
Abstract
Aims/hypothesis
Whether metformin reduces cardiovascular or cancer risk is unclear owing to concerns over immortal time bias and confounding in observational studies. This study evaluated the effect of AMP-activated protein kinase (AMPK), the target of metformin, on risk of cardiovascular disease and cancer.
Methods
This is a Mendelian randomisation design, using AMPK, the pharmacological target of metformin, to infer the AMPK pathway-dependent effects of metformin on risk of cardiovascular disease and cancer in participants of white British ancestry in the UK Biobank.
Results
A total of 391,199 participants were included (mean age 56.9 years; 54.1% women), including 26,690 cases of type 2 diabetes, 38,098 cases of coronary artery disease and 80,941 cases of overall cancer. Genetically predicted reduction in HbA1c (%) instrumented by AMPK variants was associated with a 61% reduction in risk of type 2 diabetes (OR 0.39; 95% CI 0.20, 0.78; p = 7.69 × 10−3), a 53% decrease in the risk of coronary artery disease (OR 0.47; 95% CI 0.26, 0.84; p = 0.01) and a 44% decrease in the risk of overall cancer (OR 0.56; 95% CI 0.36, 0.85; p = 7.23 × 10−3). Results were similar using median or quartiles of AMPK score, with dose–response effects (p for trend = 4.18 × 10−3 for type 2 diabetes, 4.37 × 10−3 for coronary artery disease and 4.04 × 10−3 for overall cancer).
Conclusions/interpretation
This study provides some genetic evidence that AMPK activation by metformin may protect against cardiovascular disease and cancer, which needs to be confirmed by randomised controlled trials.
Graphical abstract

Similar content being viewed by others



Introduction
Metformin is the first-line pharmacological treatment to manage hyperglycaemia in people with type 2 diabetes, and is on the World Health Organization list of essential medicines [1]. Increasing evidence suggests that metformin may differ from other classes of glucose-lowering medications in having superior safety and lower risk of cardiovascular complications [2]. Furthermore, pharmaco-epidemiological studies have suggested that metformin may reduce cardiovascular disease and cancer [3, 4], suggesting the possibility of its use for these diseases. Metformin not only impacts glycaemic traits but also other potentially relevant factors, such as growth differentiation factor 15 (GDF-15) and vascular endothelial growth factors [5]. However, pharmaco-epidemiological studies may be open to immortal time bias and confounding, which may generate spurious protective effects of metformin, in particular for cancer-related studies [6, 7]. To date, relevant randomised controlled trials of metformin in cardiovascular disease are not large enough to be definitive [8], whilst the impact of metformin on cancer has not been evaluated fully in a randomised controlled trial.
Mendelian randomisation studies, which make use of the random allocation of genetic variants at conception, are less susceptible to confounding and time-related biases than other observational studies, and are now increasingly used to infer health effects of medications, such as the use 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGCR) variants to mimic the health effects of statins [9]. Previous Mendelian randomisation studies have attempted to use genetics to infer health effects of metformin, but they were potentially underpowered and included non-specific instruments [10], or only evaluated health effects of metformin biomarkers instead of metformin itself [11]. To provide more definitive and direct evidence concerning the effects of metformin on cardiovascular disease and cancer risk, we conducted a Mendelian randomisation study using AMP-activated protein kinase (AMPK), the target of metformin, as a proxy of metformin use, in one of the largest prospective cohort studies globally.
Methods
Study design
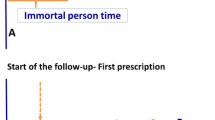
This is a Mendelian randomisation design, using AMPK, the pharmacological target of metformin, to infer the AMPK pathway-dependent effects of metformin. The study design is depicted in Fig. 1 [12].

Study design of this Mendelian randomisation study (a) and its comparison with a randomised controlled trial (b)
Study population
The UK Biobank recruited ~500,000 participants intended to be aged 39–73 years between 2006 and 2010 from 22 recruitment centres across Scotland, Wales and England in the UK. Participants provided biological samples; completed questionnaires, covering self-reported diseases and regular prescription medications; underwent assessments; and had nurse-led interviews. A blood sample for standard haematological tests was collected by venepuncture in ethylenediaminetetraacetic acid tubes, and tested at the central processing laboratory in Stockport, within 24 h of blood collection. HbA1c was measured by high performance liquid chromatography on Bio-Rad Variant II Turbo analysers (Bio-Rad Laboratories, CA, USA). Longitudinal follow-up via record linkage to all health service encounters and death is ongoing. Hospital inpatient data and cancer registries used ICD-9 and ICD-10 codes and death registries used ICD-10 codes. Genotyping was undertaken with two similar arrays, the UK Biobank Lung Exome Variant Evaluation (BiLEVE) Axiom array (49,979 participants) and the UK Biobank Axiom array (438,398 participants). Genotype imputation was based on the reference panel combining the UK10K haplotype and the Haplotype Reference Consortium reference panels. To reduce confounding by latent population structure [13], we restricted the analysis to genetically verified white British participants and further excluded participants with (1) withdrawn consent; (2) sex mismatch (genetic sex differs from reported sex); (3) putative sex chromosomes aneuploidy; (4) poor-quality genotyping (outliers in heterozygosity and missing rate >1.5%); or (5) excessive relatedness (more than ten putative third-degree relatives), as per our previous study [14]. We used genotype and phenotype data from the UK Biobank provided in February 2020.
