
Abstract
Aims/hypothesis
Glucagon-like peptide 1 receptor agonists (GLP-1 RA) such as exenatide are used as monotherapy and add-on therapy for maintaining glycaemic control in patients with type 2 diabetes mellitus. The current study investigated the safety and efficacy of once-weekly PB-119, a PEGylated exenatide injection, in treatment-naive patients with type 2 diabetes.
Methods
In this Phase II, randomised, placebo-controlled, double-blind study, we randomly assigned treatment-naive Chinese patients with type 2 diabetes in a 1:1:1:1 ratio to receive subcutaneous placebo or one of three subcutaneous doses of PB-119 (75, 150, and 200 μg) for 12 weeks. The primary endpoint was the change in HbA1c from baseline to week 12, and other endpoints were fasting plasma glucose, 2 h postprandial glucose (PPG), and proportion of patients with HbA1c < 53 mmol/mol (<7.0%) and ≤48 mmol/mol (≤6.5%) at 2, 4, 8 and 12 weeks of treatment. Safety was assessed in all patients who received at least one dose of study drug.
Results
We randomly assigned 251 patients to one of the four treatment groups (n = 62 in placebo and 63 each in PB-119 75 μg, 150 μg and 200 μg groups). At the end of 12 weeks, mean differences in HbA1c in the treatment groups were −7.76 mmol/mol (95% CI −9.23, −4.63, p < 0.001) (−0.72%, 95% CI −1.01, −0.43), −12.89 mmol/mol (95% CI −16.05, −9.72, p < 0.001) (−1.18%, 95% CI −1.47, −0.89) and −11.14 mmol/mol (95% CI −14.19, −7.97, p <0 .001) (−1.02%, 95% CI −1.30, −0.73) in the 75 μg, 150 μg and 200 μg PB-119 groups, respectively, compared with that in the placebo group after adjusting for baseline HbA1c. Similar results were also observed for other efficacy endpoints across different time points. There was no incidence of treatment-emergent serious adverse event, severe hypoglycaemia or death.
Conclusions/interpretation
All tested PB-119 doses had superior efficacy compared with placebo and were safe and well tolerated over 12 weeks in treatment-naive Chinese patients with type 2 diabetes.
Trial registration
ClinicalTrials.gov NCT03520972
Funding
The study was funded by National Major Scientific and Technological Special Project for Significant New Drugs Development and PegBio.
Graphical abstract

Similar content being viewed by others



Introduction
Type 2 diabetes mellitus affects approximately 9.3% of the world population, and is projected to increase to 10.9% by 2045 [1]. Despite the availability of multiple antidiabetic drugs, disease progression and deterioration of glycaemic control are difficult to prevent. Hence, new therapeutic drugs are being pursued [2, 3]. The incretin system is an important target for the therapeutic management of type 2 diabetes [4]. Incretins are intestinal hormones that regulate insulin production in response to oral intake of nutrients, called the ‘incretin effect’, which is lacked in patients with type 2 diabetes [5].
Glucagon-like peptide-1 receptor agonists (GLP-1 RAs) are a novel class of injectable incretin mimetics that provide glycaemic and extra-glycaemic benefits for the treatment of patients with type 2 diabetes [6]. The glycaemic effects of GLP-1 RAs are mainly mediated by the induction of glucose-dependent insulin secretion, inhibition of glucagon secretion, reduction of gastric motility and promotion of satiety [7,8,9,10,11]. Exenatide is a first-in-class GLP-1 RA, available both as a short-acting formulation (twice daily) and as a long-acting (once weekly) formulation [12, 13]. It has been used both as monotherapy with lifestyle modifications and as an add-on therapy with oral antidiabetic drugs (OADs) or insulin in addition to dipeptidyl peptidase-4 inhibitors [14, 15]. The terminal t½ of the original subcutaneous formulation of exenatide was 2.4 h, requiring twice daily injections [16]. In earlier clinical trials with the twice daily formulation, an HbA1c reduction of 1.0% to 1.5% was observed in patients who had not been well controlled with OADs, with a baseline HbA1c of 7.9% (63 mmol/mol) to 9.0% (75 mmol/mol) [17, 18].
The short t½ of exenatide had impeded the routine use of exenatide; this has prompted the use of microsphere technology to extend the t½, creating a feasible once weekly formulation [12]. Exenatide once weekly regimen dispersed by the microsphere technology has been evaluated in previous trials, both as a monotherapy and in combination with OADs and insulin [13, 19,20,21]. One of its potential limitations is the multiphasic concentration-time profile, which limits the prediction of accurate pharmacokinetic (PK) variables. Meanwhile, the need for a dispersing diluent may affect patients’ compliance [16].
