
Abstract
Background
Athina Markou and her colleagues discovered persistent changes in adult behavior following adolescent exposure to ethanol or nicotine consistent with increased risk for developing addiction. Building on Dr. Markou’s important work and that of others in the field, researchers at the Bowles Center for Alcohol Studies have found that persistent changes in behavior following adolescent stress or alcohol exposure may be linked to induction of immune signaling in brain.
Aim
This study aims to illuminate the critical interrelationship of the innate immune system (e.g., toll-like receptors [TLRs], high-mobility group box 1 [HMGB1]) in the neurobiology of addiction.
Method
This study reviews the relevant research regarding the relationship between the innate immune system and addiction.
Conclusion
Emerging evidence indicates that TLRs in brain, particularly those on microglia, respond to endogenous innate immune agonists such as HMGB1 and microRNAs (miRNAs). Multiple TLRs, HMGB1, and miRNAs are induced in the brain by stress, alcohol, and other drugs of abuse and are increased in the postmortem human alcoholic brain. Enhanced TLR-innate immune signaling in brain leads to epigenetic modifications, alterations in synaptic plasticity, and loss of neuronal cell populations, which contribute to cognitive and emotive dysfunctions. Addiction involves progressive stages of drug binges and intoxication, withdrawal-negative affect, and ultimately compulsive drug use and abuse. Toll-like receptor signaling within cortical-limbic circuits is modified by alcohol and stress in a manner consistent with promoting progression through the stages of addiction.
Similar content being viewed by others


Introduction
Several studies have found lasting changes in brain that contribute to the progression through the stages of addiction (Koob and Volkow 2010). Emerging findings suggest that signaling through known innate immune signaling systems in brain contributes to mounting negative affect and may underlie the neurobiological similarities of depression and addiction (Markou et al. 1998). Athina Markou and her colleagues discovered persistent changes in adult behavior following adolescent exposure to ethanol or nicotine consistent with increased risk for developing addiction. For example, adolescent alcohol exposure was shown to increase high-reward choices even when the probability of reward outcome was unlikely (i.e., rats exposed to ethanol during adolescence led to increased risky decision-making in adulthood; Boutros et al. 2014, 2015). Risky choice preference was found to correlate with the loss of cholinergic neurons in the basal forebrain (Boutros et al. 2014). Adolescent ethanol exposure was also found to increase adult central nucleus of the amygdala corticotropin-releasing factor (CRF) messenger RNA (mRNA) 40 days following the last ethanol dose (i.e., a persistent change in amygdalar CRF levels; Boutros et al. 2016). Similarly, 20 days after the last ethanol exposure, adolescent, but not adult rats, were found to have a long-lasting decrease in time spent in the open arms of a radial arm maze, consistent with deficits in risk-reward integration (Risher et al. 2013). Further, this study found a lasting change in sensitivity to acute ethanol disruption of working memory (Risher et al. 2013), and another study using delayed discounting found that acute ethanol weeks after adolescent exposure altered impulsivity (Mejia-Toiber et al. 2014). These studies are consistent with findings indicating that adolescent ethanol exposure alters adult reward responses to ethanol (Boutros et al. 2014). Taken together, these findings and others are consistent with adolescent ethanol causing persistent changes in brain that increase risk for addiction in adulthood (Crews et al. 2016). These persistent changes in behavior following adolescent stress or alcohol exposure may be linked to induction of immune signaling in brain.
Over the past decade, the innate immune system has emerged as a critical component in the development of alcohol use disorders and other addictions. Microglia, the resident macrophage-like innate immune cells of the central nervous system, have only recently been discovered to play a critical role in brain homeostasis with key roles in neuronal differentiation, synapse formation, and neurocircuitry. It was only recently discovered that on embryonic day 8 in mice, mesodermal progenitor yolk sac cells migrate to brain to form microglia that persist throughout life as unique brain cells, distinct from the neuro-ectodermal cells that form neurons and other glia (e.g., astrocytes and oligodendrocytes) (Ginhoux et al. 2010, 2013). Originating from the mesoderm, microglia express a multitude of innate immune signaling receptors and molecules. One microglia-associated receptor system, the toll-like receptor (TLR) superfamily (i.e., interleukin-1 receptor/toll-like receptor), mounts proinflammatory responses to pathogens by sensing large molecules containing lipids, sugars, protein, and nucleic acid components. Initial studies suggested that only microglia are involved in TLR signaling, but more recent studies suggest that all brain cell types are involved in this signaling (Crews and Vetreno 2016). Toll-like receptors activate signaling cascades that converge on nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), a key innate immune nuclear transcription factor that promotes expression of proinflammatory cytokines, including tumor necrosis factor α (TNFα), interleukin-1β (IL-1β), IL-6, and monocyte chemoattractant protein-1 (MCP-1). Recent studies in brain, which is normally a sterile environment, indicate that endogenous TLR agonists may comprise an important signaling pathway between microglia, other glia, and neurons. The ubiquitously expressed nuclear protein, high-mobility group box 1 (HMGB1), is a cytokine-like molecule expressed in all cell types that upon release can directly activate TLRs and enhance the response of other innate immune signaling molecules at their respective receptors (see Fig. 1). Human and preclinical animal studies find that stress (Frank et al. 2015), alcohol (Crews and Vetreno 2014), and other drugs of abuse (Hutchinson et al. 2010; Northcutt et al. 2015) increase expression of TLRs and other innate immune signaling molecules (e.g., HMGB1) in brain. Induction of TLRs and HMGB1, an endogenous TLR agonist, leads to activation of positive loops of amplification that result in a progressive and persistent increase in TLR and HMGB1 signaling. For example, expression of HMGB1 and multiple TLRs in the postmortem human orbitofrontal cortex are correlated with lifetime alcohol consumption in alcoholic and moderate drinking controls (Crews et al. 2013). Cycles of binge drinking may sensitize the brain to promote drinking behavior (see Fig. 2). Similarly, mechanistic preclinical studies of epilepsy reveal that neuronal activation causes the rapid release of HMGB1 and IL-1β that occur prior to seizure onset resulting in neuronal sensitization to excitation due to alterations in excitatory receptor expression as well as lowered seizure threshold leading to increased seizure frequency and severity with each event (i.e., “kindling”; Maroso et al. 2011). While the brain regions and neural circuits underlying addiction and epilepsy differ, stress and drug abuse may share a common mechanism of neurocircuitry sensitization through increased HMGB1 and TLR signaling.

High-mobility group box 1 (HMGB1) signaling involvement in addiction neuropathology. HMGB1 is actively and/or passively released from neurons and other cells leading to the activation of multiple innate immune signaling pathways. Extracellularly, HMGB1 can directly interact with toll-like receptors (TLRs) or form complexes with various ligands to enhance immune responses at various pattern recognition receptors (PPRs). In addition, HMGB1 can form complexes with nucleic acids and microRNAs (miRNAs; e.g., let-7) that are endocytosed into the cell-activating intracellular TLRs. Activation of TLRs and other PRRs leads to activation of the transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and subsequent induction of proinflammatory cytokines and oxidases that are released into the extracellular space. Evidence implicates these cascades in contributing to the progression through the stages of addiction. LPS lipopolysaccharide, RAGE receptor for advanced glycation end-products, IL-1β interleukin-1beta

Lifetime alcohol consumption and HMGB1-TLR expression in the human postmortem orbitofrontal cortex. Expression of toll-like receptors (TLRs) 2, 3, and 4 and the TLR endogenous agonist high-mobility group box 1 (HMGB1) are positively correlated with lifetime alcohol consumption (kg) (Crews et al. 2013). Moderate drinking controls are clustered along the left of the graph due to low lifetime levels of alcohol consumption and concomitant low levels of TLR-HMGB1 expression. Alcoholic subjects all consumed more alcohol than moderate drinking controls but show a 10-fold variation in lifetime alcohol consumption. Note that the x axis is broken to allow for visualization of the moderate drinking control data. Repeated cycles of binge drinking are hypothesized to induce a progressive and persistent shift in the allostatic set point for negative affect that contributes to the neurobiology of addiction (Koob and Le Moal 2005). Depicted are correlations for individual TLRs and HMGB1 and overall grouped correlations with lifetime alcohol consumption
Preclinical models reveal that ethanol increases the expression of HMGB1 and TLRs in the brain that persists through long periods of abstinence (Vetreno and Crews 2012; Vetreno et al. 2013). Human studies find increased expression of microglial markers and cytokines as well as HMGB1 and TLRs in the postmortem alcoholic brain (Crews et al. 2013; He and Crews 2008; Zou and Crews 2012) with the latter two correlating with lifetime alcohol consumption (Crews et al. 2013). Repeated stress and cycles of alcohol and drug abuse sensitize microglia consistent with the hypothesis that induction of innate immune signaling pathways contributes to the progressive increase in craving, mood dysfunction, and cognitive impairments observed in addiction. Recent discoveries indicate that ethanol leads to the induction of multiple TLRs as well as endogenous TLR agonists in brain. While poorly understood at present, studies suggest that activation and escalated signaling of this system lead to a progressive loss of behavioral control, increased impulsivity and anxiety, and negative affect and craving coupled with increasing ventral striatal responses to promote reward-seeking behaviors and increase the risk of developing alcohol use disorders.