AMPK genetic score
We created a weighted AMPK genetic score to mimic the effects of AMPK activation by metformin use based on the strength of the association of genetic variants in the relevant gene regions with HbA1c in the Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC), a genome-wide association study (GWAS) of HbA1c, with validation in the UK Biobank. Specifically, we selected genetic variants within 1 megabase pairs downstream and upstream of each of the PRKAA1, PRKAA2, PRKAB1, PRKAB2, PRKAG1, PRKAG2 and PRKAG3 genes that encode AMPK subunits [15]. We selected low-linkage disequilibrium (r2 < 0.3) variants associated with HbA1c at a nominal level of statistical significance (p ≤ 0.05) in MAGIC, restricted to people of European ancestry to minimise population stratification (n = 123,665) [16]. We then validated the associations in the UK Biobank (using multivariable linear regression, adjusted for sex, age at recruitment, genotyping array and the first 20 principal components of genetic ancestry) and only retained variants also reaching statistical significance (p ≤ 0.05) in the UK Biobank, which were used to construct the AMPK score. Electronic supplementary material (ESM) Table 1 and ESM Fig. 1 show the details regarding the 44 variants used to construct the AMPK score. A weighted AMPK score was calculated for each participant by summing the number of HbA1c-lowering alleles that a participant inherited at each variant included in the AMPK score, weighted by the effect of that variant on HbA1c measured in percentage (as estimated in MAGIC) [16]. This score was then considered as above and below the median to mimic metformin use and non-use. We also considered AMPK score in quartiles to assess whether our finding was robust to the way we consider AMPK scores.
Sensitivity analysis
As a sensitivity analysis, we used a stringent variant selection criterion by using only variants associated with HbA1c at genome-wide significance (p ≤ 5 × 10−8) in both MAGIC and UK Biobank and not in linkage disequilibrium with the other variants (r2< 0.01), which gave rs2732480 (ESM Table 1). rs2732480 was associated with lower HbA1c in both MAGIC (p = 2 × 10−9) and UK Biobank (p = 1.07 × 10−142). The effect allele was associated with lower HbA1c percentage (β −0.012; 95% CI −0.016, −0.008).
Study outcomes
The primary outcomes were coronary artery disease and overall cancer. The secondary outcomes were stroke and three main cancers, i.e. breast cancer, colorectal cancer and prostate cancer. Each disease outcome was defined based on self-reported medical conditions at baseline, or subsequent primary and secondary diagnoses of hospital episodes (ICD-9 and ICD-10), or cancer diagnosis (ICD-9 and ICD-10), or underlying and contributing causes of death (ICD-10).
Positive control outcomes
We included type 2 diabetes and HbA1c as positive control outcomes given that these are the expected effects of metformin use. Type 2 diabetes was ascertained using a validated algorithm [17]. Specifically, the criteria included (1) self-reported type 2 diabetes at baseline; (2) indication of type 2 diabetes based on diagnostic codes (ICD-9250 and ICD-10 E11); (3) diabetes medications (metformin, sodium-glucose cotransporter 2 inhibitors, glucagon-like peptide 1 receptor agonists, dipeptidyl peptidase 4 inhibitors, sulfonylureas and thiazolidinediones); and (4) hyperglycaemic blood result (either HbA1c ≥6.5% or 48 mmol/mol, or random glucose ≥11.1 mmol/l). The algorithmic definitions are described in ESM Table 2. HbA1c was measured in mmol/mol (International Federation of Clinical Chemistry [IFCC] unit), and was converted to percentage (National Glycohemoglobin Standardization Program [NGSP] unit) using the equation: NGSP = (0.09148 × IFCC) + 2.152 [18].
