwater fishes. Annu Rev Physiol 59:325–347
Ramasamy RK, Ramasamy S et al (2014) STRUCTURE PLOT: a program for drawing elegant STRUCTURE bar plots in user friendly interface. Springerplus 3:431
Sakamoto T, McCormick SD (2006) Prolactin and growth hormone in fish osmoregulation. Gen Comp Endocrinol 147(1):24–30
Sampaio LA, Freitas LS et al (2007) Effects of salinity on Brazilian flounder Paralichthys orbignyanus from fertilization to juvenile settlement. Aquaculture 262(2–4):340–346
Seehausen O, Terai Y et al (2008) Speciation through sensory drive in cichlid fish. Nature 455(7213):620–626
Shi ZH, Huang XX et al (2008) Salinity stress on embryos and early larval stages of the pomfret Pampus punctatissimus. Aquaculture 275(1–4):306–310
Sinha AK, Liew HJ et al (2012) Combined effects of high environmental ammonia, starvation and exercise on hormonal and ion-regulatory response in goldfish (Carassius auratus L.). Aquat Toxicol 114:153–164
Song L, Liu B et al (2001) Molecular phylogeny and species identification of pufferfish of the genus Takifugu (Tetraodontiformes, Tetraodontidae). Mar Biotechnol (NY) 3(4):398–406
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313
Wang PJ, Yang WK et al (2017) FXYD8, a novel regulator of renal Na(+)/K(+)-ATPase in the euryhaline teleost, Tetraodon nigroviridis. Front Physiol 8:576
Wang J, Hou X et al (2019) Interactive effects of temperature and salinity on the survival, oxidative stress, and Na(+)/K(+)-ATPase activity of newly hatched obscure puffer (Takifugu obscurus) larvae. Fish Physiol Biochem 45(1):93–103
Xiong Y, Dong S et al (2020) Growth, osmoregulatory response, adenine nucleotide contents, and liver transcriptome analysis of steelhead trout (Oncorhynchus mykiss) under different salinity acclimation methods. Aquaculture 520:734937
Xu J, Li Q et al (2013) Gene expression changes leading extreme alkaline tolerance in Amur ide (Leuciscus waleckii) inhabiting soda lake. BMC Genomics 2013:14
Xu J, Li JT et al (2017) Genomic basis of adaptive evolution: the survival of amur ide (Leuciscus waleckii) in an extremely alkaline environment. Mol Biol Evol 34(1):145–159
Yamaguchi Y, Breves JP et al (2018) Acute salinity tolerance and the control of two prolactins and their receptors in the Nile tilapia (Oreochromis niloticus) and Mozambique tilapia (O. mossambicus): a comparative study. Gen Comp Endocrinol 257:168–176
Yamanoue Y, Miya M et al (2009) Explosive speciation of Takifugu: another use of fugu as a model system for evolutionary biology. Mol Biol Evol 26(3):623–629
Yan M, Li Z et al (2005) Preliminary results on osmolality response of pufferfish Takifugu obscurus to sudden salinity change. J Appl Ichthyol 21(2):156–159
Yang Z, Chen YF (2003) Induced ovulation using LHRHa in anadromous obscure puffer Takifugu obscurus cultured entirely in freshwater. Fish Physiol Biochem 29(4):323–326
Yang J, Lee SH et al (2011) GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 88(1):76–82
Zhang YB, He SP (2008) Investigations into the perplexing interrelationship of the Genus Takifugu Abe, 1949 (Tetraodontiformes, Tetraodontidae). Chin Sci Bull 53(2):233–244
Zhang X, Mizukoshi M et al (2018) Ultrahigh-density linkage map construction using low-coverage whole-genome sequencing of a doubled haploid population: case study of Torafugu (Takifugu rubripes). Genes (Basel) 9:3
Acknowledgements
This work was supported by the Central Public-interest Scientific Institution Basal Research Fund, CAFS (nos. 2017B003, 2018B003 and 2016HY-JC0301) and the Special Scientific Research Funds for Central Non-profit Institutes, Chinese Academy of Fishery Sciences (2020TD24) for genome re-sequencing. The work was supported by the National Infrastructure of Fishery Germplasm Resources of China (no. 2019DKA30470), the Beijing Natural Science Foundation (no. 6202034) and the National Natural Science Foundation of China (no. 31502151) for DNA extraction and quality control. The work was supported by the China Agriculture Research System (CARS-47-01) for sample collection.
Author information
Authors and Affiliations
Contributions
JX initiated and coordinated the research project. HZ and JX conducted the analysis, and drafted the manuscript. JH, HL, HZ and GX engaged in sample collection and DNA extraction. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Responsible Editor: T. Reusch.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Reviewed by M. Heckwolf and undisclosed experts.
Electronic supplementary material
Below is the link to the electronic supplementary material.
227_2020_3705_MOESM1_ESM.pdf
Supplementary Fig. 1 Genetic structure of pufferfishes for K=2, 3, and 5. Each individual has been indicated as a colored segment representing the proportion contributed by its ancestral population. (PDF 631 kb)
227_2020_3705_MOESM2_ESM.pdf
Supplementary Fig. 2 Pie plot of the enriched pathways of osmotic regulation related genes between the Saline and T. obscurus populations. (PDF 745 kb)
227_2020_3705_MOESM3_ESM.pdf
Supplementary Fig. 3 Box plot of the Tajima’s D differences between selective sweep regions and the whole genome. Boxes denote the values between the 25th and 75th percentiles and the black transverse line inside the box denotes the median. Black vertical lines denote the values within 1.5 times of quartile values. Outliers are shown as black dots. (A) T. rubripes vs T. obscurus. (B) T. flavidus vs T. obscurus. (C) T. bimaculatus vs T. obscurus. (D) T. niphobles vs T. obscurus. (PDF 1225 kb)
227_2020_3705_MOESM4_ESM.xlsx
Supplementary Table 1-12. Summary of sample information and re-sequencing data, LD decay and block size, gene lists under selective sweeps, GO and KEGG enrichment, and potential candidate genes related to osmoregulation. (XLSX 877 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, H., Hou, J., Liu, H. et al. Adaptive evolution of low-salinity tolerance and hypoosmotic regulation in a euryhaline teleost, Takifugu obscurus. Mar Biol 167, 90 (2020). https://doi.org/10.1007/s00227-020-03705-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00227-020-03705-x