The innate immune system
While early studies characterized innate immune receptors based on their response to specific pathogens (e.g., bacteria and viruses), more recent “sterile inflammation” studies in brain led to the identification of these receptors as pattern recognition receptors (PRRs). Pattern recognition receptors have revolutionized our understanding of innate immune system signaling in brain. These receptors recognize and respond not only to specific molecular patterns present on foreign (exogenous) pathogens (i.e., pathogen-associated molecular patterns [PAMPS]) but also to endogenous signaling molecules associated with cell damage, degeneration, or stress (i.e., danger-associated molecular patterns [DAMPs]) (Bianchi 2007). To date, five classes of PRRs have been characterized: (1) TLRs, (2) C-type lectin receptors, (3) nucleotide-binding oligomerization domain-like receptors (NOD-like receptors), (4) RIG-I-like receptors, and (5) AIM2-like receptors (Brubaker et al. 2015). Although all of these receptors are important for host defense, the TLR family of PRRs has most thoroughly been characterized using inflammagens, such as endotoxin/lipopolysaccharide at TLR4 and viral RNA at TLR3 and TLR7. Toll-like receptors share an extracellular N-terminal leucine-rich repeat sequence and an intracellular toll/interleukin-1 receptor/resistance motif (TIR; Takeuchi and Akira 2010). Currently, 10 TLRs have been identified in humans and 12 TLRs have been identified in mice (Brubaker et al. 2015). All of these PRRs recognize a variety of PAMPs, including bacterial endotoxin and viral RNA as well as DAMPs such as mammalian HMGB1, microRNAs, and heat shock proteins (Vabulas et al. 2002). Interestingly, DAMP activation of TLRs has been identified as a contributing factor in several non-infectious neurological disorders, including alcoholism. For instance, expressions of TLR2, TLR3, and TLR4 as well as the PRR receptor for advanced glycation end-products (RAGE) and the TLR/RAGE endogenous agonist HMGB1 are each increased in the postmortem human alcoholic orbitofrontal cortex (Crews et al. 2013; Vetreno et al. 2013). Preclinical models of adolescent binge ethanol treatment find long-term upregulation of TLR3, TLR4, RAGE, and proinflammatory cytokines in the adult frontal cortex (Vetreno and Crews 2012) as well as several TLRs (TLR1–TLR10 except for TLR9) in the adult cerebellum (see Fig. 3). Immune signaling through TLRs and other immune signaling receptors induce additional TLRs and other innate immune signaling agonists and their respective receptors, which further induce immune signals, creating positive loops of activation that likely amplify and contribute to the persistence of drug-induced immune signaling. Similarly, other studies report that chronic ethanol exposure increases expression of TLR2 in the mouse brain (Fernandez-Lizarbe et al. 2013; Oak et al. 2006). Thus, both human and animal studies find that alcohol upregulates the expression of multiple TLRs.

Adolescent intermittent ethanol (AIE) treatment upregulates expression of toll-like receptors (TLRs) in the adult cerebellum. From postnatal day (P)25 to P55, AIE-treated male Wistar rats received a single daily intragastric dose of ethanol (5.0 g/kg, 20% ethanol, w/v) on a 2-day on/off schedule, and CON subjects received comparable volumes of water as described previously (Vetreno et al. 2016). Following a 25-day abstinence period, cerebellar tissue samples were collected on P80. Tissue samples were processed for qPCR and TLR mRNAs assessed as previously described (Vetreno and Crews 2012). Data are presented as mean ± SEM. *p < 0.05
In order for these innate immune signaling molecules to remain elevated for extended periods of abstinence, signaling amplification of these signaling molecules likely occurs. Ethanol causes activation of the transcription factor NF-κB, leading to increased PRR expression, cytokine induction, and DAMP release. This signaling system leads to amplification of the innate immune response in brain through an autocrine and paracrine process such that secreted cytokines, DAMPs, and microRNAs activate receptors on the cells of origin as well as surrounding cells. In addition, ethanol causes generation of reactive oxygen species that further activates NF-κB transcription of cytokines and DAMPs in an autocrine and paracrine fashion (Pietri et al. 2005; Qin and Crews 2012b; Thakur et al. 2007). Thus, the gradual induction of inflammatory processes that accompanies repeated binge-drinking episodes, particularly during adolescence, leads to a progressive and persistent activation of the innate immune system and might underlie the transition from alcohol abuse to alcoholism.
As alluded to above, TLR signaling pathways are quite complex. Following recognition of a ligand by a specific TLR, the adaptor protein TIRAP/MyD88, which interacts with all of the TLRs except TLR3, initiates intracellular signaling cascades leading to downstream activation of IL-1 receptor-associated kinases (IRAKs) and TNF receptor-associated factor 6 (TRAF6), which cause IκB and MAPK activation. Activation of IκB and MAPK in turn leads to downstream activation of the transcription factor NF-κB and subsequent cytokine induction (Crews et al. 2013; Lippai et al. 2013b). NF-κB is a critical modulator of innate immune function, and exposure to stress and drugs of abuse result in NF-κB activation. For instance, psychosocial stress induces NF-κB activation in human blood mononuclear cells (Bierhaus et al. 2003). In rodents, restraint stress increases expressions of NF-κB, proinflammatory cytokines, and the proinflammatory oxidase cyclooxygenase-2 (COX-2) in brain (Madrigal et al. 2002, 2003). Ethanol also activates NF-κB in rat and mouse brain (Qin and Crews 2012b; Ward et al. 1996), and NF-κB target genes are upregulated in the postmortem human alcohol prefrontal cortex (Okvist et al. 2007). Cocaine exposure also leads to activation of NF-κB in the nucleus accumbens and is required for the establishment of cocaine-induced, conditioned place preference (Ang et al. 2001a; Russo et al. 2009). Thus, both stress and drugs of abuse lead to activation of the innate immune transcription factor NF-κB in the brain that might contribute to innate immune induction.
Innate immunity, TLR signaling, and addiction
There is evidence consistent with the hypothesis that stress- and drug-induced innate immune upregulation inactivates the prefrontal cortex and sensitizes limbic circuitry. This suggests that the progressive and persistent increase of HMGB1-TLR signaling occurring with each cycle of stress and drug abuse is the mechanism underlying addiction (Crews et al. 2011; Vetreno and Crews 2014). Induction of innate immune signaling is known to cause cognitive and emotive dysfunctions in both preclinical (Dantzer et al. 2008; Hanke and Kielian 2011; Okun et al. 2010; Yirmiya and Goshen 2011) and human studies (Brites and Fernandes 2015; Critchley and Harrison 2013; Harrison et al. 2015; Rea et al. 2016). Alcohol and other drugs of abuse promote innate immune induction (Crews et al. 2016; He and Crews 2008; Qin et al. 2008) that is associated with alterations in executive function, reinforcement, and affective processes that promote alcohol abuse and addiction (Vetreno and Crews 2014). Indeed, ethanol induction of innate immune activation and TLR signaling contribute to brain alterations and the development of addiction-like behaviors (Liu et al. 2011b; Montesinos et al. 2016; Pascual et al. 2011, 2015). For instance, Guerri and colleagues (Pascual et al. 2011) found that binge ethanol-induced innate immune activation in mice impairs novel object recognition memory and increases anxiety-like behavior, an effect that is not found in TLR4 knockout mice. Interestingly, the observed behavioral impairments in this study were accompanied by reduced H3 and H4 histone acetylation as well as diminished histone acetyltransferase activity in the frontal cortex, striatum, and hippocampus, which was also not observed in TLR4 knockout mice. The finding that mice lacking TLR4 do not evidence ethanol-induced changes in epigenetic markers suggests that TLR-innate immune signaling contributes to drug-induced epigenetic changes leading to alterations in neurotransmitter signaling related to addiction. Similarly, adolescent binge ethanol exposure increases both ethanol preference and cocaine-conditioned place preference in young adult rodents, which was not observed in TLR4 knockout mice (Montesinos et al. 2016) or adult rats following treatment with trichostatin A, a histone deacetylase inhibitor (Pandey et al. 2015). These findings suggest that TLRs, specifically TLR4 in these experiments, are required for chronic ethanol-induced behavioral and histone acetylation changes in brain. TLR-innate immune induction appears to drive epigenetic and behavioral modifications that likely play a role in the dysfunctional cognitive, emotive, and reinforcement processes observed in addiction, since mice lacking TLR4 do not show these ethanol responses (Montesinos et al. 2016). Although the epigenetic mechanism underlying these effects is unclear, the data suggest that these changes might be due in part to increased innate immunity and diminished neurotropic expression that leads to alterations in synaptic plasticity (see Fig. 4). Indeed, adolescent intermittent ethanol (AIE) -induced activation of astrocytes, which are an important component of the innate immune system (Farina et al. 2007), is associated with increased expression of thrombospondins, which regulate synapse formation (Christopherson et al. 2005) in vitro and in the adult hippocampus (Risher et al. 2015). Thus, alcohol exposure may increase neuroplasticity in brain regions associated with addiction. An additional inflammatory pathway mediated by RAGE might also contribute to addiction and the cognitive impairments associated with alcoholism. Studies in the postmortem human alcoholic brain and AIE model find increased expression of RAGE in the adult prefrontal cortex (Vetreno et al. 2013), and this receptor has been implicated in memory impairments associated with Alzheimer’s disease (Arancio et al. 2004; Fang et al. 2010; Maczurek et al. 2008; Wilson et al. 2009). Together, these data connect TLRs, RAGE, HMGB1, and other innate immune signaling molecules with epigenetic modifications to drug- and stress-induced changes in neurobiology that are related to addiction.

Simplified schematic depicting mechanisms of alcohol-induced alterations to brain plasticity. Alcohol binge drinking and abuse increases expression of the endogenous innate immune receptor agonist high-mobility group box 1 (HMGB1) leading to activation of the transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and subsequent induction of proinflammatory cytokines and oxidases (Crews et al. 2013; Qin and Crews 2012a; Vetreno and Crews 2012, 2015; Vetreno et al. 2016). This also leads to increased expression of toll-like receptors (TLRs) and the receptor for advanced glycation end-products (RAGE) (Crews et al. 2013; Vetreno and Crews 2012, 2015; Vetreno et al. 2013), which are receptors for HMGB1. Activation of this signaling pathways leads to the establishment of positive loops of amplification that persist during abstinence from alcohol (Crews and Vetreno 2016) and alter synapse formation through increased expression of thrombospondins (Risher et al. 2015) and alterations in extracellular matrix proteins (Coleman et al. 2014). Concomitantly, innate immune induction causes epigenetic modifications that induce further release of HMGB1 (Zou and Crews 2014) that contribute to positive loops of amplification as well as alterations in synaptic plasticity molecules (Pandey et al. 2015; Sakharkar et al. 2016) that contribute to alterations in cognitive and emotive functioning. Innate immune driven epigenetic modifications might also lead to microglial alterations resulting in the reprogramming of the innate immune system. Innate immune induction also causes neurodegeneration (Braun and Crews 2000; Crews et al. 2000) and the loss of neuron-specific cell populations (Boutros et al. 2015; Vetreno et al. 2014; Vetreno and Crews 2015; Vetreno et al. 2016) that might also contribute to the dysfunctional cognitive and affective states observed in addiction (Crews and Vetreno 2016)
Although TLRs are capable of sensing and responding to a multitude of ligands, HMGB1 has been identified as a critical mediator of innate immune induction in alcohol abuse. HMGB1 is a nuclear protein involved in chromatin stabilization and acts as a chaperone for various transcription factors (Thomas and Stott 2012). Following extracellular release, HMGB1 binds to TLR4 or RAGE depending on its redox state causing induction of innate immune responses (Janko et al. 2014). Specifically, the Cys106 thiol/Cys23-Cys45 disulfide form of HMGB1 acts at TLR4 while the fully reduced form of HMGB1 is active at the RAGE receptor (Venereau et al. 2012; Yang et al. 2012). Ethanol has been found to cause nuclear translocation of HMGB1 through an epigenetic process leading to cellular release in ex vivo slice culture (Zou and Crews 2014). Expression of HMGB1 is increased in the prefrontal cortex of postmortem human alcoholics and adult rats following adolescent binge ethanol exposure (Crews et al. 2013; Vetreno and Crews 2012; Vetreno et al. 2013). In addition to direct receptor binding, extracellular HMGB1 can form heterodimers with a number of molecules, including lipoglycans, endotoxin, cytokines (e.g., IL-1β), and nucleic acids (e.g., microRNAs) (Bianchi 2009; Hreggvidsdottir et al. 2009), resulting in increased potency of immune responses. Further, HMGB1 is necessary for the induction of immune responses by endosome nucleic acid-binding TLRs (i.e., TLR3, TLR7, and TLR9) (Yanai et al. 2009), although the mechanism remains to be fully elucidated. Thus, alcohol-induced release of HMGB1 might activate multiple TLRs contributing to innate immune induction in brain.
