
Abstract
Fabry disease is an X-linked lysosomal storage disease due to deficient α-galactosidase A (α-Gal A) activity and the resultant lysosomal accumulation of globotriaosylceramide (Gb3) and related lipids primarily in blood vessels, kidney, heart, and other organs. The renal distribution of stored glycolipid species in the α-Gal A knockout mouse model was compared to that in mice to assess relative distribution and absolute amounts of accumulated sphingolipid isoforms. Twenty isoforms of five sphingolipid groups were visualized by mass spectrometry imaging (MSI), and their distribution was compared with immunohistochemical (IHC) staining of Gb3, the major stored glycosphingolipid in consecutive tissue sections. Quantitative bulk lipid analysis of tissue sections was assessed by electrospray ionization with tandem mass spectrometry (ESI-MS/MS). In contrast to the findings in wild-type mice, all three analytical techniques (MSI, IHC, and ESI-MS/MS) revealed increases in Gb3 isoforms and ceramide dihexosides (composed mostly of galabiosylceramides), respectively. To our knowledge, this is the first report of the distribution of individual molecular species of Gb3 and galabiosylceramides in kidney sections in Fabry disease mouse. In addition, the spatial distribution of ceramides, ceramide monohexosides, and sphingomyelin forms in renal tissue is presented and discussed in the context of their biosynthesis.

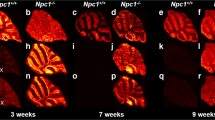
Immunohistochemical images of a wild type (left) and Fabry mouse kidney (right)






Similar content being viewed by others


References
Poupetova H, Ledvinova J, Berna L, Dvorakova L, Kozich V, Elleder M (2010) The birth prevalence of lysosomal storage disorders in the Czech Republic: comparison with data in different populations. J Inherit Metab Dis 33:387–396
Desnick RJ, Ioannou YA, Eng CM (eds) (2001) Alpha-galactosidase A deficiency: Fabry disease. The metabolic and molecular bases of inherited disease. McGraw-Hill, New York
Rombach SM, Dekker N, Bouwman MG, Linthorst GE, Zwinderman AH, Wijburg FA, Kuiper S, Weerrnan MAB, Groener JEM, Poorthuis BJ, Hollak CEM, Aerts JMFG (2010) Plasma globotriaosylsphingosine: diagnostic value and relation to clinical manifestations of Fabry disease. Biochim Biophys Acta-Mol Basis Dis 1802:741–748
Elleder M, Bradova V, Smid F, Budesinsky M, Harzer K, Kustermannkuhn B, Ledvinova J, Belohlavek, Kral V, Dorazilova V (1990) Cardiocyte storage and hypertrophy as a sole manifestation of Fabry disease—report on a case simulating hypertrophic nonobstructive cardiomyopathy. Virchows Archiv A: Pathol Anat Histopathol 417:449–455
Touboul D, Roy S, Germain D, Baillet A, Brion F, Prognon P, Chaminade P, Laprevote O (2005) Fast fingerprinting by MALDI-TOF mass spectrometry of urinary sediment glycosphingolipids in Fabry disease. Anal Bioanal Chem 382:1209–1216
Kuchar L, Ledvinova J, Hrebicek M, Myskova H, Dvorakova L, Berna L, Chrastina P, Asfaw B, Elleder M, Petermoeller M, Mayrhofer H, Staudt M, Kraegeloh-Mann I, Paton BC, Harzer K (2009) Prosaposin deficiency and saposin B deficiency (activator-deficient metachromatic leukodystrophy): report on two patients detected by analysis of urinary sphingolipids and carrying novel PSAP gene mutations. Am J Med Genet A 149A:613–621
Fuller M, Sharp PC, Rozaklis T, Whitfield PD, Blacklock D, Hopwood JJ, Meikle PJ (2005) Urinary lipid profiling for the identification of Fabry hemizygotes and heterozygotes. Clin Chem 51:688–694
Kitagawa T, Ishige N, Suzuki K, Owada M, Ohashi T, Kobayashi M, Eto Y, Tanaka A, Mills K, Winchester B, Keutzer J (2005) Non-invasive screening method for Fabry disease by measuring globotriaosylceramide in whole urine samples using tandem mass spectrometry. Mol Genet Metab 85:196–202