Covalent attachment of polyethylene glycol (PEG) to peptide drugs such as exenatide increases the relative molecular mass and reduces the renal clearance rate, prolonging retention in the circulation [22]. It also reduces the immunogenicity, thereby preventing adverse immunological reactions. The study drug of this study, PEGylated exenatide injection (PB-119), has been previously evaluated for safety, tolerability, and PK effects in a Phase 1 study with 70 healthy volunteers, and the study recommended a once-weekly PB-119 injection of 2 to 200 μg, showing this dose to be safe and well tolerated [23]. In this study, we assessed the efficacy, tolerability and safety of different doses of PB-119 as a monotherapy, compared with placebo, in treatment-naive Chinese patients with type 2 diabetes.
Methods
Study design
This Phase II, randomised, multiple doses, double-blind, parallel, placebo-controlled, four-arm study was conducted in 31 clinical centres in China (ClinicalTrials.gov registration no: NCT03520972). The protocol was approved by the institutional review board of the participating study centres. The study was conducted in accordance with the Declaration of Helsinki and other local regulatory guidelines governing the conduct of clinical studies in China. All patients provided written informed consent to participate before study initiation.
Study population
Eligible patients were men or women with type 2 diabetes (according to 1999 WHO type 2 diabetes diagnostic criteria) who were aged 18 to 70 years, with a BMI of 18.5 to 35 kg/m2, and were on a diet and exercise regimen. All patients were previously untreated for 3 months with any antidiabetic drug except short-term insulin treatment (≤7 days). At the time of randomisation, the patients were required to have HbA1c between 58.5 mmol/mol (7.5%), and 91.3 mmol/mol (11.0%) and fasting plasma glucose (FPG) between 4.4 and 13.3 mmol/l.
Key exclusion criteria were pregnancy, clinical diagnosis of type 1 diabetes, acute complications of diabetes, previous incidence of severe hypoglycaemia within 6 months before the study, a severe cardiovascular event within 6 months before screening, any type of malignancy, uncontrolled high BP (systolic BP >160 mmHg or diastolic BP >100 mmHg), haemoglobin concentration of <12 mg/l for men and <10 mg/l for women, severe gastrointestinal diseases, history or ongoing symptoms or signs of severe allergy or hypersensitivity, triacylglycerol concentration >5.65 mmol/l or lipid-lowering drugs used within 3 months before screening, and renal dysfunction (GFR <45 ml min−1 [1.73 m]−2 according to the Modification of Diet in Renal Disease formula).
Randomisation and masking
Eligible patients were enrolled into a single-blind run-in period of 2 weeks during which placebo was administered. The patients were then assessed for eligibility again at the end of run-in period. Patients who met the eligibility criteria were randomised by the randomisation plan devised by the SAS 9.4 statistical tool in a 1:1:1:1 to one of the three different doses of PB-119 (75, 150 and 200 μg subcutaneous injection, once weekly) or placebo group. The random grouping information was maintained in the central randomisation system, and each drug was given a specific number. The dosages of PB-119 or placebo were delivered by a weekly, subcutaneous abdominal injection at a dose of 0.5 mg/ml during any time of the day. The investigators, site personnel, patients, and sponsors were masked to treatment assignment, and the anonymised data were stored in a secured directory that was accessible only after the study was unblinded. The participants were distributed across 31 research centres.
Procedures and data collection
Demographic data were collected during the screening period. Different laboratory variables including HbA1c and FPG were assessed prior to randomisation. Patients were treated for 12 weeks and HbA1c, FPG, and 2 h postprandial glucose (PPG) were measured at 4, 8 and 12 weeks.
Outcomes and endpoints
The primary efficacy endpoint was change in HbA1c between baseline (day 1) and at the end of 12 weeks. The secondary efficacy endpoints included the proportion of participants with HbA1c <53 mmol/mol (<7.0%) and ≤48 mmol/mol (≤6.5%) at the end of 4, 8 and 12 weeks; change in FPG from baseline at 2, 4, 8 and 12 weeks; and 2 h PPG at 4, 8 and 12 weeks. Changes in blood pressure, body weight and lipid profile were also evaluated.
Safety outcomes were assessed by the incidence of adverse events (AE) as defined by the MedDRA version 22.0. The different AEs included incidence of hypoglycaemic events (blood glucose <3.9 mmol/l), clinical findings in the physical examination, vital signs, 12-lead ECG and clinical laboratory tests.
Statistical analysis
The sample size was determined based on an assumed change in HbA1c after 12 weeks of treatment from a baseline of −8 mmol/mol (−0.8%), −12 mmol/mol (−1.1%) and −12 mmol/mol (−1.1%) in the three treatment groups and 0 mmol/mol (0%) in the placebo group, and the combined SD was assumed to be 13 mmol/mol (1.2%). The two-sided α level was set at 0.05, the Bonferroni method was used to adjust the multiple comparison, and the ratio of participants in the four groups was 1:1:1:1. On the basis of these variables, the number of participants required for 80% statistical power was determined to be 50 patients in each group. Considering a dropout rate of approximately 20%, 240 participants were planned to be enrolled in this study.