In addition to HMGB1-TLR signaling pathways, ethanol also modulates innate immune function through the release of microRNAs (miRNAs), which are small, non-coding RNAs (∼22 nucleotides) that regulate innate immunity (Thounaojam et al. 2013). At present, miRNAs are thought to regulate innate immunity through two processes that involve (1) the stabilization of target mRNAs in the cytosol (Ambros 2004; Bartel 2004; Czech and Hannon 2011) and (2) extracellular release of miRNAs (Ambros 2004; Bartel 2004; Czech and Hannon 2011). Previous studies have found that upregulation of miRNAs is associated with psychiatric disorders (O’Connor et al. 2016) and addiction (Dreyer 2010), although the precise mechanism and impact on innate immune signaling are uncertain. In the extracellular milieu, miRNAs are either bound to lipoprotein chaperones or encapsulated in extracellular vesicles (EVs) that can be endocytosed by neighboring cells, thereby modulating their function. Ethanol exposure has been found to alter miRNA expression in the brain. Encapsulated miRNAs from ethanol-treated monocytes were found to modulate the activation state of naïve monocytes (Saha et al. 2016). Further, alcohol alters the expression profile of multiple miRNAs in the frontal cortex of postmortem human alcoholics and mice (Lewohl et al. 2011; Nunez and Mayfield 2012; Nunez et al. 2013). Interestingly, the miRNAs let-7 and miR-21 were recently identified as endogenous endosomal TLR7 agonists (Lehmann et al. 2012; Yelamanchili et al. 2015), and our laboratory found that ethanol potentiates TLR7-mediated innate immune induction through the release of let-7 in microvesicles (Coleman et al. 2017). The miRNA miR-155 is also found in vesicles and promotes TLR4-associated innate immune responses following chronic ethanol exposure (Lippai et al. 2013a). Although poorly understood at present, the release of miRNAs is an additional mechanism through which ethanol activates TLRs and the innate immune system, thereby contributing to the stages of addiction.
Animal studies provide compelling evidence that the innate immune system modulates alcohol consumptive behaviors. Analysis of genetically paired rats and mice reveals that high ethanol-drinking animals evidenced higher levels of NF-κB and other proinflammatory genes relative to their low-drinking litter mates (Mulligan et al. 2006). Indeed, expression of β2-microglobulin, a NF-κB target gene involved in immune signaling (Pahl 1999), was elevated in high ethanol-preferring brain transcriptomes (Mulligan et al. 2006). Studies in transgenic mouse lines provide further support for the hypothesis that the innate immune system regulates ethanol-drinking behaviors. Across multiple ethanol-drinking paradigms, transgenic mice with deletion of a single immune gene, such as CD14 and IL-6, consumed significantly less ethanol than matched wild-type controls (Blednov et al. 2005b, 2012). Further, Blednov and colleagues (Blednov et al. 2011) found that a single dose of LPS, the prototypical TLR4 agonist, produced a delayed but long-lasting increase of ethanol self-administration in mice. Similarly, site-specific microinjections of a GABAAα2 small interfering RNA (siRNA) vector into the central nucleus of the amygdala of alcohol-preferring rats diminished binge drinking, which was associated with reduced expression of TLR4 (Liu et al. 2011a). As GABAAα2 receptors are located on neurons, these data suggest that involvement of TLR4 in binge drinking is at least partially mediated by neurons. Indeed, both postmortem human and rodent studies find that TLR2–TLR4, RAGE, and HMGB1 are expressed on neurons (Crews et al. 2013; Vetreno and Crews 2012; Vetreno et al. 2013). Thus, there is accumulating data for a neuromodulatory role of the innate immune system in driving alcohol preference and consumption.
The frontal cortex mediates executive functions, including motivation, planning and goal setting, and behavioral flexibility. In non-alcoholic social drinking humans, the heaviest binge drinkers report more negative mood and perform worse on executive functioning tasks (Townshend and Duka 2003; Weissenborn and Duka 2003), suggesting that diminished frontal cortical function occurs with binge drinking. In astrocytes, ethanol causes NF-κB transcription and downstream induction of proinflammatory innate immune genes (Pascual et al. 2007; Zou and Crews 2006, 2010) as well as a loss of astrocyte glutamate transport (Zou and Crews 2005). Elevated levels of extracellular glutamate result in increased neuronal excitation, microglial activation, and excitotoxicity (Ward et al. 2009; Zou and Crews 2006). In the frontal cortex, ethanol-induced glutamate excitotoxicity results in increased caspase-3 and COX-2 expressions that require TLR4 signaling (Alfonso-Loeches et al. 2010; Knapp and Crews 1999). Similar glutamate hyperexcitability also occurs in the stimulant-addicted brain (Reissner and Kalivas 2010). Activation of the innate immune system reduces glutamate transporters, causing hyperexcitability that reduces frontal cortical executive function, thereby contributing to the neurobiology of addiction (Crews et al. 2006a, 2011). Indeed, frontal cortical dysfunction is common in the alcoholic brain (Crews and Boettiger 2009) and is manifest in impulsivity and impaired behavioral flexibility. In preclinical models, reversal learning tasks are used to assess frontal cortical function. Reversal learning provides a measure of behavioral flexibility, which refers to the animals’ ability to adopt a new behavioral response as a consequence of a shift in task demands. Typically, impairments in reversal learning are associated with increased perseveration of previously learned behaviors or the inability to break previously learned responses. Studies from our laboratory have consistently revealed persistent reversal learning deficits in adult rats following binge ethanol exposure (Obernier et al. 2002c) as well as in adult rats and mice following AIE treatments (Coleman et al. 2011; Vetreno and Crews 2012). Interestingly, we found that measures of reversal learning and perseveration were correlated with expression of TLRs, RAGE, and HMGB1 in the adult prefrontal cortex following AIE (Vetreno and Crews 2012; Vetreno et al. 2013) (see Fig. 5a). Additionally, deficits in reversal learning are observed in rats that self-administer cocaine or are exposed to passive cocaine injections (Calu et al. 2007; Schoenbaum et al. 2004), suggesting that behavioral flexibility deficits are common across addiction. Involvement of the frontal cortex in reversal learning is further supported by lesion-induced reversal learning deficits that are similar in nature to chronic drug abuse-induced dysfunction (Schoenbaum et al. 2006). Adolescent binge drinking is associated with increased impulsivity (White et al. 2011) and risky decision-making (Goudriaan et al. 2007). In preclinical models, risk-based decision-making is assessed using the probability-discounting task, in which rats must select either a small reward (single food pellet) that is always delivered (i.e., safe reward) or a large reward (four food pellets) that is delivered with decreasing frequency (i.e., risky choice) across trials. Elegant studies by Markou and colleagues (Boutros et al. 2015) investigated risk-based decision-making in rats using a probability-discounting task, in which rats must select either a small reward (single food pellet) that is always delivered (i.e., safe reward) or a large reward (four food pellets) that is delivered with decreased frequency (i.e., risky choice) across trials. Control animals switched from the high-reward level when more responses were required, reducing pellets, whereas AIE-treated animals demonstrated an increased preference for the risky large reward, even when many more responses were required. This suggests that AIE increased adult risky decisions (Boutros et al. 2015). Interestingly, increased risky decision-making was negatively correlated with expression of choline acetyltransferase, a marker of cholinergic neurons, in the basal forebrain, also reduced by AIE (see Fig. 5b). Basal forebrain cholinergic neurons provide acetylcholine inputs to the frontal cortex, which are important modulators of frontal cortical executive function (Baxter and Chiba 1999) and impulsivity (Sarter and Paolone 2011). Treatment of rats with LPS, the TLR4 agonist, causes a reduction of cholinergic neurons similar to the effects of AIE on cholinergic populations in adulthood. These data implicate innate immune activation through TLR4 in the loss of specific neuron populations that might contribute to behavioral deficits commonly observed in addiction. Taken together, considerable evidence is emerging that supports a role for HMGB1/TLR4 signaling, innate immune gene induction, and epigenetic alterations as culminating in the neurobiology of addiction (Crews et al. 2011; Cui et al. 2015; Vetreno and Crews 2014).