External validation
To validate our findings from the UK Biobank, we conducted an external validation study for the outcomes using summary statistics for type 2 diabetes (12,171 cases and 56,862 control participants) from the DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) consortium [19], for coronary artery disease (60,801 cases and 123,504 control participants) from the Coronary ARtery DIsease Genome wide Replication and Meta-analysis plus The Coronary Artery Disease Genetics (CARDIoGRAMplusC4D) consortium [20], for stroke (40,585 cases of stroke and 406,111 control participants) from the MEGASTROKE consortium [21], for breast cancer (122,977 cases and 105,974 control participants) from the Breast Cancer Association Consortium (BCAC) [22] and for prostate cancer (79,148 cases and 61,106 control participants) from the Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome (PRACTICAL) consortium [23]. All participants were of predominantly European ancestry and non-overlapping with the participants in the UK Biobank to avoid bias due to population structure and any potential bias due to participant overlap for a weak instrument [24].
Statistical analysis
To assess the assumption of independence of the genetic instruments (AMPK groups) from potential confounders, we assessed the association of AMPK groups with confounders (age at recruitment, BMI, smoking status, alcohol drinking status, education level, Townsend deprivation index) using χ2 tests or ANOVA. To demonstrate that AMPK had the expected effect on HbA1c, we assessed the differences in HbA1c and random glucose between each group using ANOVA. We assessed the association of AMPK categories with HbA1c using multivariable linear regression. We assessed the association of AMPK categories with risk of type 2 diabetes, cardiovascular diseases and cancers using multivariable logistic regression. All regression analyses were adjusted for sex (if relevant), age at recruitment, genotyping array and the first 20 principal components of genetic ancestry. As per previous studies, we also assessed the impact of genetically predicted reduction in HbA1c (%) instrumented by AMPK variants on risk of type 2 diabetes, coronary artery disease and overall cancer [25].
For the external validation, we performed a standard Mendelian randomisation analysis. We obtained the summary statistics of each variant included in the AMPK score on risk of type 2 diabetes, coronary artery disease, stroke, breast cancer and prostate cancer as reported by each consortium. We obtained the Wald ratio for each variant (the ratio of the genetic association with outcome to the genetic association of exposure), and then combined them using weighted generalised linear regression in an inverse variance-weighted manner and accounted for the correlation between variants [26]. The correlations between variants were obtained in 503 participants of European ancestry from the 1000 Genomes Project (Phase 3). Since variants are from multiple gene regions that may have different mechanisms of effect, a random-effects model was used [26]. We aligned the effect allele of each variant to the HbA1c decreasing allele. We used the Cochran’s Q statistic to assess heterogeneity of the Wald ratios [27], where high heterogeneity may indicate the presence of invalid genetic variants [28].
Exploring the association of HbA1c with cardiovascular disease and cancer risk using Mendelian randomisation
To preclude the possibility that the observed effects of AMPK activation, a target of metformin, are due to lowering HbA1c, we also assessed the association of genetically predicted lower HbA1c on cardiovascular disease and cancer risk. As previously, we obtained 38 independent genetic variants strongly related to HbA1c (p ≤ 5 × 10−8) from MAGIC (ESM Table 3), and applied them to the relevant outcomes in the UK Biobank using inverse variance-weighted, MR-Egger and a weighted median method [14].
The AMPK score was generated using PLINK 2.0 (https://www.cog-genomics.org/plink/2.0/) [29]. Mendelian randomisation analyses were performed with the MendelianRandomisation package version 0.4.2 [30] and all analyses were performed using R software, version 3.5.1 (R Foundation for Statistical Computing, Vienna, Austria) [31]. A two-tailed p value less than 0.05 was considered statistically significant.
Ethical approval
The UK Biobank received ethical approval from the North West Multi-centre Research Ethics Committee (11/NW/0382), and all participants provided written, informed consent. No ethics approval was acquired for the analyses using summary statistics. The contributing studies to the consortium received ethical approval from their specific institutional review boards, and written, informed consent was obtained from all participants.
Results
Participant characteristics
A total of 391,199 participants were included in the main analysis (mean age, 56.9 years; 54.1% women). For type 2 diabetes, there were 26,690 cases. For cardiovascular disease, there were 38,098 cases of coronary artery disease and 11,358 cases of stroke. For cancer, there were 80,941 cases of overall cancer, 9251 cases of breast cancer, 5861 cases of colorectal cancer and 8970 cases of prostate cancer. In Table 1, HbA1c, glucose, insulin therapy users and metformin therapy users were significantly lower in the high AMPK group than in the low group. No other significant differences in baseline characteristics between the two groups were found.