The full analysis set (FAS) included all patients who were randomised and who received at least a single dose of the study drug after the run-in phase, with data from at least one post-baseline data. Efficacy analysis was based on the intention-to-treat principle and included all patients who received at least one dose of the study medication and had at least one post-baseline assessment of the primary endpoint. Safety analysis included all randomised patients who received at least one dose of study medication. We assessed the primary endpoint using an ANCOVA model, with study treatment as a fixed effect and baseline HbA1c as a covariate. Least squares mean (LSM), SE and the corresponding 95% CI for each treatment were calculated by the ANCOVA model. The within-group change in HbA1c was assessed by a paired t test. Change in HbA1c at 2, 4 and 8 weeks; change in FPG at 2, 4, 8 and 12 weeks; and change in 2 h PPG at 4, 8 and 12 weeks were also analysed by the ANCOVA model and t test. The efficacy indicators including observational indicators were imputed from last available post-baseline follow-up data in the case of missing patients/dropouts. The proportion of patients with HbA1c <53 mmol/mol (<7.0%) and ≤48 mmol/mol (≤6.5%) at 4, 8 and 12 weeks was analysed by Fisher’s test.
Descriptive statistics (number of patients, median [IQR], and mean [SD]) were used to summarise continuous variables. Sensitivity analysis for the main outcome with the original observation values was performed using a mixed-effect model repeated measure (MMRM) model. Baseline HbA1c value, group, visit, interaction between group and visit, and interaction between centre, centre and group were included in the MMRM model. Treatment-emergent AEs were assessed in the safety dataset, which included all patients who received at least one dose of the study drug after the run-in phase. All statistical analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC, USA). All the statistical tests were done at a two-sided α level of 0.05.
Results
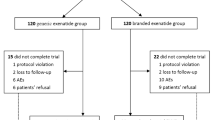
Between June 2018 and July 2019, we screened 394 participants, of whom 287 eligible patients entered the run-in phase, and 251 patients were randomly assigned to receive 75 μg PB-119 once weekly (n = 63), 150 μg PB-119 once weekly (n = 63), 200 μg PB-119 once weekly (n = 63), and matching placebo group (n = 62). Of the 251 randomised patients, except one patient in the 75 μg PB-119 group who did not receive the study drug, all received at least one study dose and entered the FAS. Of the 250 randomised participants, 222 patients completed the study with a minimum of one time point post-baseline follow-up data. A total of 29 patients were withdrawn from the study mainly due to hyperglycaemia (20.7%) and withdrawal of informed consent (10.3%) (Fig. 1). Demographic and disease characteristics at baseline in the FAS are provided in Table 1.

Patient disposition in the study
Treatment compliance
The number of patients with a treatment compliance of 80%–100% were: 56 patients (90.3%) in the placebo group, 54 patients (87.1%) in the PB-119 75 μg group, 57 patients (90.5%) in the 150 μg PB-119 group and 56 patients (88.9%) in the 200 μg PB-119 group.
Efficacy
After 12 weeks of treatment, the LSM change in HbA1c from baseline was −4.26 mmol/mol (95% CI −6.55, −2.07) (−0.39%, 95% CI −0.60, −0.19) in the placebo group, −12.02 mmol/mol (95% CI −14.3, −9.86) (−1.11%, 95% CI −1.32, −0.91) in the 75 μg PB-119 group, −17.15 mmol/mol (95% CI −19.45, −14.97) (−1.57%, 95% CI −1.78, −1.37) in the 150 μg PB-119 group and −15.40 mmol/mol (95% CI −17.59, −13.10) (−1.41%, 95% CI −1.61, −1.20) in the 200 μg PB-119 group. The placebo-adjusted difference in LSM change in HbA1c from baseline to 12 weeks was −7.76 mmol/mol (95% CI −9.23, −4.63) (−0.72%, 95% CI −1.01, −0.43) in the 75 μg PB-119 group, −12.89 mmol/mol (95% CI −16.05, −9.72) (−1.18%, 95% CI −1.47, −0.89) in the 150 μg PB-119 group, and −11.14 mmol/mol (95% CI −14.19, −7.97) (−1.02%, 95% CI −1.30, −0.73) in the 200 μg PB-119 group (Table 2). Although there was a dose-dependent decrease in HbA1c from the 75 μg to 150 μg PB-119 group (LSM difference: −5.13 mmol/mol, 95% CI −8.53, −1.89; −0.46%, 95% CI −0.74, −0.17; p = 0.002), there was an increase in HbA1c from the 150 μg to 200 μg PB-119 group (LSM difference: 1.75 mmol/mol, 95% CI −1.24, 4.63; 0.17%, 95% CI, −0.12, 0.45; p = 0.261; Table 2). Compared with the placebo group, HbA1c was significantly reduced in all the three dose groups of PB-119 (p < 0.001). Similar findings were also observed after 2, 4 and 8 weeks of treatment (Fig. 2a).

