
Abstract
Aims/hypothesis
Skeletal muscle accounts for >80% of insulin-stimulated glucose uptake; dysfunction of this process underlies insulin resistance and type 2 diabetes. Insulin sensitivity is impaired in mice deficient in the double C2 domain β (DOC2B) protein, while whole-body overexpression of DOC2B enhances insulin sensitivity. Whether insulin sensitivity in the skeletal muscle is affected directly by DOC2B or is secondary to an effect on other tissues is unknown; the underlying molecular mechanisms also remain unclear.
Methods
Human skeletal muscle samples from non-diabetic or type 2 diabetic donors were evaluated for loss of DOC2B during diabetes development. For in vivo analysis, new doxycycline-inducible skeletal-muscle-specific Doc2b-overexpressing mice fed standard or high-fat diets were evaluated for insulin and glucose tolerance, and insulin-stimulated GLUT4 accumulation at the plasma membrane (PM). For in vitro analyses, a DOC2B-overexpressing L6-GLUT4-myc myoblast/myotube culture system was coupled with an insulin resistance paradigm. Biochemical and molecular biology methods such as site-directed mutagenesis, co-immunoprecipitation and mass spectrometry were used to identify the molecular mechanisms linking insulin stimulation to DOC2B.
Results
We identified loss of DOC2B (55% reduction in RNA and 40% reduction in protein) in the skeletal muscle of human donors with type 2 diabetes. Furthermore, inducible enrichment of DOC2B in skeletal muscle of transgenic mice enhanced whole-body glucose tolerance (AUC decreased by 25% for female mice) and peripheral insulin sensitivity (area over the curve increased by 20% and 26% for female and male mice, respectively) in vivo, underpinned by enhanced insulin-stimulated GLUT4 accumulation at the PM. Moreover, DOC2B enrichment in skeletal muscle protected mice from high-fat-diet-induced peripheral insulin resistance, despite the persistence of obesity. In L6-GLUT4-myc myoblasts, DOC2B enrichment was sufficient to preserve normal insulin-stimulated GLUT4 accumulation at the PM in cells exposed to diabetogenic stimuli. We further identified that DOC2B is phosphorylated on insulin stimulation, enhancing its interaction with a microtubule motor protein, kinesin light chain 1 (KLC1). Mutation of Y301 in DOC2B blocked the insulin-stimulated phosphorylation of DOC2B and interaction with KLC1, and it blunted the ability of DOC2B to enhance insulin-stimulated GLUT4 accumulation at the PM.
Conclusions/interpretation
These results suggest that DOC2B collaborates with KLC1 to regulate insulin-stimulated GLUT4 accumulation at the PM and regulates insulin sensitivity. Our observation provides a basis for pursuing DOC2B as a novel drug target in the muscle to prevent/treat type 2 diabetes.
Similar content being viewed by others



Introduction
Insulin resistance, characterised by a lack of target tissue response to insulin stimulation, is a key factor in progression towards the metabolic syndrome and type 2 diabetes [1]. Skeletal muscle has been reported to account for 80–90% of insulin-stimulated glucose uptake [2], with uptake facilitated by the insulin-responsive glucose transporter GLUT4. As GLUT4 vesicles must translocate from the cell interior to the plasma membrane (PM) to facilitate uptake [3], strategies to promote GLUT4 translocation and improve insulin sensitivity in skeletal muscle are in demand as a means to prevent and treat type 2 diabetes.
Double C2 domain β (DOC2B) is a calcium-sensitive protein that positively regulates the insulin-stimulated translocation of GLUT4 to the PM in adipocytes [4]. In addition, DOC2B enhances insulin secretion in pancreatic beta cells [5, 6] and increases spontaneous and asynchronous neurotransmitter release from neuronal cells [7, 8]. Doc2b-knockout mice show insulin resistance and impaired glucose uptake in skeletal muscle [9, 10] and ‘global’ overexpression of Doc2b enhances peripheral insulin sensitivity [11]. While an association between DOC2B deficiency in skeletal muscle and diabetes susceptibility exists in rodents [12], it is not known whether skeletal muscle DOC2B is associated with human type 2 diabetes. Furthermore, it is not known whether skeletal-muscle-specific enrichment of DOC2B is sufficient to enhance whole-body insulin sensitivity.