Association of AMPK score with glycaemic traits and type 2 diabetes
Compared with participants with a low AMPK score (below median favouring higher HbA1c), participants with a high AMPK score (above median) had lower HbA1c (β −0.032%; 95% CI −0.035, −0.028; p = 2.34 × 10−64) and lower random blood glucose (β −0.013 mmol/l; 95% CI −0.022, −0.005, p = 1.09 × 10−3). High AMPK score (above median) was also associated with a decreased risk of type 2 diabetes (OR 0.96; 95% CI 0.94, 0.99; p = 4.16 × 10−3), as shown in Fig. 2a. AMPK quartiles were associated with a stepwise decrease in HbA1c (quartile 2, −0.011%; 95% CI −0.016, −0.006; p = 2.70 × 10−5; quartile 3, −0.028%; 95% CI −0.033, −0.022; p = 2.02 × 10−25; and quartile 4, −0.047%; 95% CI −0.052, −0.042; p = 4.67 × 10−70), and a corresponding stepwise decrease in the risk of type 2 diabetes (p for trend = 4.18 × 10−3, Fig. 2a). Genetically predicted reduction in HbA1c (%) instrumented by AMPK variants was associated with a 61% decrease in the risk of type 2 diabetes (OR 0.39 per % reduction; 95% CI 0.20, 0.78; p = 7.69 × 10−3) (Fig. 3).

Association of AMPK score with risk of type 2 diabetes (a), coronary heart disease (b) and overall cancer (c) in the UK Biobank. Boxes represent ORs and lines represent 95% CIs

The impact of genetically predicted reduction in HbA1c (%) instrumented by AMPK variants on risk of type 2 diabetes, coronary artery disease and overall cancer in the UK Biobank. Boxes represent ORs and lines represent 95% CIs
Association of AMPK score with cardiovascular diseases
High AMPK score (above median) was associated with a 3% lower risk of coronary artery disease (OR 0.97; 95% CI 0.95, 0.99; p = 5.69 × 10−3; Fig. 2b), but not stroke (ESM Fig. 2a). AMPK quartile was associated with a stepwise decrease in the risk of coronary artery disease (p for trend = 4.37 × 10−3; Fig. 2b). Genetically predicted reduction in HbA1c (%) instrumented by AMPK variants was associated with a 53% decrease in the risk of coronary artery disease (OR 0.47 per % reduction; 95% CI 0.26, 0.84; p = 0.01) (Fig. 3).
Association of AMPK score with cancer
High AMPK score (above median) was associated with lower risk of overall cancer (OR 0.98; 95% CI 0.96, 1.00; p = 0.01 and p for trend = 4.04 × 10−3; Fig. 2c), but not with prostate cancer, breast cancer or colorectal cancer (ESM Fig. 2b–d). AMPK quartile was associated with prostate cancer (quartile 2, OR 0.91; 95% CI 0.85, 0.96; p = 1.61 × 10−3; quartile 3, OR 0.93; 95% CI 0.88, 0.99; p = 0.02), although the dose–response was unclear (ESM Fig. 2b). Genetically predicted reduction in HbA1c (%) instrumented by AMPK variants was associated with a 44% decrease in the risk of overall cancer (OR 0.56 per % reduction; 95% CI 0.36, 0.85; p = 7.23 × 10−3) (Fig. 3).
Sensitivity analysis by using a more stringent variant selection criterion
A 1% reduction in HbA1c instrumented by rs2732480 was associated with a decreased risk of type 2 diabetes (OR 0.11; 95% CI 0.02, 0.50; p = 4.08 × 10−3) and coronary artery disease (OR 0.22; 95% CI 0.06, 0.81; p = 0.02). The direction with overall cancer was consistent with the main analysis but with wider CI (OR 0.45; 95% CI 0.17, 1.14; p = 0.09) (ESM Table 4).
External validation
In external replication analyses, genetically predicted lower HbA1c instrumented by AMPK variants was associated with decreased risk of type 2 diabetes (OR 0.11 per % reduction; 95% CI 0.04, 0.35; p = 1.78 × 10−4) and coronary artery disease (OR 0.48 per % reduction; 95 CI 0.33, 0.72; p = 2.89 × 10−4), but not with stroke, breast cancer or prostate cancer (ESM Table 5 and ESM Fig. 3a–e). The Q statistic suggested possible heterogeneity for the associations with type 2 diabetes, coronary artery disease, stroke and breast cancer.
Association of HbA1c with cardiovascular disease and cancer risk in the UK Biobank using Mendelian randomisation
ESM Table 6 shows that genetically predicted higher HbA1c was associated with higher risk of coronary artery disease (OR 1.41; 95% CI 1.03, 1.93; p = 0.03), and possibly with lower risk of overall cancer (OR 0.84; 95% CI 0.70, 1.01; p = 0.07), but not with stroke, or any cancer subtype.