Adolescent intermittent ethanol (AIE) treatment impairs reversal learning and increases risky decision-making in adulthood. a From postnatal day (P)25 to P55, AIE-treated male Wistar rats received a single daily intragastric (i.g.) dose of ethanol (5.0 g/kg, 20% ethanol, w/v) on a 2-day on/off schedule, and CON subjects received comparable volumes of water as previously described (Vetreno and Crews 2012). Following a 25-day abstinence period, spatial and reversal learning were assessed on the Barnes maze. While AIE treatment did not impair spatial learning, reversal learning, which provides a measure of behavioral flexibility, was impaired in adulthood as evidenced by increased latency to the reversal goal and increased perseveration. Following behavioral testing, brain tissue was collected and tissue stained for innate immune markers. Expression of toll-like receptor 4 (TLR4) was positive correlated with latency to the reversal goal, indicating that innate immune induction by AIE contributes to deficits in adult deficits in behavioral flexibility. Adopted from Vetreno and Crews (2012). Data are presented as mean ± SEM. *p < 0.05. b From P28 to P53, male Wistar rats received three daily i.g. doses of ethanol (5.0 g/kg, 25% ethanol, w/v) on a 2-day on/off schedule, and CON subjects received comparable volumes of water. Following an abstinent period on P63, training and testing were performed on the probability-discounting task. Prior AIE treatment significantly increased risky responses at reducing reward probabilities, relative to CON subjects. Expression of choline acetyltransferase (ChAT), a marker of cholinergic neurons, was reduced in the basal forebrain of adult AIE-treated animals that was negatively correlated with probability-discounting behavior. Adopted from Markou and colleagues (Boutros et al. 2015). Data are presented as mean ± SEM. *p < 0.05
Addiction, stress, and innate immune signaling
Cycles of exposure to drugs of abuse and stress interact, contributing to a progressive sequence of stages leading to addiction (Volkow et al. 2016). Both drugs of abuse and stress activate innate immune proinflammatory responses. Stress-induced innate immune responses can have beneficial or maladaptive consequences on brain function. Physical stressors, such as injury and infection, induce proinflammatory cytokines and other signaling molecules that trigger adaptive behavioral changes known as “sickness behavior.” Sickness behaviors, which include social withdrawal, decreased activity, somnolence/sleepiness, and anhedonia, help facilitate energy conservation and recovery from illness (Dantzer et al. 2008). Interestingly, even psychological stressors (e.g., social stress) can induce inflammatory responses (Steptoe et al. 2007). The adaptive response of sickness behavior to acute stress and inflammation is presumably beneficial; however, prolonged or chronic exposure to stress and inflammation can become maladaptive, leading to neuropsychiatric disease, such as depression. Indeed, the similarities between sickness behavior and depression are well known (Maes et al. 2012). Chronic innate immune activation following prolonged alcohol and drug abuse likely results in similar psychopathologies. Thus, persistent innate immune induction caused by chronic alcoholism might lead to maladaptive sickness behavior that manifests as cognitive and emotive dysfunctions.
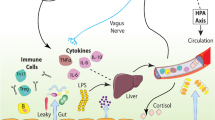
Recent studies find that stress and alcohol contribute to inflammation through a similar mechanism of increased intestinal permeability. Stress causes intestinal permeability and bacterial translocation from the gut lumen (Garate et al. 2013). In response to the leaked bacteria, the peripheral immune system mounts a proinflammatory immune response. At binge levels, alcohol (i.e., >2 g/kg; Ferrier et al. 2006) also causes disruption of gut tight junctions, allowing for the translocation of gut bacteria into the periphery (Adachi et al. 1995). The bacteria enter portal circulation leading to inflammation of the liver and release of proinflammatory cytokines into systemic circulation (Crews and Vetreno 2014; Mayfield et al. 2013) (see Fig. 6). This has also been observed in humans, with increased bacterial endotoxin and 16S RNA after alcohol (Bala et al. 2014). The peripheral inflammatory response can then impact the brain and behavior through several routes (Critchley and Harrison 2013; Miller and Raison 2016). Systemic changes in cytokines, such as IL-1β, activate IL-1 receptors located on the vagus nerve (Ek et al. 1998). Vagal activation is transmitted to neural centers within the brain to promote the induction of sickness behaviors. Indeed, vagotomy reduces the sickness behavioral response to a peripheral injection of LPS, the TLR4 agonist (Bluthe et al. 1994). Secondly, peripheral cytokines can influence brain and behavior through transport in the blood. Proinflammatory cytokines in the blood can diffuse through permeable regions of the blood-brain barrier (BBB), such as the circumventricular organs, or be transported across the blood-brain barrier (Banks and Erickson 2010; Qin et al. 2007). TNFα transporters on the BBB are essential for systemic inflammation to cause brain inflammation (Qin et al. 2007). Similarly, in humans, circulating TNFα alters availability of the brain serotonin transporter (Krishnadas et al. 2016) and activation of immune signaling following vaccination causes confusion, fatigue, and loss of motivation (Harrison et al. 2009b). Fatigue following experimental inflammation in humans induces altered cortical metabolism and microstructure of the insular cortex (Harrison et al. 2015). Activated immune cells such as monocytes can be trafficked into the brain; however, this mechanism has been questioned with recent discoveries on microglial origination (Ginhoux et al. 2013). Once activated in brain, the innate immune response persists (Qin et al. 2008), thereby contributing to the neurobiology of addiction through the induction of alcohol consumptive behaviors as well as cognitive and emotive dysfunctions.

Mechanisms of stress- and ethanol-induced innate immune activation. Alcohol and stress activate the peripheral and central immune systems in multiple ways. Alcohol and stress cause microbiome dysbiosis and disrupt gut tight junctions, leading to permeability of the gut and release of bacteria and endotoxin that enter portal circulation-inducing inflammatory responses in the liver. The consequent release of inflammatory cytokines from the liver activates the innate immune system in brain through direct transport via cytokine receptors and activation of the vagus nerve. Stress and alcohol exposure leads to innate immune activation in brain that induces sickness-like behaviors that contribute to the stages of addiction. These pathways of ethanol- and stress-induced activation of HMGB1-LTR signaling likely contribute to the persistent and progressive stages of addiction
Innate immunity may contribute to progressive stages of addiction
Current theories conceptualize addiction as encompassing a three-stage cycle that includes (1) binge/intoxication, (2) withdrawal/negative affect, and (3) preoccupation/craving (Koob and Volkow 2010) that contribute to the maintenance of substance use disorders. Accumulating evidence suggests that the innate immune system plays a role in each stage of the addiction cycle. With regards to the binge/intoxication stage, blockade of cocaine-induced upregulation of IL-1β in the ventral tegmental area (VTA) with the (+)-isomer of naloxone, a TLR4 antagonist, prevented the subsequent increase of dopamine release in the nucleus accumbens and diminished cocaine self-administration (Northcutt et al. 2015). Interestingly, this effect may be mediated in part by TLR4 signaling in microglia as treatment with minocycline, a microglial inhibitor, blocked cocaine-induced, conditioned place preference, supporting a role for the innate immune system in the rewarding effects of drugs of abuse. Similarly, self-administration of ethanol in the drinking in the dark paradigm in mice has been shown to increase expression of IL-1β in the basolateral amygdala. Injections of IL-1-ra, an IL-1 receptor antagonist, into the basolateral amygdala reduced ethanol self-administration in these animals (Marshall et al. 2016). Further, delivery of siRNAs for TLR4 or MCP-1 into either the VTA or central nucleus of the amygdala blunted ethanol self-administration in alcohol-preferring rats (June et al. 2015). Across multiple ethanol-drinking paradigms, transgenic mice with deletion of a single immune gene, such as CD14 and IL-6, consumed significantly less ethanol than matched wild-type controls (Blednov et al. 2005a, 2012). Conversely, stimulation of the innate immune system has been found to elicit ethanol consumptive behaviors. Indeed, systemic administration of LPS to adult mice produced a delayed but long-lasting increase in ethanol self-administration in mice (Blednov et al. 2011). Similarly, ventricular infusions of MCP-1 increase ethanol self-administration in rats (Valenta and Gonzales 2016). Together, these data provide evidence that the innate immune system, including TLRs, is involved in the binge/intoxication stage of addiction.
There is also evidence implicating innate immune activation in the withdrawal/negative affect stage of addiction. Substance abuse is associated with the development of negative affective states, such as anxiety and dysphoria (Koob and Le Moal 2005), which are commonly observed during the withdrawal stage of addiction. Indeed, the negative states associated with withdrawal are likely a driving force behind continued substance use, thereby contributing to cycles of abuse that might lead to dependence. Evidence indicates that it is during ethanol withdrawal that proinflammatory cytokine induction increases in brain. Freeman et al. (2012) found that expressions of MCP-1, NOS-2, TNFα, and the TNFα receptor are increased in the central nucleus of the amygdala following 48 h of withdrawal. Administration of LPS and cytokines sensitize the anxiety- and depressive-like behaviors associated with ethanol withdrawal (Breese et al. 2008). Knockout of TLR4 protects against ethanol withdrawal associated anxiety-like behavior and memory impairment (Pascual et al. 2011). These data implicate TLR4 and the innate immune system in ethanol withdrawal and the development of negative affect, although the mechanism remains to be fully determined. A probable point of convergence of withdrawal-induced innate immune upregulation and negative affect is hippocampal neurogenesis. Hippocampal neurogenesis is involved in regulation of mood and affective states (Malberg et al. 2000), and ethanol and LPS treatments reduce neurogenesis (see, e.g., Vetreno and Crews 2015). Importantly, reductions in hippocampal neurogenesis are observed during ethanol withdrawal that was associated with increased depression-like behavior on the forced swim test in mice (Stevenson et al. 2009). Further, the effects of ethanol withdrawal were prevented by anti-depressant treatment.
Stress causes induction of innate immune signaling in brain and mounting evidence indicates that immune activation can lead to alterations in affective states. As mentioned in the previous section, stress sensitizes microglia to inflammation in an HMGB1-dependent manner (Weber et al. 2015), and chronic stress activates microglia in multiple brain regions (Tynan et al. 2010) and causes depression-like behavior. Administration of microglial-modulating agents can block the development of depression-like behavior in rodents (Kreisel et al. 2014). Similarly, human studies also provide support for the involvement of stress and inflammation in affective disorders. Patients receiving systemic interferon-α for the treatment of hepatitis C can develop depression-like symptoms in the absence of a prior history of depression (Bonaccorso et al. 2002). Individuals with increased plasma levels of C-reactive protein (CRP), an acute phase marker of inflammation, are at greater risk for the development of depression (Young et al. 2014), and increased plasma level of CRP in adolescents is predictive of addiction later in life (Costello et al. 2013). Further, peripheral inflammation, which can increase innate immune signaling in brain, has been found to contribute to negative affect (Harrison et al. 2014). Human PET neuroimaging studies also find that expression levels of translocator protein (18 kDa), a marker of reactive microglia, are increased in the prefrontal cortex, insula, and anterior cingulate cortex of individuals during a major depressive episode (Setiawan et al. 2015). In depressed individuals who commit suicide, expression levels of microglial markers (i.e., Iba-1 and CD45) and the proinflammatory cytokine MCP-1 are increased in the cingulate cortex (Torres-Platas et al. 2014). These studies suggest that systemic inflammation leads to increased innate immune signaling in brain, contributing to the development of affective disorders.