Kitagawa T, Suzuki K, Ishige N, Ohashi T, Kobayashi M, Eto Y, Tanaka A, Odaka H, Owada M (2008) Non-invasive high-risk screening for Fabry disease hemizygotes and heterozygotes. Pediatr Nephrol 23:1461–1471
Dobrovolny R, Dvorakova L, Ledvinova J, Magage S, Bultas J, Lubanda JC, Elleder M, Karetova D, Pavlikova M, Hrebicek M (2005) Relationship between X-inactivation and clinical involvement in Fabry heterozygotes. Eleven novel mutations in the alpha-galactosidase A gene in the Czech and Slovak population. J Mol Med 83:647–654
Gold H, Mirzaian M, Dekker N, Ferraz MJ, Lugtenburg J, Codee JDC, van der Marel GA, Overkleeft HS, Linthorst GE, Groener JEM, Aerts JM, Poorthuis B (2013) Quantification of globotriaosylsphingosine in plasma and urine of Fabry patients by stable isotope ultraperformance liquid chromatography-tandem mass spectrometry. Clin Chem 59:547–556
Niemann M, Rolfs A, Giese A, Mascher H, Breunig F, Ertl G, Wanner C, Weidemann F (2013) Lyso-Gb3 indicates that the alpha-galactosidase A mutation D313Y is not clinically relevant for Fabry disease. In: Zschocke J, Gibson KM, Brown G, Morava E, Peters V (eds) Jimd reports—case and research reports, 2012/4, vol 7, JIMD Reports. Springer, Berlin, pp 99–102. doi:10.1007/8904_2012_154
Touboul D, Roy S, Germain DP, Chaminade P, Brunelle A, Laprevote O (2007) MALDI-TOF and cluster-TOF-SIMS imaging of Fabry disease biomarkers. Int J Mass Spectrom 260:158–165
Onoue K, Zaima N, Sugiura Y, Isojima T, Okayama S, Horii M, Akai Y, Uemura S, Takemura G, Sakuraba H, Sakaguchi Y, Setou M, Saito Y (2011) Using imaging mass spectrometry to accurately diagnose Fabry’s disease. Circ J 75:221–223
Najafian B, Mauer M, Hopkin RJ, Svarstad E (2013) Renal complications of Fabry disease in children. Pediatr Nephrol 28:679–687
Ioannou YA, Zeidner KM, Gordon RE, Desnick RJ (2001) Fabry disease: preclinical studies demonstrate the effectiveness of alpha-galactosidase A replacement in enzyme-deficient mice. Am J Hum Genet 68:14–25
Vyberg M, Nielsen S (1998) Dextran polymer conjugate two-step visualization system for immunohistochemistry—a comparison of EnVision + with two three-step avidin-biotin techniques. Appl Immunohistochem 6:3–10
Strohalm M, Kavan D, Novak P, Volny M, Havlicek V (2010) MMass 3: a cross-platform software environment for precise analysis of mass spectrometric data. Anal Chem 82:4648–4651
Hulkova H, Ledvinova J, Kuchar L, Smid F, Honzikova J, Elleder M (2012) Glycosphingolipid profile of the apical pole of human placental capillaries: the relevancy of the observed data to Fabry disease. Glycobiology 22:725–732
Kuchar L, Rotkova J, Asfaw B, Lenfeld J, Horak D, Korecka L, Bilkova Z, Ledvinova J (2010) Semisynthesis of C17:0 isoforms of sulphatide and glucosylceramide using immobilised sphingolipid ceramide N-deacylase for application in analytical mass spectrometry. Rapid Commun Mass Spectrom 24:2393–2399
Durant B, Forni S, Sweetman L, Brignol N, Meng XL, Benjamin ER, Schiffmann R, Shen JS (2011) Sex differences of urinary and kidney globotriaosylceramide and lyso-globotriaosylceramide in Fabry mice. J Lipid Res 52:1742–1746
Zhang Y, Wang Y, Guo S, Guo Y, Liu H, Li Z (2013) Ammonia-treated N-(1-naphthyl) ethylenediamine dihydrochloride as a novel matrix for rapid quantitative and qualitative determination of serum free fatty acids by matrix-assisted laser desorption/ionization-Fourier transform ion cyclotron resonance mass spectrometry. Anal Chim Acta 794:82–89
Elleder M (2010) Subcellular, cellular and organ pathology of Fabry disease. In: Elstein et al. (eds) Fabry disease. Springer: Berlin, p 39-79. doi:10.1007/978-90-481-9033-1_3