Discussion
To the best of our knowledge, this is one of the first Mendelian randomisation studies to ascertain the effects of metformin, based on AMPK variants, on cardiovascular diseases and cancer. Using a design more robust to immortal time biases and confounding, our study is consistent with previous pharmaco-epidemiological studies suggesting that metformin use may reduce coronary artery disease and overall cancer risk. We added some genetic evidence that the putative cancer-protective effect of metformin via AMPK pathways is unlikely to be associated with glycaemic control.
A protective effect of metformin on cardiovascular health was observed in small randomised controlled trials using surrogate outcomes [32], the UK Prospective Diabetes Study post-trial analysis [33] and a recent meta-analysis [8], which were consistent with our findings. Although a genetically predicted reduction in HbA1c is protective against coronary artery disease [14], it is apparent that metformin’s protective effect is not solely due to its improvement in glycaemic profile given that these benefits are not clearly observed for all other classes of glucose-lowering medications [8], such as sulfonylureas and insulin [2, 34]. Metformin increases GDF-15, a stress-responsive cytokine which suppresses appetite and promotes weight loss [35], hence providing a potential mechanistic pathway by which metformin reduces cardiovascular disease risk. However, changes in GDF-15 were not clearly associated with coronary artery disease risk based on our previous Mendelian randomisation study [11]. On the contrary, sulfonylureas and insulin may lead to cardiotoxicity via weight gain, hypoglycaemia [34] or alteration of hormone levels [36].
The relation of metformin use with cancer risk is more controversial given the concern over immortal time bias [6]. Our study, where the start of ‘exposure’ is at birth, effectively removes this bias. As such, our study adds by showing that immortal time bias alone may not have explained the inverse relation of metformin use with cancer risk. Given that previous studies generally have ruled out the causal role of glycaemic traits in cancer risk [10, 37], possible mechanisms underlying the anti-cancer property of metformin are likely via pre-cursors of glycaemic traits or of glycaemic-independent pathways [38]. People without growth hormone appear to be protected against both diabetes and cancer [39]. This may suggest a possible pathway via growth hormone or the closely related insulin like growth factor-1 [40]. Glycaemic-independent pathways may include inhibition of tumour-mesothelial cell interaction by suppressing hypoxia-inducible factor 1α and TGF-β signalling [41], immune-mediated responses via metabolic reprogramming of tumour-specific T cells [42] and GDF-15 overexpressing fibroblasts promoting the growth of tumour xenografts [43]. The examination of these potential mechanisms can be explored in further studies. Big data approaches, such as metabolomics, may also be warranted to better understand the full spectrum of effects of metformin, and hence to help identify the main pathways in which metformin confers the additional benefits on cardiovascular disease and cancer [44].
Although our study is more robust to confounding and immortal time bias than previous observational studies, there are limitations. First, whilst our study suggested that AMPK activation by metformin may protect against coronary artery disease, and possibly cancer, the estimates from this study cannot be used directly to infer the health impact of metformin given the differences in exposure time, where randomised controlled trials often consider short-term pharmacological treatment in contrast to the effects of lifelong exposures estimated by Mendelian randomisation [12]. Moreover, our study using AMPK variants may only predict the effect of metformin which acts on the AMPK activation pathways, and metformin may also have AMPK-independent pathways that could be explored in additional studies to fully capture the overall effect of metformin on cardiovascular disease and cancer [45]. Second, we used a lower threshold than genome-wide statistical significance to select AMPK variants as a proxy of metformin use to maximise total prediction of AMPK function by the genetic score. We reduced the possibility of false positives by cross-checking the variants’ associations with HbA1c in two independent studies. We also repeated the analysis with stringent variant selection criteria which gave a consistent conclusion. However, this may compromise the generalisability of the genetic score in other studies [46]. Third, we cannot rule out selection bias resulting from the recruitment of generally healthier participants and survivors in the UK Biobank, which may bias the estimate towards the null. We also cannot rule out selection bias from competing risk before recruitment for diseases which share risk factors with other diseases that typically occur at younger ages, which could have biased estimates for stroke and prostate cancer to the null. Forth, the Q statistic suggested possible heterogeneity in some analyses. These heterogeneities may imply that multiple gene regions encoding subunits of AMPK may have different mechanisms of influencing the outcomes, and should be explored in future studies [28]. Fifth, given the pleiotropic effects of metformin and its association with multiple non-glycaemic makers [5], it would be difficult to identify a suitable negative control outcome. Nevertheless, we also assessed the impact of HbA1c on these outcomes and found that HbA1c unlikely explained all of the observed associations related to AMPK. Lastly, we could not exclude the possibility that metformin may reduce subtypes of cancer as the number of cases was not large enough for adequate statistical power, although the directions of effect for some cancer subtypes are similar to overall cancer. Few AMPK genetic variants were available for prostate cancer and breast cancer in the consortia and we were unable to create an overall genetic score in the associated analyses to increase statistical power. Together with possible selection biases embedded in these GWAS [47], these factors might explain the discrepancies between the estimates from the UK Biobank and from external consortia. Further investigations in large consortia on specific cancers may help verify the potential anti-cancer property of metformin.
