
Abstract.
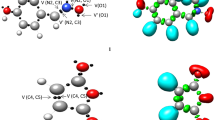
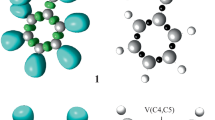
Hybrid quantum mechanical/molecular mechanical electronic structure calculations reveal the transition state for C–H bond cleavage in [(LCu)2 (μ-O)2]2+ (L=1,4,7-tribenzyl-1,4,7-triazacyclononane) to be consistent with a hydrogen-atom-transfer mechanism from carbon to oxygen. At the MPW1K/double-zeta effective core potential(+)|univeral force field level, 0 K activation enthalpies for the parent, p-CF3, and p-OH substituted benzyl systems are predicted to be 8.8, 9.5, and 7.8 kcal/mol. Using a one-dimensional Eckart potential to estimate quantum effects on the reaction coordinate, reaction in the unsubstituted system is predicted to proceed with a primary kinetic isotope effect of 22 at 233 K. Structural parameters associated with the hydrogen-atom transfer are consistent with the Hammond postulate.
Similar content being viewed by others

Author information
Authors and Affiliations
Additional information
Received: 10 October 2000 / Accepted: 3 November 2000 / Published online: 3 April 2001
Rights and permissions
About this article
Cite this article
Cramer, C., Pak, Y. Transition state for intramolecular C–H bond cleavage in [(LCu)2(μ-O)2]2+ (L = 1,4,7-tribenzyl-1,4,7-triazacyclononane). Theor Chem Acc 105, 477–480 (2001). https://doi.org/10.1007/s002140000238
Issue Date:
DOI: https://doi.org/10.1007/s002140000238