Evidence supports an involvement of the innate immune system in affective disorders, but the mechanism remains to be elucidated fully. The literature provides evidence that a potential mechanism for innate immune involvement in the development of affective disorders involves alterations in the activity of neural circuitry that modulates affect. Administration of a typhoid vaccine was found to increase circulating levels of the proinflammatory cytokine IL-6 and significantly reduce mood (Harrison et al. 2009a). Interestingly, functional magnetic resonance imaging in these same participants revealed that the inflammation-induced reduction in mood was correlated with increased activity in the subgenual anterior cingulate cortex, a region implicated in the etiology of depression, and reduced connectivity of this region with the amygdala, prefrontal cortex, and nucleus accumbens (Harrison et al. 2009a). Studies also find that a variety of cytokine receptors, such as those for TNFα, IL-1β, IL-6, and the interferons, are expressed on neurons (Khairova et al. 2009), suggesting that cytokines act directly on neurons to influence their activity. Indeed, innate immune induction can reduce populations of serotonin (5-HT)-producing neurons in the raphe nucleus, and the serotonergic system has also been implicated in the etiology of affective disorders (Kelley et al. 2011). We found that AIE treatment persistently reduces populations of 5-HT-immunopositive neurons in the adult raphe nucleus, an effect that was mimicked by treatment with the TLR4 agonist LPS. Inflammation also increases activity of indolamine 2,3-dioxygenase (IDO), an enzyme that converts tryptophan (a precursor of 5-HT) to kynurenine, thereby reducing the synthesis of 5-HT (Wichers and Maes 2004). Interestingly, kynurenine has also been shown to induce depression-like behavior, and pharmacological inhibition of IDO blocks microglial activation and the development of depression-like behavior (O’Connor et al. 2009). Induction of innate immunity can also disrupt hippocampal neurogenesis, the process whereby new neurons are continuously added to the existing hippocampal neurocircuitry. Although heavily implicated in learning and memory processes, neurogenesis is also involved in mood and affective states (Malberg et al. 2000). Further, the ability of anti-depressants to alleviate depression-like behavior appears to be dependent upon hippocampal neurogenesis (Santarelli et al. 2003). Both inflammation (Ryan and Nolan 2016) and chronic stress (Kreisel et al. 2014) reduce neurogenesis and cause depression-like behavior. Further, stress induces IL-1β in the hippocampus, which decreases neurogenesis and contributes to depression, while inhibition of IL-1β blocks stress-induced decreases in neurogenesis and depression-like behavior (Koo and Duman 2008). While these data provide compelling evidence that the innate immune system modulates affect, the mechanism likely involves alterations to discrete nuclei as well as modifications to connectivity between regions. Together, these data implicate the TLR/innate immune system in the withdrawal/negative affect stage of addiction.
Evidence also implicates the TLR-innate immune system in the third stage of addiction involving preoccupation with drug taking and craving, which is associated with a heightened likelihood of relapse. Plasma levels of the proinflammatory cytokines IL-8 and IL-1β were positively correlated with indices of alcohol craving in human alcohol-dependent subjects (Leclercq et al. 2014). As mentioned above, stress (Madrigal et al. 2002, 2003) and drugs of abuse (Ang et al. 2001b; Crews and Vetreno 2014, 2016; Vetreno and Crews 2014) activate and sensitize (Frank et al. 2007; Qin and Crews 2012a) the innate immune system in brain. The innate immune system modulates alcohol consumptive behaviors (Blednov et al. 2005b, 2011; 2012; Liu et al. 2011b), and stressful events during abstinence might interact with the primed innate immune system leading to the craving to consume alcohol and other drugs of abuse. Treatment of abstinent human alcoholics with naltrexone, a TLR4 antagonist, reduced subjective reporting of craving (Nava et al. 2006). Similarly, treatment with the (+)-isomer of naltrexone, which is inactive at the opioid receptor, blocked heroin-seeking behavior in dependent rats (Theberge et al. 2013). Thus, TLR4 and innate immune signaling molecules contribute to the preoccupation/craving stage of addiction. Taken together, the evidence suggests that activation of the innate immune system in brain contributes to each stage of addiction.
Summary
The discovery that innate immune mechanisms contribute to the neurobiology of addiction provides a novel approach for the treatment of alcohol use disorders. While innate immune activation contributes to alcohol consumptive behaviors, it also contributes to alcohol-induced neurodegeneration (Crews et al. 2004, 2006b; Obernier et al. 2002a, b; Qin et al. 2013). It is not clear how innate immune signaling alters specific neurocircuits. Innate immune signaling involves positive loops since cytokine and TLRs converge on NF-κB inducing additional cytokines and their receptors. However, HMGB1 and TLR activation could be the initial key signaling event. The current studies find innate immune signaling in most brain diseases with little evidence distinguishing activation in mental diseases and neurodegenerative diseases (e.g., addiction, Alzheimer’s disease, and depression). It remains to be determined whether therapies that target the innate immune system would be of benefit for the prevention of the progression to addiction and if these therapies could improve recovery from alcohol use disorders. Some studies have found that systemic anti-inflammatory agents reduce symptoms, but little is known on how brain inflammatory gene expression is impacted. Indeed, neurodegeneration would likely not be reversed, although interventions might allow for the recovery of normal synaptic function. Minocycline, a tetracycline antibiotic and microglial inhibitor (Plane et al. 2010), prevents ethanol induced-microglial activation and reduces alcohol self-administration (Agrawal et al. 2011; Qin and Crews 2012a). While studies are currently underway, further research is clearly warranted to explore the therapeutic potential of immune interventions in the treatment of alcohol use disorders.
References
Adachi Y, Moore LE, Bradford BU, Gao W, Thurman RG (1995) Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology 108:218–224
Agrawal RG, Hewetson A, George CM, Syapin PJ, Bergeson SE (2011) Minocycline reduces ethanol drinking. Brain Behav Immun 25(Suppl 1):S165–S169
Alfonso-Loeches S, Pascual-Lucas M, Blanco AM, Sanchez-Vera I, Guerri C (2010) Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. J Neurosci Off J Soc Neurosci 30:8285–8295
Ambros V (2004) The functions of animal microRNAs. Nature 431:350–355
Ang E, Chen J, Zagouras P, Magna H, Holland J, Schaeffer E, Nestler EJ (2001a) Induction of nuclear factor-kappaB in nucleus accumbens by chronic cocaine administration. J Neurochem 79:221–224
Arancio O, Zhang HP, Chen X, Lin C, Trinchese F, Puzzo D, Liu S, Hegde A, Yan SF, Stern A, Luddy JS, Lue LF, Walker DG, Roher A, Buttini M, Mucke L, Li W, Schmidt AM, Kindy M, Hyslop PA, Stern DM, Du Yan SS (2004) RAGE potentiates Abeta-induced perturbation of neuronal function in transgenic mice. EMBO J 23:4096–4105
Bala S, Marcos M, Gattu A, Catalano D, Szabo G (2014) Acute binge drinking increases serum endotoxin and bacterial DNA levels in healthy individuals. PLoS One 9:e96864
Banks WA, Erickson MA (2010) The blood-brain barrier and immune function and dysfunction. Neurobiol Dis 37:26–32
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Baxter MG, Chiba AA (1999) Cognitive functions of the basal forebrain. Curr Opin Neurobiol 9:178–183
Bianchi ME (2007) DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol 81:1–5
Bianchi ME (2009) HMGB1 loves company. J Leukoc Biol 86:573–576
Bierhaus A, Wolf J, Andrassy M, Rohleder N, Humpert PM, Petrov D, Ferstl R, von Eynatten M, Wendt T, Rudofsky G, Joswig M, Morcos M, Schwaninger M, McEwen B, Kirschbaum C, Nawroth PP (2003) A mechanism converting psychosocial stress into mononuclear cell activation. Proc Natl Acad Sci U S A 100:1920–1925
Blednov YA, Bergeson SE, Walker D, Ferreira VM, Kuziel WA, Harris RA (2005a) Perturbation of chemokine networks by gene deletion alters the reinforcing actions of ethanol. Behav Brain Res 165:110–125
Blednov YA, Bergeson SE, Walker D, Ferreira VM, Kuziel WA, Harris RA (2005b) Perturbation of chemokine networks by gene deletion alters the reinforcing actions of ethanol. Behav Brain Res 165:110–125
Blednov YA, Benavidez JM, Geil C, Perra S, Morikawa H, Harris RA (2011) Activation of inflammatory signaling by lipopolysaccharide produces a prolonged increase of voluntary alcohol intake in mice. Brain Behav Immun 25(Suppl 1):S92–S105
Blednov YA, Ponomarev I, Geil C, Bergeson S, Koob GF, Harris RA (2012) Neuroimmune regulation of alcohol consumption: behavioral validation of genes obtained from genomic studies. Addict Biol 17:108–120
Bluthe RM, Walter V, Parnet P, Laye S, Lestage J, Verrier D, Poole S, Stenning BE, Kelley KW, Dantzer R (1994) Lipopolysaccharide induces sickness behaviour in rats by a vagal mediated mechanism. C R Acad Sci III 317:499–503
Bonaccorso S, Marino V, Biondi M, Grimaldi F, Ippoliti F, Maes M (2002) Depression induced by treatment with interferon-alpha in patients affected by hepatitis C virus. J Affect Disord 72:237–241
Boutros N, Semenova S, Liu W, Crews FT, Markou A (2014) Adolescent intermittent ethanol exposure is associated with increased risky choice and decreased dopaminergic and cholinergic neuron markers in adult rats. Int J Neuropsychopharmacol 18