(a) Change in HbA1c from baseline at 2, 4, 8 and 12 weeks; (b) change in FPG from baseline at 2, 4, 8 and 12 weeks; (c) change in 2 h PPG from baseline at 4, 8 and 12 weeks
The proportion of patients with HbA1c <53 mmol/mol (<7.0%) at 12 weeks was 8.1% in the placebo group, 46.8% in the 75 μg PB-119 group, 47.6% in the 150 μg PB-119 group and 41.3% in the 200 μg PB-119 group. The OR of achieving HbA1c of <53 mmol/mol (<7.0%) was 10.02 (95% CI 3.54, 28.38), 10.36 (95% CI 3.67, 29.30) and 8.01 (95% CI 2.82, 22.73) in the 75 μg, 150 μg and 200 μg PB-119 groups, respectively, compared with the placebo group (p < 0.001). Similar results were also observed for 4 and 8 weeks (Table 3). The proportion of participants with HbA1c ≤ 48 mmol/mol (≤6.5%) at 12 weeks was 1.6% in the placebo group, 27.4% in the 75 μg PB-119 group, 30.2% in the 150 μg PB-119 group and 19.0% in the 200 μg PB-119 group. The OR of achieving HbA1c ≤ 48 mmol/mol (≤6.5%) was 23.04 (95% CI 2.96, 179.59; p < 0.001), 26.34 (95% CI 3.40, 204.19; p < 0.001) and 14.35 (95% CI 1.80, 114.16; p = 0.002) in the 75 μg, 150 μg, and 200 μg PB-119 groups, respectively, compared with the placebo group (p < 0.001).
After 12 weeks of treatment, the LSM change in FPG was −0.23 mmol/l (−0.66, 0.20) in the placebo group, −1.20 mmol/l (−1.63, −0.77) in the 75 μg PB-119 group, −2.31 mmol/l (−2.73, −1.89) in the 150 μg PB-119 group and −2.22 mmol/l (−2.65, −1.79) in the 200 μg PB-119 group. The placebo-adjusted difference in LSM change in FPG from baseline to 12 weeks of treatment was −0.97 mmol/l (−1.58, −0.36; p = 0.002) in the 75 μg PB-119 group, −2.08 mmol/l (−2.68, −1.47; p < 0.001) in the 150 μg PB-119 group, and −1.99 mmol/l (−2.59, −1.39; p < 0.001) in the 200 μg PB-119 group. Similar findings were also observed after 2, 4 and 8 weeks of treatment (Fig. 2b). After 12 weeks of treatment, the LSM change in 2 h PPG was −0.76 mmol/l (−1.50, −0.02) in the placebo group, −1.80 mmol/l (−2.53, −1.06) in the 75 μg PB-119 group, −3.96 mmol/l (−4.69, −3.23) in the 150 μg PB-119 group and −3.61 mmol/l (−4.35, −2.87) in the 200 μg PB-119 group. The placebo-adjusted difference in LSM change in 2 h PPG from baseline to 12 weeks of treatment was −1.04 mmol/l (−2.09, 0.02) in the 75 μg PB-119 group, −3.20 mmol/l (−4.25, −2.15) in the 150 μg PB-119 group and −2.85 mmol/l (−3.88, −1.82) in the 200 μg PB-119 group. The reduction in 2 h PPG was significantly higher in the 150 μg and 200 μg PB-119 groups compared with the placebo group (p < 0.001) and approaching significance in the 75 μg PB-119 group (p = 0.054). Similar findings were also observed after 4 and 8 weeks of treatment (Fig. 2c).
Changes in BP, body weight and lipid profile
The mean change in systolic and diastolic BP after 12 weeks, were −2.4, −1.5, −2.7 and −2.9 mmHg and −0.7, −0.9, −1.3 and −2.7 mmHg in placebo, 75, 150 and 200 μg PB-119 groups, respectively. The mean changes in body weight and lipids are provided in ESM Table 1.
Safety
A total of 250 patients received at least one dose of the study drug and constituted the safety dataset. Most of the AEs were mild to moderate. During the treatment period, the incidence of AEs was 69.4% (43 participants) in the placebo group, 77.4% (48 participants) in the 75 μg PB-119 group, 81% (51 participants) in the 150 μg PB-119 group and 82.5% (52 participants) in the 200 μg PB-119 group. The number of AEs in the four groups was 116 in the placebo group, 126 in the 75 μg PB-119 group, 220 in the 150 μg PB-119 group, and 298 in the 200 μg PB-119 group. There were totally 377 events of drug-related AEs reported in 86 patients, with 14 events in six patients in the placebo group, 39 events in 20 patients in the 75 μg PB-119 group, 144 events in 29 patients in the 150 μg PB-119 group and 180 events in 31 patients in the 200 μg PB-119 group (Table 4). There were no deaths or drug-related serious AEs reported in any of the groups. Hypoglycaemia related to the study drug occurred in six (9.7%) patients in the 75 μg PB-119 group, seven (11.1%) patients in the 150 μg PB-119 group and four (6.3%) patients in the 200 μg PB-119 group. Severe hypoglycaemia was not reported in any of the groups. No clinically significant abnormalities in laboratory variables, 12-lead ECG, physical examination or vital signs were observed in any treatment groups.