Vylet’al P, Hulkova H, Zivna M, Berna L, Novak P, Elleder M, Kmoch S (2008) Abnormal expression and processing of uromodulin in Fabry disease reflects tubular cell storage alteration and is reversible by enzyme replacement therapy. J Inherit Metab Dis 31:508–517
Meehan SM, Junsanto T, Rydel JJ, Desnick RJ (2004) Fabry disease: renal involvement limited to podocyte pathology and proteinuria in a septuagenarian cardiac variant. Pathologic and therapeutic implications. Am J Kidney Dis 43:164–171
Ohshima T, Murray GJ, Swaim WD, Longenecker G, Quirk JM, Cardarelli CO, Sugimoto Y, Pastan I, Gottesman MM, Brady RO, Kulkarni AB (1997) Alpha-galactosidase A deficient mice: a model of Fabry disease. Proc Natl Acad Sci U S A 94:2540–2544
Ohshima T, Schiffmann R, Murray GJ, Kopp J, Quirk JM, Stahl S, Chan CC, Zerfas P, Tao-Cheng JH, Ward JM, Brady RO, Kulkarni AB (1999) Aging accentuates and bone marrow transplantation ameliorates metabolic defects in Fabry disease mice. Proc Natl Acad Sci U S A 96:6423–6427
Valbuena C, Oliveira JP, Carneiro F, Relvas S, Ganhao M, Clara Sa-Miranda M, Rodrigues LG (2011) Kidney histologic alterations in alpha-galactosidase-deficient mice. Virchows Arch 458:477–486
Slotte JP (2013) Molecular properties of various structurally defined sphingomyelins—correlation of structure with function. Prog Lipid Res 52:206–219
Slotte JP (2013) Biological functions of sphingomyelins. Prog Lipid Res 52:424–437
Groesch S, Schiffmann S, Geisslinger G (2012) Chain length-specific properties of ceramides. Prog Lipid Res 51:50–62
Klinkert I, McDonnell LA, Luxembourg SL, Altelaar AFM, Amstalden ER, Piersma SR, Heeren RMA (2007) Tools and strategies for visualization of large image data sets in high-resolution imaging mass spectrometry. Rev Sci Instrum 78:053716
Authors’ contributions
The manuscript was written through contributions of all authors. All authors have given their approval to the final version of the manuscript. RJD generated the FKO mouse. RD participated in mouse colony management, sample collection, preparation, and coordination of the study. LKr and HF were responsible for the MSI data acquisition; LKu collected and evaluated the quantitative MS data. JR and BA participated in preparation of lipid samples and protein analysis. MS and MV analyzed and evaluated the MSI data. HH was responsible for the IHC and in situ studies. LKry performed the immunohistochemical analyses. JL, LKu, HH, KL, RJD, and VH wrote the paper.
Funding sources
The authors acknowledge the major direct support from the Ministry of Education, Youth and Sports of the Czech Republic (COST-CZ-LD13038, PRVOUK-P24/LF1/3, COST-CZ-LD13005 and UNCE 204011), Ministry of Health of the Czech Republic (Grant IGA MZ NT14015-3/2013), and Czech Science Foundation (P206/12/1150). Access to instrumental and other facilities was also supported by EU (COST BM1104, Operational Program Prague—Competitiveness project CZ.2.16/3.1.00/24023) and IMIC institutional research concept RVO61388971.
Conflict of interest
RJD serves as a consultant to Amicus Therapeutics and Genzyme Corp, holds shares of Amicus Therapeutics, receives grants from Genzyme Corp., and receives royalties from Genzyme Corp. and Shire HGT.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the topical collection Mass Spectrometry Imaging with guest editors Andreas Römpp and Uwe Karst.
Ladislav Kuchar and Helena Faltyskova contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 13442 kb)
Rights and permissions
About this article

Cite this article
Kuchar, L., Faltyskova, H., Krasny, L. et al. Fabry disease: renal sphingolipid distribution in the α-Gal A knockout mouse model by mass spectrometric and immunohistochemical imaging. Anal Bioanal Chem 407, 2283–2291 (2015). https://doi.org/10.1007/s00216-014-8402-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-014-8402-7