Boutros N, Semenova S, Liu W, Crews FT, Markou A (2015) Adolescent intermittent ethanol exposure is associated with increased risky choice and decreased dopaminergic and cholinergic neuron markers in adult rats. Int J Neuropsychopharmacol 18
Boutros N, Der-Avakian A, Semenova S, Lee S, Markou A (2016) Risky choice and brain CRF after adolescent ethanol vapor exposure and social stress in adulthood. Behav Brain Res 311:160–166
Braun CJ, Crews FT (2000) Binge ethanol treatment causes greater neurodegeneration in alcohol preferring (P) rats as compared to non-alcohol preferring (NP) rats. Alcohol Clin Exp Res 24:65A
Breese GR, Knapp DJ, Overstreet DH, Navarro M, Wills TA, Angel RA (2008) Repeated lipopolysaccharide (LPS) or cytokine treatments sensitize ethanol withdrawal-induced anxiety-like behavior. Neuropsychopharmacology 33:867–876
Brites D, Fernandes A (2015) Neuroinflammation and depression: microglia activation, extracellular microvesicles and microRNA dysregulation. Front Cell Neurosci 9:476
Brubaker SW, Bonham KS, Zanoni I, Kagan JC (2015) Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol 33:257–290
Calu DJ, Roesch MR, Stalnaker TA, Schoenbaum G (2007) Associative encoding in posterior piriform cortex during odor discrimination and reversal learning. Cereb Cortex 17:1342–1349
Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA (2005) Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120:421–433
Coleman LG Jr, He J, Lee J, Styner M, Crews FT (2011) Adolescent binge drinking alters adult brain neurotransmitter gene expression, behavior, brain regional volumes, and neurochemistry in mice. Alcohol Clin Exp Res 35:671–688
Coleman LG Jr, Liu W, Oguz I, Styner M, Crews FT (2014) Adolescent binge ethanol treatment alters adult brain regional volumes, cortical extracellular matrix protein and behavioral flexibility. Pharmacol Biochem Behav 116:142–151
Coleman Jr LG, Zou J, Crews FT (2017) Microglial-derived miRNA Let-7 and HMGB1 contribute to ethanol-induced neurotoxicity via TLR7. J Neuroinflammation
Costello EJ, Copeland WE, Shanahan L, Worthman CM, Angold A (2013) C-reactive protein and substance use disorders in adolescence and early adulthood: a prospective analysis. Drug Alcohol Depend 133:712–717
Crews FT, Boettiger CA (2009) Impulsivity, frontal lobes and risk for addiction. Pharmacol Biochem Behav 93:237–247
Crews FT, Vetreno RP (2014) Neuroimmune basis of alcoholic brain damage. Int Rev Neurobiol 118:315–357
Crews FT, Vetreno RP (2016) Mechanisms of neuroimmune gene induction in alcoholism. Psychopharmacology 233:1543–1557
Crews FT, Braun CJ, Hoplight B, Switzer RC 3rd, Knapp DJ (2000) Binge ethanol consumption causes differential brain damage in young adolescent rats compared with adult rats. Alcohol Clin Exp Res 24:1712–1723
Crews FT, Collins MA, Dlugos C, Littleton J, Wilkins L, Neafsey EJ, Pentney R, Snell LD, Tabakoff B, Zou J, Noronha A (2004) Alcohol-induced neurodegeneration: when, where and why? Alcohol Clin Exp Res 28:350–364
Crews F, Nixon K, Kim D, Joseph J, Shukitt-Hale B, Qin L, Zou J (2006a) BHT blocks NF-kappaB activation and ethanol-induced brain damage. Alcohol Clin Exp Res 30:1938–1949
Crews F, Nixon K, Kim D, Joseph J, Shukitt-Hale B, Qin L, Zou J (2006b) BHT blocks NF-kappaB activation and ethanol-induced brain damage. Alcohol Clin Exp Res 30:1938–1949
Crews FT, Zou J, Qin L (2011) Induction of innate immune genes in brain create the neurobiology of addiction. Brain Behav Immun 25(Suppl 1):S4–S12
Crews FT, Qin L, Sheedy D, Vetreno RP, Zou J (2013) High mobility group box 1/toll-like receptor danger signaling increases brain neuroimmune activation in alcohol dependence. Biol Psychiatry 73:602–612
Crews FT, Vetreno RP, Broadwater MA, Robinson DL (2016) Adolescent alcohol exposure persistently impacts adult neurobiology and behavior. Pharmacol Rev 68:1074–1109
Critchley HD, Harrison NA (2013) Visceral influences on brain and behavior. Neuron 77:624–638
Cui C, Noronha A, Warren KR, Koob GF, Sinha R, Thakkar M, Matochik J, Crews FT, Chandler LJ, Pfefferbaum A, Becker HC, Lovinger D, Everitt BJ, Egli M, Mandyam CD, Fein G, Potenza MN, Harris RA, Grant KA, Roberto M, Meyerhoff DJ, Sullivan EV (2015) Brain pathways to recovery from alcohol dependence. Alcohol 49:435–452
Czech B, Hannon GJ (2011) Small RNA sorting: matchmaking for Argonautes. Nat Rev Genet 12:19–31
Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW (2008) From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9:46–56
Dreyer JL (2010) New insights into the roles of microRNAs in drug addiction and neuroplasticity. Genome Med 2:92
Ek M, Kurosawa M, Lundeberg T, Ericsson A (1998) Activation of vagal afferents after intravenous injection of interleukin-1beta: role of endogenous prostaglandins. J Neurosci 18:9471–9479
Fang F, Lue LF, Yan S, Xu H, Luddy JS, Chen D, Walker DG, Stern DM, Schmidt AM, Chen JX, Yan SS (2010) RAGE-dependent signaling in microglia contributes to neuroinflammation, Abeta accumulation, and impaired learning/memory in a mouse model of Alzheimer’s disease. FASEB J 24:1043–1055
Farina C, Aloisi F, Meinl E (2007) Astrocytes are active players in cerebral innate immunity. Trends Immunol 28:138–145
Fernandez-Lizarbe S, Montesinos J, Guerri C (2013) Ethanol induces TLR4/TLR2 association, triggering an inflammatory response in microglial cells. J Neurochem 126:261–273
Ferrier L, Berard F, Debrauwer L, Chabo C, Langella P, Bueno L, Fioramonti J (2006) Impairment of the intestinal barrier by ethanol involves enteric microflora and mast cell activation in rodents. Am J Pathol 168:1148–1154
Frank MG, Baratta MV, Sprunger DB, Watkins LR, Maier SF (2007) Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav Immun 21:47–59
Frank MG, Weber MD, Watkins LR, Maier SF (2015) Stress sounds the alarmin: the role of the danger-associated molecular pattern HMGB1 in stress-induced neuroinflammatory priming. Brain Behav Immun 48:1–7
Freeman K, Brureau A, Vadigepalli R, Staehle MM, Brureau MM, Gonye GE, Hoek JB, Hooper DC, Schwaber JS (2012) Temporal changes in innate immune signals in a rat model of alcohol withdrawal in emotional and cardiorespiratory homeostatic nuclei. J Neuroinflammation 9:97
Garate I, Garcia-Bueno B, Madrigal JL, Caso JR, Alou L, Gomez-Lus ML, Mico JA, Leza JC (2013) Stress-induced neuroinflammation: role of the toll-like receptor-4 pathway. Biol Psychiatry 73:32–43
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330:841–845
Ginhoux F, Lim S, Hoeffel G, Low D, Huber T (2013) Origin and differentiation of microglia. Front Cell Neurosci 7:45
Goudriaan AE, Grekin ER, Sher KJ (2007) Decision making and binge drinking: a longitudinal study. Alcohol Clin Exp Res 31:928–938
Hanke ML, Kielian T (2011) Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clin Sci 121:367–387
Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Critchley HD (2009a) Inflammation causes mood changes through alterations in subgenual cingulate activity and mesolimbic connectivity. Biol Psychiatry 66:407–414
Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Critchley HD (2009b) Inflammation causes mood changes through alterations in subgenual cingulate activity and mesolimbic connectivity. Biol Psychiatry 66:407–414
Harrison NA, Doeller CF, Voon V, Burgess N, Critchley HD (2014) Peripheral inflammation acutely impairs human spatial memory via actions on medial temporal lobe glucose metabolism. Biol Psychiatry 76:585–593
Harrison NA, Cooper E, Dowell NG, Keramida G, Voon V, Critchley HD, Cercignani M (2015) Quantitative magnetization transfer imaging as a biomarker for effects of systemic inflammation on the brain. Biol Psychiatry 78:49–57
He J, Crews FT (2008) Increased MCP-1 and microglia in various regions of the human alcoholic brain. Exp Neurol 210:349–358
Hreggvidsdottir HS, Ostberg T, Wahamaa H, Schierbeck H, Aveberger AC, Klevenvall L, Palmblad K, Ottosson L, Andersson U, Harris HE (2009) The alarmin HMGB1 acts in synergy with endogenous and exogenous danger signals to promote inflammation. J Leukoc Biol 86:655–662
Hutchinson MR, Zhang Y, Shridhar M, Evans JH, Buchanan MM, Zhao TX, Slivka PF, Coats BD, Rezvani N, Wieseler J, Hughes TS, Landgraf KE, Chan S, Fong S, Phipps S, Falke JJ, Leinwand LA, Maier SF, Yin H, Rice KC, Watkins LR (2010) Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain Behav Immun 24:83–95
Janko C, Filipovic M, Munoz LE, Schorn C, Schett G, Ivanovic-Burmazovic I, Herrmann M (2014) Redox modulation of HMGB1-related signaling. Antioxid Redox Signal 20:1075–1085
June HL, Liu J, Warnock KT, Bell KA, Balan I, Bollino D, Puche A, Aurelian L (2015) CRF-amplified neuronal TLR4/MCP-1 signaling regulates alcohol self-administration. Neuropsychopharmacology 40:1549–1559
Kelley KW, Dantzer, Robert (2011) Alcoholism and inflammation: neuroimmunology of behavioral and mood disorders. Brain Behav Immun
Khairova RA, Machado-Vieira R, Du J, Manji HK (2009) A potential role for pro-inflammatory cytokines in regulating synaptic plasticity in major depressive disorder. Int J Neuropsychopharmacol 12:561–578
Knapp DJ, Crews FT (1999) Induction of cyclooxygenase-2 in brain during acute and chronic ethanol treatment and ethanol withdrawal. Alcohol Clin Exp Res 23:633–643
Koo JW, Duman RS (2008) IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc Natl Acad Sci U S A 105:751–756
Koob GF, Le Moal M (2005) Plasticity of reward neurocircuitry and the “dark side” of drug addiction. Nat Neurosci 8:1442–1444
Koob GF, Volkow ND (2010) Neurocircuitry of addiction. Neuropsychopharmacology 35:217–238
Kreisel T, Frank MG, Licht T, Reshef R, Ben-Menachem-Zidon O, Baratta MV, Maier SF, Yirmiya R (2014) Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol Psychiatry 19:699–709