Sensitivity analysis
Sensitivity analysis by MMRM revealed a statistically significant difference in placebo-adjusted change in HbA1c levels in all three PB-119 treatment groups (p < 0.001). The change in HbA1c from baseline was not significantly different between the 200 μg PB-119, 75 μg PB-119 (p = 0.181), as well as 200 μg PB-119 group and 150 μg PB-119 (p = 0.052) groups. The effect estimates and the corresponding 95% CIs are provided in ESM Table 2. Similar results in MMRM and ANCOVA model confirms the robustness of the analysis.
Immunogenicity
At baseline, PB-119 antibody was identified in 3.2% (two patients) and 1.6% (one patient) of participants in the placebo group and 75 μg PB-119 group, respectively. After 12 weeks of treatment or termination of visit, the positive rates of PB-119 antibody were 3.2% (2 patients) in the placebo group, 22.6% (14 patients) in the 75 μg group, 25.4% (16 patients) in the 150 μg group and 34.9% (22 patients) in the 200 μg group.
Discussion
The main objective of the current study was to assess the efficacy and safety of PB-119 in treatment-naive patients with type 2 diabetes and to identify the relative efficacies of three different doses of PB-119. The results revealed the superior efficacy of PB-119 compared with placebo, and we also observed dose-dependent efficacy up to 150 μg of PB-119, but there was no significant improvement in efficacy at 200 μg of PB-119 compared with 150 μg of PB-119. The results were consistent across different glycaemic endpoints. We found a favourable safety profile after 12 weeks of treatment with no incidence of drug-related serious AEs or severe hypoglycaemia.
In a previous Phase I study assessing the safety and PK/pharmacodynamics of PB-119, the mean peak retention time was found to be between 20 and 40 h, with an elimination t½ of 45–64 h, supporting the once-a-week administration. There was also not much difference in t½ and retention time among the different doses tested in healthy volunteers without any sex-based difference. The clinical laboratory variables, vital signs, ECG and AEs showed that a single dose of 2–200 μg was safe and tolerable. Even after 6 weeks of administration, PB-119 had a long t½ in vivo with a strong correlation between dose and pharmacokinetic variables. After a single subcutaneous dose of 25–400 μg, the maximum serum concentrations achieved ranged from 7 ng/ml to 99 ng/ml with a time to maximum concentration ranging from 19 h to 34 h [23]. In the case of exenatide twice daily, the maximum concentration that was achieved after a dose of 2.5–5 μg was 0.056–0.085 ng/ml with a time to reach maximum serum concentration of 2 h [24]. In the case of PB-119, the steady state concentrations were reached after 2 weeks while with exenatide once weekly and once monthly suspension, steady state concentrations were reported after 6 weeks [9]. The clinical laboratory parameters, vital signs, ECG and AEs showed that a single dose of PB-119 within the range of 2–200 μg was safe and tolerable.
The findings of our study were consistent with previous placebo-controlled studies with exenatide twice daily and exenatide once weekly (microsphere technology). In the current study, LSM difference of change in HbA1c with reference to the placebo arm was significantly better in the PB-119-treated groups, ranging from −7.76 mmol/mol (−0.72%) to −12.89 mmol/mol (−1.18%). In the early clinical trial with exenatide twice daily, the placebo-adjusted change in HbA1c from baseline ranged from −0.98% to −0.58% in patients previously treated with OADs [17]. The results of our study suggest that PB-119 may have retained similar if not superior glycaemic control. Currently, Phase II dose-finding studies for PB-119 in combination with metformin in patients with HbA1c > 58 mmol/mol (>7.5%) after treatment with metformin have been completed (NCT03604419).
The main advantage of exenatide once weekly is the improved patient adherence because of less frequent dosing. In addition, previous studies have established the superior glycaemic control of exenatide once weekly. In a previous study by Drucker et al., after 30 weeks of treatment in treatment-naive patients with type 2 diabetes, exenatide once weekly had significantly greater change in HbA1c from baseline than exenatide twice daily did (−1.9% vs −1.5%; p = 0.0023). Glycaemic control with respect to the proportion of patients with HbA1c < 53 mmol/mol (<7.0%) was also significantly higher in the exenatide once weekly group (77% vs 61%; p = 0.0039). Further, the activity of exenatide once weekly was also found to be higher than that of exenatide twice daily in patients with baseline HbA1c > 75 mmol/mol (>9.0%) [19]. The superior efficacy of exenatide once weekly both as monotherapy and as an add-on therapy to OADs and basal insulin was confirmed in the DURATION trials [13]. Considering the mechanism of action of PB-119, which is similar to exenatide QW, PB-119 could also reveal superior efficacy in comparison with available OADs.