Conclusion
This Mendelian randomisation study provides some genetic evidence that AMPK activation by metformin may reduce coronary artery disease risk, and possibly overall cancer risk. Whether metformin can be repurposed for coronary artery disease and cancer should be explored in large randomised controlled trials.
Data availability
The data generated and analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AMPK:
-
AMP-activated protein kinase
- GDF-15:
-
Growth differentiation factor 15
- GWAS:
-
Genome-wide association study
- IFCC:
-
International Federation of Clinical Chemistry
- MAGIC:
-
Meta-Analyses of Glucose and Insulin-related traits Consortium
- NGSP:
-
National Glycohemoglobin Standardization Program
References
Bailey CJ (2017) Metformin: historical overview. Diabetologia 60(9):1566–1576. https://doi.org/10.1007/s00125-017-4318-z
Douros A, Dell’Aniello S, Yu OHY, Filion KB, Azoulay L, Suissa S (2018) Sulfonylureas as second line drugs in type 2 diabetes and the risk of cardiovascular and hypoglycaemic events: population based cohort study. BMJ 362:k2693. https://doi.org/10.1136/bmj.k2693
Heckman-Stoddard BM, DeCensi A, Sahasrabuddhe VV, Ford LG (2017) Repurposing metformin for the prevention of cancer and cancer recurrence. Diabetologia 60(9):1639–1647. https://doi.org/10.1007/s00125-017-4372-6
Rena G, Lang CC (2018) Repurposing metformin for cardiovascular disease. Circulation 137(5):422–424. https://doi.org/10.1161/CIRCULATIONAHA.117.031735
Gerstein HC, Pare G, Hess S et al (2017) Growth differentiation factor 15 as a novel biomarker for metformin. Diabetes Care 40(2):280–283. https://doi.org/10.2337/dc16-1682
Suissa S, Azoulay L (2012) Metformin and the risk of cancer: time-related biases in observational studies. Diabetes Care 35(12):2665–2673. https://doi.org/10.2337/dc12-0788
Farmer RE, Ford D, Mathur R et al (2019) Metformin use and risk of cancer in patients with type 2 diabetes: a cohort study of primary care records using inverse probability weighting of marginal structural models. Int J Epidemiol 48(2):527–537. https://doi.org/10.1093/ije/dyz005
Zhu J, Yu X, Zheng Y et al (2020) Association of glucose-lowering medications with cardiovascular outcomes: an umbrella review and evidence map. Lancet Diabetes Endocrinol 8(3):192–205. https://doi.org/10.1016/S2213-8587(19)30422-X
Ference BA, Robinson JG, Brook RD et al (2016) Variation in PCSK9 and HMGCR and risk of cardiovascular disease and diabetes. N Engl J Med 375(22):2144–2153. https://doi.org/10.1056/NEJMoa1604304
Au Yeung SL, Schooling CM (2019) Impact of glycemic traits, type 2 diabetes and metformin use on breast and prostate cancer risk: a Mendelian randomization study. BMJ Open Diabetes Res Care 7(1):e000872. https://doi.org/10.1136/bmjdrc-2019-000872
Au Yeung SL, Luo S, Schooling CM (2019) The impact of GDF-15, a biomarker for metformin, on the risk of coronary artery disease, breast and colorectal cancer, and type 2 diabetes and metabolic traits: a Mendelian randomisation study. Diabetologia 62(9):1638–1646. https://doi.org/10.1007/s00125-019-4913-2
Ference BA (2018) How to use Mendelian randomization to anticipate the results of randomized trials. Eur Heart J 39(5):360–362. https://doi.org/10.1093/eurheartj/ehx462
Haworth S, Mitchell R, Corbin L et al (2019) Apparent latent structure within the UK Biobank sample has implications for epidemiological analysis. Nat Commun 10(1):333. https://doi.org/10.1038/s41467-018-08219-1
Au Yeung SL, Luo S, Schooling CM (2018) The impact of glycated hemoglobin (HbA1c) on cardiovascular disease risk: a Mendelian randomization study using UK Biobank. Diabetes Care 41(9):1991–1997. https://doi.org/10.2337/dc18-0289
Xiao B, Heath R, Saiu P et al (2007) Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 449(7161):496–500. https://doi.org/10.1038/nature06161
Wheeler E, Leong A, Liu CT et al (2017) Impact of common genetic determinants of hemoglobin A1c on type 2 diabetes risk and diagnosis in ancestrally diverse populations: a transethnic genome-wide meta-analysis. PLoS Med 14(9):e1002383. https://doi.org/10.1371/journal.pmed.1002383
Eastwood SV, Mathur R, Atkinson M et al (2016) Algorithms for the capture and adjudication of prevalent and incident diabetes in UK Biobank. PLoS One 11(9):e0162388. https://doi.org/10.1371/journal.pone.0162388