Krishnadas R, Nicol A, Sassarini J, Puri N, Burden AD, Leman J, Combet E, Pimlott S, Hadley D, McInnes IB, Cavanagh J (2016) Circulating tumour necrosis factor is highly correlated with brainstem serotonin transporter availability in humans. Brain Behav Immun 51:29–38
Leclercq S, De Saeger C, Delzenne N, de Timary P, Starkel P (2014) Role of inflammatory pathways, blood mononuclear cells, and gut-derived bacterial products in alcohol dependence. Biol Psychiatry 76:725–733
Lehmann SM, Kruger C, Park B, Derkow K, Rosenberger K, Baumgart J, Trimbuch T, Eom G, Hinz M, Kaul D, Habbel P, Kalin R, Franzoni E, Rybak A, Nguyen D, Veh R, Ninnemann O, Peters O, Nitsch R, Heppner FL, Golenbock D, Schott E, Ploegh HL, Wulczyn FG, Lehnardt S (2012) An unconventional role for miRNA: let-7 activates toll-like receptor 7 and causes neurodegeneration. Nat Neurosci 15:827–835
Lewohl JM, Nunez YO, Dodd PR, Tiwari GR, Harris RA, Mayfield RD (2011) Up-regulation of microRNAs in brain of human alcoholics. Alcohol Clin Exp Res 35:1928–1937
Lippai D, Bala S, Csak T, Kurt-Jones EA, Szabo G (2013a) Chronic alcohol-induced microRNA-155 contributes to neuroinflammation in a TLR4-dependent manner in mice. PLoS One 8:e70945
Lippai D, Bala S, Petrasek J, Csak T, Levin I, Kurt-Jones EA, Szabo G (2013b) Alcohol-induced IL-1beta in the brain is mediated by NLRP3/ASC inflammasome activation that amplifies neuroinflammation. J Leukoc Biol 94:171–182
Liu J, Yang AR, Kelly T, Puche A, Esoga C, June HL Jr, Elnabawi A, Merchenthaler I, Sieghart W, June HL Sr, Aurelian L (2011a) Binge alcohol drinking is associated with GABAA alpha2-regulated toll-like receptor 4 (TLR4) expression in the central amygdala. Proc Natl Acad Sci U S A 108:4465–4470
Liu J, Yang AR, Kelly T, Puche A, Esoga C, June HL Jr, Elnabawi A, Merchenthaler I, Sieghart W, June HL Sr, Aurelian L (2011b) Binge alcohol drinking is associated with GABAA alpha2-regulated toll-like receptor 4 (TLR4) expression in the central amygdala. Proc Natl Acad Sci U S A 108:4465–4470
Maczurek A, Shanmugam K, Munch G (2008) Inflammation and the redox-sensitive AGE-RAGE pathway as a therapeutic target in Alzheimer’s disease. Ann N Y Acad Sci 1126:147–151
Madrigal JL, Moro MA, Lizasoain I, Lorenzo P, Leza JC (2002) Stress-induced increase in extracellular sucrose space in rats is mediated by nitric oxide. Brain Res 938:87–91
Madrigal JL, Garcia-Bueno B, Moro MA, Lizasoain I, Lorenzo P, Leza JC (2003) Relationship between cyclooxygenase-2 and nitric oxide synthase-2 in rat cortex after stress. Eur J Neurosci 18:1701–1705
Maes M, Berk M, Goehler L, Song C, Anderson G, Galecki P, Leonard B (2012) Depression and sickness behavior are Janus-faced responses to shared inflammatory pathways. BMC Med 10:66
Malberg JE, Eisch AJ, Nestler EJ, Duman RS (2000) Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci 20:9104–9110
Markou A, Kosten TR, Koob GF (1998) Neurobiological similarities in depression and drug dependence: a self-medication hypothesis. Neuropsychopharmacology 18:135–174
Maroso M, Balosso S, Ravizza T, Liu J, Bianchi ME, Vezzani A (2011) Interleukin-1 type 1 receptor/toll-like receptor signalling in epilepsy: the importance of IL-1beta and high-mobility group box 1. J Intern Med 270:319–326
Marshall SA, Casachahua JD, Rinker JA, Blose AK, Lysle DT, Thiele TE (2016) IL-1 receptor signaling in the basolateral amygdala modulates binge-like ethanol consumption in male C57BL/6J mice. Brain Behav Immun 51:258–267
Mayfield J, Ferguson L, Harris RA (2013) Neuroimmune signaling: a key component of alcohol abuse. Curr Opin Neurobiol 23:513–520
Mejia-Toiber J, Boutros N, Markou A, Semenova S (2014) Impulsive choice and anxiety-like behavior in adult rats exposed to chronic intermittent ethanol during adolescence and adulthood. Behav Brain Res 266:19–28
Miller AH, Raison CL (2016) The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol 16:22–34
Montesinos J, Pascual M, Rodriguez-Arias M, Minarro J, Guerri C (2016) Involvement of TLR4 in the long-term epigenetic changes, rewarding and anxiety effects induced by intermittent ethanol treatment in adolescence. Brain Behav Immun 53:159–171
Mulligan MK, Ponomarev I, Hitzemann RJ, Belknap JK, Tabakoff B, Harris RA, Crabbe JC, Blednov YA, Grahame NJ, Phillips TJ, Finn DA, Hoffman PL, Iyer VR, Koob GF, Bergeson SE (2006) Toward understanding the genetics of alcohol drinking through transcriptome meta-analysis. Proc Natl Acad Sci U S A 103:6368–6373
Nava F, Premi S, Manzato E, Lucchini A (2006) Comparing treatments of alcoholism on craving and biochemical measures of alcohol consumption. J Psychoactive Drugs 38:211–217
Northcutt AL, Hutchinson MR, Wang X, Baratta MV, Hiranita T, Cochran TA, Pomrenze MB, Galer EL, Kopajtic TA, Li CM, Amat J, Larson G, Cooper DC, Huang Y, O’Neill CE, Yin H, Zahniser NR, Katz JL, Rice KC, Maier SF, Bachtell RK, Watkins LR (2015) DAT isn’t all that: cocaine reward and reinforcement require toll-like receptor 4 signaling. Mol Psychiatry 20:1525–1537
Nunez YO, Mayfield RD (2012) Understanding alcoholism through microRNA signatures in brains of human alcoholics. Front Genet 3:43
Nunez YO, Truitt JM, Gorini G, Ponomareva ON, Blednov YA, Harris RA, Mayfield RD (2013) Positively correlated miRNA-mRNA regulatory networks in mouse frontal cortex during early stages of alcohol dependence. BMC Genomics 14:725
Oak S, Mandrekar P, Catalano D, Kodys K, Szabo G (2006) TLR2- and TLR4-mediated signals determine attenuation or augmentation of inflammation by acute alcohol in monocytes. J Immunol 176:7628–7635
Obernier JA, Bouldin TW, Crews FT (2002a) Binge ethanol exposure in adult rats causes necrotic cell death. Alcohol Clin Exp Res 26:547–557
Obernier JA, White AM, Swartzwelder HS, Crews FT (2002b) Cognitive deficits and CNS damage after a 4-day binge ethanol exposure in rats. Pharmacol Biochem Behav 72:521–532
Obernier JA, White AM, Swartzwelder HS, Crews FT (2002c) Cognitive deficits and CNS damage after a 4-day binge ethanol exposure in rats. Pharmacol Biochem Behav 72:521–532
O’Connor JC, Lawson MA, Andre C, Moreau M, Lestage J, Castanon N, Kelley KW, Dantzer R (2009) Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol Psychiatry 14:511–522
O’Connor RM, Gururajan A, Dinan TG, Kenny PJ, Cryan JF (2016) All roads lead to the miRNome: miRNAs have a central role in the molecular pathophysiology of psychiatric disorders. Trends Pharmacol Sci 37:1029–1044
Okun E, Griffioen K, Barak B, Roberts NJ, Castro K, Pita MA, Cheng A, Mughal MR, Wan R, Ashery U, Mattson MP (2010) Toll-like receptor 3 inhibits memory retention and constrains adult hippocampal neurogenesis. Proc Natl Acad Sci U S A 107:15625–15630
Okvist A, Johansson S, Kuzmin A, Bazov I, Merino-Martinez R, Ponomarev I, Mayfield RD, Harris RA, Sheedy D, Garrick T, Harper C, Hurd YL, Terenius L, Ekstrom TJ, Bakalkin G, Yakovleva T (2007) Neuroadaptations in human chronic alcoholics: dysregulation of the NF-kappaB system. PLoS One 2:e930
Pahl HL (1999) Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18:6853–6866
Pandey SC, Sakharkar AJ, Tang L, Zhang H (2015) Potential role of adolescent alcohol exposure-induced amygdaloid histone modifications in anxiety and alcohol intake during adulthood. Neurobiol Dis 82:607–619
Pascual M, Blanco AM, Cauli O, Minarro J, Guerri C (2007) Intermittent ethanol exposure induces inflammatory brain damage and causes long-term behavioural alterations in adolescent rats. Eur J Neurosci 25:541–550
Pascual M, Balino P, Alfonso-Loeches S, Aragon CM, Guerri C (2011) Impact of TLR4 on behavioral and cognitive dysfunctions associated with alcohol-induced neuroinflammatory damage. Brain Behav Immun 25(Suppl 1):S80–S91
Pascual M, Balino P, Aragon CM, Guerri C (2015) Cytokines and chemokines as biomarkers of ethanol-induced neuroinflammation and anxiety-related behavior: role of TLR4 and TLR2. Neuropharmacology 89:352–359
Pietri M, Schneider B, Mouillet-Richard S, Ermonval M, Mutel V, Launay JM, Kellermann O (2005) Reactive oxygen species-dependent TNF-alpha converting enzyme activation through stimulation of 5-HT2B and alpha1D autoreceptors in neuronal cells. FASEB J 19:1078–1087
Plane JM, Shen Y, Pleasure DE, Deng W (2010) Prospects for minocycline neuroprotection. Arch Neurol 67:1442–1448
Qin L, Crews FT (2012a) Chronic ethanol increases systemic TLR3 agonist-induced neuroinflammation and neurodegeneration. J Neuroinflammation 9:130
Qin L, Crews FT (2012b) NADPH oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration. J Neuroinflammation 9:5
Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT (2007) Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55:453–462
Qin L, He J, Hanes RN, Pluzarev O, Hong JS, Crews FT (2008) Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J Neuroinflammation 5:10
Qin L, Liu Y, Hong JS, Crews FT (2013) NADPH oxidase and aging drive microglial activation, oxidative stress, and dopaminergic neurodegeneration following systemic LPS administration. Glia 61:855–868
Rea K, Dinan TG, Cryan JF (2016) The microbiome: a key regulator of stress and neuroinflammation. Neurobiol Stress 4:23–33
Reissner KJ, Kalivas PW (2010) Using glutamate homeostasis as a target for treating addictive disorders. Behav Pharmacol 21:514–522
Risher ML, Fleming RL, Boutros N, Semenova S, Wilson WA, Levin ED, Markou A, Swartzwelder HS, Acheson SK (2013) Long-term effects of chronic intermittent ethanol exposure in adolescent and adult rats: radial-arm maze performance and operant food reinforced responding. PLoS One 8:e62940
Risher ML, Sexton HG, Risher WC, Wilson WA, Fleming RL, Madison RD, Moore SD, Eroglu C, Swartzwelder HS (2015) Adolescent intermittent alcohol exposure: dysregulation of thrombospondins and synapse formation are associated with decreased neuronal density in the adult hippocampus. Alcohol Clin Exp Res 39:2403–2413