In the current study, all PB-119 doses led to clinically meaningful improvement in glycaemic control. Unlike exenatide once weekly and exenatide once-monthly dosing regimens [9], dose-dependent improvement in glycaemic control with PB-119 once weekly seems to attain a plateau at a dose of 150 μg. This suggests a dose of 150 μg to be optimum for Phase III studies. PB-119 also did not lead to significant drug-related hypoglycaemic events, so adjustments in drug dose may not be required for administration, and it could be administered at any time of the day, irrespective of whether the patients are in fasting or fed condition. The AEs with PB-119 suggested that drug titrations is not necessary in patients with different comorbidities. Similar to exenatide once weekly, PB-119 could also be made available as single-use disposable cartridges that may improve patient compliance. PB-119 was reported to be absorbed slowly and have a longer retention time (t½ of ⁓64 h) with low predicted immunogenicity. This is facilitated by the PEGylation technology, which converts small peptides into peptides of larger size, leading to slower renal clearance rates. The utility of PEGylation technology in the treatment of type 2 diabetes was previously explored in the PEGylation of basal insulin [25].
The most well-established AEs in patients treated with GLP-1RAs, as per previous studies were gastrointestinal symptoms and injection-site reactions [26, 27]. In previous studies with GLP-1RAs, nausea was the most predominant AE, which improved with the continuation of study drug [28, 29]. In the current study, gastrointestinal AEs were more common in the 200 μg group, which could be due to the dose-dependent effect. In a previous meta-analysis, the odds of incidence of nausea was higher in patients receiving exenatide 10 μg twice daily than in patients receiving exenatide 5 μg twice daily (OR: 2.28) and exenatide once weekly (OR: 2.78) [30]. This substantiates the dose-dependent increase in gastrointestinal AEs in patients treated with higher doses of exenatide. This increase in nausea may also reduce treatment compliance, which consequently may affect the efficacy of exenatide. This could also contribute to the lack of dose-dependent changes in efficacy from 150 μg to 200 μg PB-119 [31]. The incidence of treatment discontinuation due to AEs was 0% in the placebo group and 1.6% each in 75 μg, 150 μg and 200 μg PB-119 groups, which is much lower than the reported treatment discontinuation rates due to AEs in previous studies with exenatide (up to 4%) [31].
Treatment with PB-119 also significantly reduced total cholesterol and triacylglycerols (150 μg and 200 μg groups, p< 0.05), body weight and LDL-C (200 μg group, p < 0.05). These findings need to be further evaluated in larger Phase III trials. Further, in the current study, 20–30% of the patients were also positive for anti-exenatide antibodies. In a previous study, 45% of the patients treated with exenatide once weekly were found to be positive for low-titre antibodies, which is much higher than reported in the current study. Nevertheless, apart from injection-site reactions, anti-exenatide antibodies have not been reported to affect the safety or efficacy of the drug [26]. The immunogenicity of PB-119 needs to be substantiated in Phase III trials. In the current study, the benefits of PB-119 in patients with different baseline HbA1c levels (subgroups) were also not assessed and should be assessed in Phase III trials. Further, this study was conducted in Chinese patients, and the efficacy and safety results might not be generalisable to other geographic regions. Moreover, the sample size in the current study was determined for the primary endpoint (change in HbA1c), so evaluating PB-119 in a larger patient population over a longer treatment duration may provide further insights. Similarly, the safety events reported in the current study were after a short treatment duration of 12 weeks. Hence further studies with long-term follow-up are required to substantiate our results. Nevertheless, PB-119 showed superior glycaemic control compared with placebo, and we identified 150 μg PB-119 once weekly to be the minimum effective dose with an acceptable safety profile. This needs to be further substantiated in larger Phase III studies investigating PB-119 as a monotherapy in drug-naive patients or as an add-on therapy for patients on OAD and insulin treatment.
To conclude, the tested doses of subcutaneous PB-119 once weekly were found to be an effective treatment option in treatment-naive patients with type 2 diabetes, providing glycaemic benefits with good overall safety and tolerance. The ease of use without titration requirement may provide additional advantages in real-world settings.
Data availability
Study data will be made available by the corresponding author on reasonable request.