English E, Lenters-Westra E (2018) HbA1c method performance: the great success story of global standardization. Crit Rev Cl Lab Sci 55(6):408–419. https://doi.org/10.1080/10408363.2018.1480591
Morris AP, Voight BF, Teslovich TM et al (2012) Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 44(9):981–990. https://doi.org/10.1038/ng.2383
Nikpay M, Goel A, Won HH et al (2015) A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet 47(10):1121–1130. https://doi.org/10.1038/ng.3396
Malik R, Chauhan G, Traylor M et al (2018) Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet 50(4):524–537. https://doi.org/10.1038/s41588-018-0058-3
Michailidou K, Lindstrom S, Dennis J et al (2017) Association analysis identifies 65 new breast cancer risk loci. Nature 551(7678):92–94. https://doi.org/10.1038/nature24284
Schumacher FR, Al Olama AA, Berndt SI et al (2018) Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat Genet 50(7):928–936. https://doi.org/10.1038/s41588-018-0142-8
Burgess S, Davies NM, Thompson SG (2016) Bias due to participant overlap in two-sample Mendelian randomization. Genet Epidemiol 40(7):597–608. https://doi.org/10.1002/gepi.21998
Yarmolinsky J, Bull CJ, Vincent EE et al (2020) Association between genetically proxied inhibition of HMG-CoA reductase and epithelial ovarian cancer. JAMA 323(7):646–655. https://doi.org/10.1001/jama.2020.0150
Burgess S, Dudbridge F, Thompson SG (2016) Combining information on multiple instrumental variables in Mendelian randomization: comparison of allele score and summarized data methods. Stat Med 35(11):1880–1906. https://doi.org/10.1002/sim.6835
Greco MF, Minelli C, Sheehan NA, Thompson JR (2015) Detecting pleiotropy in Mendelian randomisation studies with summary data and a continuous outcome. Stat Med 34(21):2926–2940. https://doi.org/10.1002/sim.6522
Burgess S, Butterworth A, Thompson SG (2013) Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol 37(7):658–665. https://doi.org/10.1002/gepi.21758
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4:7. https://doi.org/10.1186/s13742-015-0047-8
Yavorska OO, Burgess S (2017) MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int J Epidemiol 46(6):1734–1739. https://doi.org/10.1093/ije/dyx034
R Core Team (2013) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Larsen AH, Jessen N, Norrelund H et al (2019) A randomised, double-blind, placebo-controlled trial of metformin on myocardial efficiency in insulin-resistant chronic heart failure patients without diabetes. Eur J Heart Fail. https://doi.org/10.1002/ejhf.1656
Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA (2008) 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 359(15):1577–1589. https://doi.org/10.1056/NEJMoa0806470
Ferrannini E, DeFronzo RA (2015) Impact of glucose-lowering drugs on cardiovascular disease in type 2 diabetes. Eur Heart J 36(34):2288–2296. https://doi.org/10.1093/eurheartj/ehv239
Coll AP, Chen M, Taskar P et al (2020) GDF15 mediates the effects of metformin on body weight and energy balance. Nature 578(7795):444–448. https://doi.org/10.1038/s41586-019-1911-y
Zhao JV, Luo S, Schooling CM (2019) Sex-specific Mendelian randomization study of genetically predicted insulin and cardiovascular events in the UK Biobank. Commun Biol 2(1):332. https://doi.org/10.1038/s42003-019-0579-z
Shu X, Wu L, Khankari NK et al (2019) Associations of obesity and circulating insulin and glucose with breast cancer risk: a Mendelian randomization analysis. Int J Epidemiol 48(3):795–806. https://doi.org/10.1093/ije/dyy201
Vancura A, Bu PL, Bhagwat M, Zeng J, Vancurova I (2018) Metformin as an anticancer agent. Trends Pharmacol Sci 39(10):867–878. https://doi.org/10.1016/j.tips.2018.07.006
Guevara-Aguirre J, Rosenbloom AL (2015) Obesity, diabetes and cancer: insight into the relationship from a cohort with growth hormone receptor deficiency. Diabetologia 58(1):37–42. https://doi.org/10.1007/s00125-014-3397-3
Laron Z (2008) The GH-IGF1 axis and longevity. The paradigm of IGF1 deficiency. Hormones (Athens) 7(1):24–27. https://doi.org/10.14310/horm.2002.1111034
Hart PC, Kenny HA, Grassl N et al (2019) Mesothelial cell HIF1α expression is metabolically downregulated by metformin to prevent oncogenic tumor-stromal crosstalk. Cell Rep 29(12):4086–4098 e4086. https://doi.org/10.1016/j.celrep.2019.11.079