Russo SJ, Wilkinson MB, Mazei-Robison MS, Dietz DM, Maze I, Krishnan V, Renthal W, Graham A, Birnbaum SG, Green TA, Robison B, Lesselyong A, Perrotti LI, Bolanos CA, Kumar A, Clark MS, Neumaier JF, Neve RL, Bhakar AL, Barker PA, Nestler EJ (2009) Nuclear factor kappa B signaling regulates neuronal morphology and cocaine reward. J Neurosci Off J Soc Neurosci 29:3529–3537
Ryan SM, Nolan YM (2016) Neuroinflammation negatively affects adult hippocampal neurogenesis and cognition: can exercise compensate? Neurosci Biobehav Rev 61:121–131
Saha B, Momen-Heravi F, Kodys K, Szabo G (2016) MicroRNA cargo of extracellular vesicles from alcohol-exposed monocytes signals naive monocytes to differentiate into M2 macrophages. J Biol Chem 291:149–159
Sakharkar AJ, Vetreno RP, Zhang H, Kokare DM, Crews FT, Pandey SC (2016) A role for histone acetylation mechanisms in adolescent alcohol exposure-induced deficits in hippocampal brain-derived neurotrophic factor expression and neurogenesis markers in adulthood. Brain Struct Funct
Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, Weisstaub N, Lee J, Duman R, Arancio O, Belzung C, Hen R (2003) Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301:805–809
Sarter M, Paolone G (2011) Deficits in attentional control: cholinergic mechanisms and circuitry-based treatment approaches. Behav Neurosci 125:825–835
Schoenbaum G, Saddoris MP, Ramus SJ, Shaham Y, Setlow B (2004) Cocaine-experienced rats exhibit learning deficits in a task sensitive to orbitofrontal cortex lesions. Eur J Neurosci 19:1997–2002
Schoenbaum G, Roesch MR, Stalnaker TA (2006) Orbitofrontal cortex, decision-making and drug addiction. Trends Neurosci 29:116–124
Setiawan E, Wilson AA, Mizrahi R, Rusjan PM, Miler L, Rajkowska G, Suridjan I, Kennedy JL, Rekkas PV, Houle S, Meyer JH (2015) Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry 72:268–275
Steptoe A, Hamer M, Chida Y (2007) The effects of acute psychological stress on circulating inflammatory factors in humans: a review and meta-analysis. Brain Behav Immun 21:901–912
Stevenson JR, Schroeder JP, Nixon K, Besheer J, Crews FT, Hodge CW (2009) Abstinence following alcohol drinking produces depression-like behavior and reduced hippocampal neurogenesis in mice. Neuropsychopharmacology 34:1209–1222
Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140:805–820
Thakur V, McMullen MR, Pritchard MT, Nagy LE (2007) Regulation of macrophage activation in alcoholic liver disease. J Gastroenterol Hepatol 22(Suppl 1):S53–S56
Theberge FR, Li X, Kambhampati S, Pickens CL, St Laurent R, Bossert JM, Baumann MH, Hutchinson MR, Rice KC, Watkins LR, Shaham Y (2013) Effect of chronic delivery of the toll-like receptor 4 antagonist (+)-naltrexone on incubation of heroin craving. Biol Psychiatry 73:729–737
Thomas JO, Stott K (2012) H1 and HMGB1: modulators of chromatin structure. Biochem Soc Trans 40:341–346
Thounaojam MC, Kaushik DK, Basu A (2013) MicroRNAs in the brain: it’s regulatory role in neuroinflammation. Mol Neurobiol 47:1034–1044
Torres-Platas SG, Cruceanu C, Chen GG, Turecki G, Mechawar N (2014) Evidence for increased microglial priming and macrophage recruitment in the dorsal anterior cingulate white matter of depressed suicides. Brain Behav Immun
Townshend JM, Duka T (2003) Mixed emotions: alcoholics’ impairments in the recognition of specific emotional facial expressions. Neuropsychologia 41:773–782
Tynan RJ, Naicker S, Hinwood M, Nalivaiko E, Buller KM, Pow DV, Day TA, Walker FR (2010) Chronic stress alters the density and morphology of microglia in a subset of stress-responsive brain regions. Brain Behav Immun 24:1058–1068
Vabulas RM, Braedel S, Hilf N, Singh-Jasuja H, Herter S, Ahmad-Nejad P, Kirschning CJ, Da Costa C, Rammensee HG, Wagner H, Schild H (2002) The endoplasmic reticulum-resident heat shock protein Gp96 activates dendritic cells via the toll-like receptor 2/4 pathway. J Biol Chem 277:20847–20853
Valenta JP, Gonzales RA (2016) Chronic intracerebroventricular infusion of monocyte chemoattractant protein-1 leads to a persistent increase in sweetened ethanol consumption during operant self-administration but does not influence sucrose consumption in long-Evans rats. Alcohol Clin Exp Res 40:187–195
Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, Liu J, Antonelli A, Preti A, Raeli L, Shams SS, Yang H, Varani L, Andersson U, Tracey KJ, Bachi A, Uguccioni M, Bianchi ME (2012) Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med 209:1519–1528
Vetreno RP, Crews FT (2012) Adolescent binge drinking increases expression of the danger signal receptor agonist HMGB1 and toll-like receptors in the adult prefrontal cortex. Neuroscience 226:475–488
Vetreno RP, Crews FT (2014) Current hypotheses on the mechanisms of alcoholism. Handb Clin Neurol 125:477–497
Vetreno RP, Crews FT (2015) Binge ethanol exposure during adolescence leads to a persistent loss of neurogenesis in the dorsal and ventral hippocampus that is associated with impaired adult cognitive functioning. Front Neurosci 9:35
Vetreno RP, Qin L, Crews FT (2013) Increased receptor for advanced glycation end product expression in the human alcoholic prefrontal cortex is linked to adolescent drinking. Neurobiol Dis 59:52–62
Vetreno RP, Broadwater M, Liu W, Spear LP, Crews FT (2014) Adolescent, but not adult, binge ethanol exposure leads to persistent global reductions of choline acetyltransferase expressing neurons in brain. PLoS One 9:e113421
Vetreno RP, Patel Y, Patel U, Jordan Walter T, Crews FT (2016) Adolescent intermittent ethanol reduces serotonin expression in the adult raphe nucleus and upregulates innate immune expression that is prevented by exercise. Brain Behav Immun
Volkow ND, Koob GF, McLellan AT (2016) Neurobiologic advances from the brain disease model of addiction. N Engl J Med 374:363–371
Ward RJ, Zhang Y, Crichton RR, Piret B, Piette J, de Witte P (1996) Identification of the nuclear transcription factor NFkappaB in rat after in vivo ethanol administration. FEBS Lett 389:119–122
Ward RJ, Colivicchi MA, Allen R, Schol F, Lallemand F, de Witte P, Ballini C, Corte LD, Dexter D (2009) Neuro-inflammation induced in the hippocampus of “binge drinking” rats may be mediated by elevated extracellular glutamate content. J Neurochem 111:1119–1128
Weber MD, Frank MG, Tracey KJ, Watkins LR, Maier SF (2015) Stress induces the danger-associated molecular pattern HMGB-1 in the hippocampus of male Sprague-Dawley rats: a priming stimulus of microglia and the NLRP3 inflammasome. J Neurosci 35:316–324
Weissenborn R, Duka T (2003) Acute alcohol effects on cognitive function in social drinkers: their relationship to drinking habits. Psychopharmacology 165:306–312
White HR, Marmorstein NR, Crews FT, Bates ME, Mun EY, Loeber R (2011) Associations between heavy drinking and changes in impulsive behavior among adolescent boys. Alcohol Clin Exp Res 35:295–303
Wichers MC, Maes M (2004) The role of indoleamine 2,3-dioxygenase (IDO) in the pathophysiology of interferon-alpha-induced depression. J Psychiatry Neurosci 29:11–17
Wilson JS, Mruthinti S, Buccafusco JJ, Schade RF, Mitchell MB, Harrell DU, Gulati NK, Miller LS (2009) Anti-RAGE and Abeta immunoglobulin levels are related to dementia level and cognitive performance. J Gerontol A Biol Sci Med Sci 64:264–271
Yanai H, Ban T, Wang Z, Choi MK, Kawamura T, Negishi H, Nakasato M, Lu Y, Hangai S, Koshiba R, Savitsky D, Ronfani L, Akira S, Bianchi ME, Honda K, Tamura T, Kodama T, Taniguchi T (2009) HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature 462:99–103
Yang H, Lundback P, Ottosson L, Erlandsson-Harris H, Venereau E, Bianchi ME, Al-Abed Y, Andersson U, Tracey KJ, Antoine DJ (2012) Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol Med 18:250–259
Yelamanchili SV, Lamberty BG, Rennard DA, Morsey BM, Hochfelder CG, Meays BM, Levy E, Fox HS (2015) MiR-21 in extracellular vesicles leads to neurotoxicity via TLR7 signaling in SIV neurological disease. PLoS Pathog 11:e1005032
Yirmiya R, Goshen I (2011) Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun 25:181–213
Young JJ, Bruno D, Pomara N (2014) A review of the relationship between proinflammatory cytokines and major depressive disorder. J Affect Disord 169:15–20
Zou JY, Crews FT (2005) TNF alpha potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: neuroprotection by NF kappa B inhibition. Brain Res 1034:11–24
Zou J, Crews F (2006) CREB and NF-kappaB transcription factors regulate sensitivity to excitotoxic and oxidative stress induced neuronal cell death. Cell Mol Neurobiol 26:385–405
Zou J, Crews F (2010) Induction of innate immune gene expression cascades in brain slice cultures by ethanol: key role of NF-kappaB and proinflammatory cytokines. Alcohol Clin Exp Res 34:777–789
Zou J, Crews FT (2012) Inflammasome-IL-1beta signaling mediates ethanol inhibition of hippocampal neurogenesis. Front Neurosci 6:77
Zou JY, Crews FT (2014) Release of neuronal HMGB1 by ethanol through decreased HDAC activity activates brain neuroimmune signaling. PLoS One 9:e87915
Acknowledgements
This work was supported in part by funding from the National Institutes of Health, National Institute on Alcoholism and Alcohol Abuse through the Neurobiology of Adolescent Drinking in Adulthood (NADIA) consortium (AA020024, AA020023), the Bowles Center for Alcohol Studies (AA011605), and the U54 collaborative partnership between NCCU and UNC (AA019767). The authors thank Kathy Neal for the help with preparation and submission of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Crews, F.T., Walter, T.J., Coleman, L.G. et al. Toll-like receptor signaling and stages of addiction. Psychopharmacology 234, 1483–1498 (2017). https://doi.org/10.1007/s00213-017-4560-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-017-4560-6