Change history
26 May 2021
A Correction to this paper has been published: https://doi.org/10.1007/s00125-021-05460-0
Abbreviations
- AE:
-
Adverse events
- FAS:
-
Full analysis set
- FPG:
-
Fasting plasma glucose
- GLP-1 RAs:
-
Glucagon-like peptide-1 receptor agonists
- LSM:
-
Least squares mean
- MMRM:
-
Mixed-effect model repeated measure
- OADs:
-
Oral antidiabetic drugs
- PB-119:
-
PEGylated exenatide injection
- PEG:
-
Polyethylene glycol
- PK:
-
Pharmacokinetic
- PPG:
-
Postprandial glucose
References
International Diabetes Federation (2019) IDF Diabetes Atlas, 9th edn. International Diabetes Federation, Brussels, Belgium
Upadhyay J, Polyzos SA, Perakakis N et al (2018) Pharmacotherapy of type 2 diabetes: An update. Metab Clin Exp 78:13–42. https://doi.org/10.1016/j.metabol.2017.08.010
Scheen AJ (2016) Investigational insulin secretagogues for type 2 diabetes. Expert Opin Investig Drugs 25(4):405–422. https://doi.org/10.1517/13543784.2016.1152260
Baggio LL, Drucker DJ (2007) Biology of incretins: GLP-1 and GIP. Gastroenterology 132(6):2131–2157. https://doi.org/10.1053/j.gastro.2007.03.054
Hinnen D (2017) Glucagon-like peptide 1 receptor agonists for type 2 diabetes. Diabetes Spectr 30(3):202–210. https://doi.org/10.2337/ds16-0026
Kalra S, Das AK, Sahay RK et al (2019) Consensus recommendations on GLP-1 RA use in the management of type 2 diabetes mellitus: South Asian Task Force. Diabetes Ther 10(5):1645. https://doi.org/10.1007/s13300-019-0669-4
Cernea S, Raz I (2011) Therapy in the early stage: Incretins. Diabetes Care 34(Suppl 2):S264–S271. https://doi.org/10.2337/dc11-s223
Aronoff SL, Berkowitz K, Shreiner B, Want L (2004) Glucose metabolism and regulation: Beyond insulin and glucagon. Diabetes Spectrum 17(3):183–190. https://doi.org/10.2337/diaspect.17.3.183
Wysham CH, MacConell L, Hardy E (2016) Efficacy and safety of multiple doses of exenatide once-monthly suspension in patients with type 2 diabetes: A phase II randomized clinical trial. Diabetes Care 39(10):1768–1776. https://doi.org/10.2337/dc16-0238
Bhavsar S, Mudaliar S, Cherrington A (2013) Evolution of exenatide as a diabetes therapeutic. Curr Diabetes Rev 9(2):161. https://doi.org/10.2174/1573399811309020007
Gutzwiller JP, Drewe J, Göke B et al (1999) Glucagon-like peptide-1 promotes satiety and reduces food intake in patients with diabetes mellitus type 2. Am J Phys 276(5):R1541–R1544. https://doi.org/10.1152/ajpregu.1999.276.5.R1541
DeYoung MB, MacConell L, Sarin V, Trautmann M, Herbert P (2011) Encapsulation of exenatide in poly-(D,L-lactide-co-glycolide) microspheres produced an investigational long-acting once-weekly formulation for type 2 diabetes. Diabetes Technol Ther 13(11):1145–1154. https://doi.org/10.1089/dia.2011.0050
Knop FK, Brønden A, Vilsbøll T (2017) Exenatide: Pharmacokinetics, clinical use, and future directions. Expert Opin Pharmacother 18(6):555–571. https://doi.org/10.1080/14656566.2017.1282463
Association AD (2019) 9. Pharmacologic approaches to glycemic treatment: Standards of medical care in diabetes—2019. Diabetes Care 42(Supplement 1):S90–S102. https://doi.org/10.2337/dc19-S009
Tobin GS, Cavaghan MK, Hoogwerf BJ, McGill JB (2012) Addition of exenatide twice daily to basal insulin for the treatment of type 2 diabetes: Clinical studies and practical approaches to therapy. Int J Clin Pract 66(12):1147–1157. https://doi.org/10.1111/ijcp.12032
Genovese S, Mannucci E, Ceriello A (2017) A review of the long-term efficacy, tolerability, and safety of exenatide once weekly for type 2 diabetes. Adv Ther 34(8):1791–1814. https://doi.org/10.1007/s12325-017-0499-6
Buse JB, Henry RR, Han J et al (2004) Effects of exenatide (exendin-4) on glycemic control over 30 weeks in sulfonylurea-treated patients with type 2 diabetes. Diabetes Care 27(11):2628–2635. https://doi.org/10.2337/diacare.27.11.2628
DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD (2005) Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care 28(5):1092–1100. https://doi.org/10.2337/diacare.28.5.1092
Drucker DJ, Buse JB, Taylor K et al (2008) Exenatide once weekly versus twice daily for the treatment of type 2 diabetes: A randomised, open-label, non-inferiority study. Lancet 372(9645):1240–1250. https://doi.org/10.1016/S0140-6736(08)61206-4
Buse JB, Drucker DJ, Taylor KL et al (2010) DURATION-1: Exenatide once weekly produces sustained glycemic control and weight loss over 52 weeks. Diabetes Care 33(6):1255–1261. https://doi.org/10.2337/dc09-1914
Gadde KM, Vetter ML, Iqbal N, Hardy E, Öhman P (2017) Efficacy and safety of autoinjected exenatide once-weekly suspension versus sitagliptin or placebo with metformin in patients with type 2 diabetes: The DURATION-NEO-2 randomized clinical study. Diabetes Obes Metab 19(7):979–988. https://doi.org/10.1111/dom.12908
Kang JS, DeLuca PP, Lee KC (2009) Emerging PEGylated drugs. Expert Opin Emerg Drugs 14(2):363–380. https://doi.org/10.1517/14728210902907847