Bahrambeigi S, Shafiei-Irannejad V (2020) Immune-mediated anti-tumor effects of metformin; targeting metabolic reprogramming of T cells as a new possible mechanism for anti-cancer effects of metformin. Biochem Pharmacol 174:113787. https://doi.org/10.1016/j.bcp.2019.113787
Bruzzese F, Hagglof C, Leone A et al (2014) Local and systemic protumorigenic effects of cancer-associated fibroblast-derived GDF15. Cancer Res 74(13):3408–3417. https://doi.org/10.1158/0008-5472.Can-13-2259
Wurtz P, Wang Q, Soininen P et al (2016) Metabolomic profiling of statin use and genetic inhibition of HMG-CoA reductase. J Am Coll Cardiol 67(10):1200–1210. https://doi.org/10.1016/j.jacc.2015.12.060
Rena G, Hardie DG, Pearson ER (2017) The mechanisms of action of metformin. Diabetologia 60(9):1577–1585. https://doi.org/10.1007/s00125-017-4342-z
Torkamani A, Wineinger NE, Topol EJ (2018) The personal and clinical utility of polygenic risk scores. Nat Rev Genet 19(9):581–590. https://doi.org/10.1038/s41576-018-0018-x
Schooling CM, Lopez P, Yang Z, Au Yeung SL, Huang JV. Bias from competing risk before recruitment in Mendelian randomization studies of conditions with shared etiology. Available https://www.biorxiv.org/content/10.1101/716621v3. Accessed June 2020.
Acknowledgements
This research has been conducted using the UK Biobank Resource (www.ukbiobank.ac.uk) under application number 51001. Summary data on HbA1c have been contributed by MAGIC investigators and have been downloaded from http://www.magicinvestigators.org/. Summary data on type 2 diabetes have been contributed by DIAGRAM investigators and have been downloaded from http://diagram-consortium.org/. Summary data on coronary artery disease have been contributed by CARDIoGRAMplusC4D investigators and have been downloaded from http://www.cardiogramplusc4d.org/. Summary data on stroke have been contributed by the MEGASTROKE investigators and have been downloaded from http://www.megastroke.org/. The MEGASTROKE project received funding from sources specified at http://www.megastroke.org/acknowledgments.html. Summary data on breast cancer have been contributed by BCAC investigators and have been downloaded from http://bcac.ccge.medschl.cam.ac.uk/bcacdata/. Summary data on prostate cancer have been contributed by the PRACTICAL consortium, CRUK, BPC3, CAPS and PEGASUS and have been downloaded from http://practical.icr.ac.uk/blog/. All studies and funders related to BCAC and PRACTICAL are listed in the ESM.
Authors’ relationships and activities
The authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Funding
This research is funded by the Seed Fund for Basic Research, the University of Hong Kong (No. 201811159054 to SLAY). SL is supported by the Bau Tsu Zung Bau Kwan Yeu Hing Research and Clinical Fellowship (*200008682.920006.20006.400.01), the University of Hong Kong. ICKW received research funding outside the submitted work from Amgen, Bristol-Myers Squibb, Pfizer, Janssen, Bayer, GSK, Novartis, the Hong Kong Research Grant Council, the Hong Kong Health and Medical Research Fund, the National Institute for Health Research in England, the European Commission, and the National Health and Medical Research Council in Australia, and also received speaker fees from Janssen and Medice in the previous 3 years. The study sponsor/funder was not involved in the design of the study; the collection, analysis and interpretation of data; writing the report; and did not impose any restrictions regarding the publication of the report.
Author information
Authors and Affiliations
Contributions
SL and SLAY designed the study, wrote the research plan and interpreted the results. SL undertook analyses with feedback from SLAY, CMS and ICKW. SL and SLAY wrote the manuscript with critical comments from CMS and ICKW. The corresponding author attests that all listed authors meet authorship criteria and that no others meeting the criteria have been omitted. All authors gave final approval of the version to be published. SL is the guarantor of this work and, as such, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM
(PDF 543 kb)
Rights and permissions
About this article

Cite this article
Luo, S., Schooling, C.M., Wong, I.C.K. et al. Evaluating the impact of AMPK activation, a target of metformin, on risk of cardiovascular diseases and cancer in the UK Biobank: a Mendelian randomisation study. Diabetologia 63, 2349–2358 (2020). https://doi.org/10.1007/s00125-020-05243-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-020-05243-z