Cui H, Zhao C-Y, Lv Y et al (2020) Safety, tolerability and pharmacokinetics of single dose polyethylene glycolated exenatide injection (PB-119) in healthy volunteers. Eur J Drug Metab Pharmacokinet. https://doi.org/10.1007/s13318-020-00605-9
McCormack PL (2014) Exenatide twice daily: A review of its use in the management of patients with type 2 diabetes mellitus. Drugs 74(3):325–351. https://doi.org/10.1007/s40265-013-0172-6
Muñoz-Garach A, Molina-Vega M, Tinahones FJ (2017) How can a good idea fail? Basal insulin peglispro [LY2605541] for the treatment of type 2 diabetes. Diabetes Ther 8(1):9. https://doi.org/10.1007/s13300-016-0214-7
Fineman MS, Mace KF, Diamant M et al (2012) Clinical relevance of anti-exenatide antibodies: Safety, efficacy and cross-reactivity with long-term treatment. Diabetes Obes Metab 14(6):546–554. https://doi.org/10.1111/j.1463-1326.2012.01561.x
Sun F, Chai S, Yu K et al (2015) Gastrointestinal adverse events of glucagon-like peptide-1 receptor agonists in patients with type 2 diabetes: A systematic review and network meta-analysis. Diabetes Technol Ther 17(1):35–42. https://doi.org/10.1089/dia.2014.0188
Reusch J, Stewart MW, Perkins CM et al (2014) Efficacy and safety of once-weekly glucagon-like peptide 1 receptor agonist albiglutide (HARMONY 1 trial): 52-week primary endpoint results from a randomized, double-blind, placebo-controlled trial in patients with type 2 diabetes mellitus not controlled on pioglitazone, with or without metformin. Diabetes Obes Metab 16(12):1257–1264
Buse JB, Rosenstock J, Sesti G et al (2009) Liraglutide once a day versus exenatide twice a day for type 2 diabetes: A 26-week randomised, parallel-group, multinational, open-label trial (LEAD-6). Lancet 374(9683):39–47. https://doi.org/10.1016/S0140-6736(09)60659-0
Sun F, Yu K, Yang Z et al (2012) Impact of GLP-1 receptor agonists on major gastrointestinal disorders for type 2 diabetes mellitus: A mixed treatment comparison meta-analysis. Exp Diabetes Res 2012:230624. https://doi.org/10.1155/2012/230624
Ratner RE, Maggs D, Nielsen LL et al (2006) Long-term effects of exenatide therapy over 82 weeks on glycaemic control and weight in over-weight metformin-treated patients with type 2 diabetes mellitus. Diabetes Obes Metab 8(4):419–428. https://doi.org/10.1111/j.1463-1326.2006.00589.x
Acknowledgements
The authors acknowledge medical writing assistance provided by G.K. Subramanian and A. Bhat of Indegene Pvt. Ltd., Bangalore, India.
Author’s relationships and activities
YD, MX and XZ are employees of PegBio Co., Ltd. All the other authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Funding
The study was funded by National Major Scientific and Technological Special Project for Significant New Drugs Development during the thirteenth Five Year Plan Period (2016ZX09101033) and PegBio Co., Ltd. The sponsor/funder was involved in study design and data collection and funded editorial support in the preparation of this publication. The authors are individually and collectively responsible for all content and editorial decisions and received no payment from the sponsor/funder for the development of this publication.
Author information
Authors and Affiliations
Contributions
LJ, Ying D, MX, XZ, ZM, JM, Jiarui L, YL, Jingna L, YW, Jing Y, Jinkui Y, GQ, SL and LC contributed to study conception and design, analysis and interpretation of data and drafting the article. LJ, ZM, JM, Jiarui L, YL, Jingna L, YW, Jing Y, Jinkui Y, GQ, SL, LC, WS, HJ, SP, HL, PL, Jie L, MY, Wenhui L, XJ, FS, HG, HZ, JR, ML, Yinghong D, SY, QG, Wenshan L, HT and TC contributed to data acquisition and analysis, and critical revision of the article. All the authors have given approval of the version to be published. LJ is the guarantor of this work, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. LJ attests that all listed authors meet authorship criteria and that no others meeting the criteria have been omitted.
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: The following sentence in the Discussion was inappropriately changed to a definite statement during editing: ‘Considering the mechanism of action of PB-119, which is similar to exenatide QW, PB-119 also showed superior efficacy compared with available OADs.’The journal would like to revert back to the authors’ original phrasing as follows: ‘Considering the mechanism of action of PB-119, which is similar to exenatide QW, PB-119 could also reveal superior efficacy in comparison with available OADs.’
Supplementary Information
ESM Tables
(PDF 137 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ji, L., Du, Y., Xu, M. et al. Efficacy and safety of PEGylated exenatide injection (PB-119) in treatment-naive type 2 diabetes mellitus patients: a Phase II randomised, double-blind, parallel, placebo-controlled study. Diabetologia 64, 1066–1078 (2021). https://doi.org/10.1007/s00125-021-05392-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-021-05392-9