While it is generally agreed that DOC2B is vital to insulin-stimulated GLUT4 vesicle translocation [13], the precise mechanism(s) involving DOC2B remain unresolved. GLUT4 translocation starts with the binding of extracellular insulin to the insulin receptors (INSRs) on the skeletal muscle PM, which activates a phosphorylation-dependent intracellular signalling cascade. This cascade triggers the intracellular GLUT4-containing vesicles to translocate to the PM, where the vesicles are docked and fused via soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor (SNARE) proteins [13, 14], including two target-associated (t)-SNARE proteins, syntaxin 4 (STX4) and synaptosome-associated protein 23 (SNAP23), as well as one vesicle-associated (v)-SNARE protein, VAMP2 [15,16,17]. Vesicle fusion promotes the integration of GLUT4 into the PM, which facilitates glucose uptake. Several reports show that DOC2B is a soluble protein and can exist in the cytosol, consistent with DOC2B having no transmembrane domain [4, 18, 19]. DOC2B is speculated to regulate insulin-stimulated GLUT4 translocation via insulin-induced DOC2B translocation from the cytosol to the PM, where the target-associated SNARE (t-SNARE) proteins reside [4, 18, 20, 21]. At the PM, insulin stimulation increases DOC2B binding to phosphorylated syntaxin binding protein (STXBP)3, increasing SNARE complex formation [9, 11, 22]. To understand how DOC2B regulates insulin-stimulated glucose uptake, it is crucial to uncover the mechanism linking the insulin signal to DOC2B response.
DOC2B is a ubiquitously expressed protein [23,24,25] that harbours a Munc13-interacting domain (MID) and two sequential C2 domains, termed C2A and C2B [21]. Recent phospho-proteomic studies identified several putative DOC2B phosphorylation sites within the C2B domain [26,27,28]. However, whether insulin stimulates DOC2B phosphorylation remains unknown. In addition, although several studies report DOC2B interaction partners, such as dynein light chain Tctex-type 1 (DYNLT1) in adipocytes [29, 30] and STXBP1 and STXBP3 in pancreatic beta cells [31], most of these DOC2B interaction partners are components of the exocytic machinery near the PM. Cytoplasmic interaction partners of DOC2B remain unexplored, particularly in skeletal muscle. Herein, we used human skeletal muscle samples, an inducible skeletal-muscle-specific Doc2b-overexpressing mouse model, and a myoblast/myotube culture system to address the relationship between DOC2B and insulin sensitivity.
Methods
Materials, antibodies and plasmid constructs
All antibodies were validated using specific blocking peptide and titrated before experiments. For detailed source and dilution of antibody, please see the Electronic supplementary material (ESM) Methods. The green fluorescent protein (GFP)-tagged Doc2b-GFP and C2AB-GFP plasmids were kindly provided by U. Ashery (Tel-Aviv University, Tel-Aviv, Israel) as described previously [1]. hDOC2B-GFP in the pEGFP-N2 backbone was constructed by Genescripts (Piscataway, NJ, USA). Y301 of Doc2b-GFP and hDOC2B-GFP was mutated to F301 by Genescripts. Adenovirus Ad-DOC2B-wild type (WT), Ad-Y301F-GFP or Ad-GFP were generated by insertion of the hDOC2B-GFP, hDOC2B-Y301F-GFP fusion gene or GFP alone into the pAd5CMVmpA adenoviral vector by ViraQuest (North Liberty, IA, USA). Adenovirus Ad-DOC2B alone was made similarly and virus packaged with GFP (see ESM Methods).
Human skeletal muscle biopsies
Sixteen skeletal muscle samples from the leg muscles of non-diabetic and type 2 diabetic donors were purchased from the National Disease Research Interchange (NDRI). The donors were white men and women between 45 and 78 years old (ESM Table 1). The procurement of human skeletal muscle biopsies from NDRI was approved by the Institutional Review Board at the University of Pennsylvania. The participants gave informed consent. The investigation was carried out in accordance with the principles of the Declaration of Helsinki as revised in 2008. The samples were snap-frozen and kept at 80°C until mRNA and protein extraction.
Animals and in vivo experiments
Animals were maintained in ventilated cages with a 12 h light/dark cycle and access to food and water ad libitum under protocols approved by the City of Hope or Indiana University School of Medicine Institutional Animal Care and Use Committees and in accordance with Guidelines for the Use and Care of Laboratory Animals as well as other applicable local and national regulations. Rat Doc2b cDNA was subcloned into the 5′ Pme1 and 3′ BamH1 restriction sites in the pTRE-IRES-nGFP vector provided by S. Afelik and J. Jenssen (Cleveland Clinic, Cleveland, OH, USA) and microinjected into B6 oocytes to produce a tetracycline-responsive element (TRE)-Doc2b−/+ founder, as described previously [32]. Skeletal-muscle-specific muscle creatine kinase (Mck [also known as Ckm]) promoter reverse tetracycline transactivator (rtTA) mice (Mck-rtTA−/+) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). TRE-Doc2b−/+ offspring crossed to Mck-rtTA−/+ mice produced doxycycline (Dox)-inducible skeletal muscle-selective expression in the double transgenic offspring (skmDoc2b-dTg) mice. Male and female B6 skmDoc2b-dTg mice (4–5 months old) were administered Dox-treated (2 mg/ml) or standard drinking water for 3 weeks to induce Doc2b overexpression prior to phenotyping assessment. A high-fat diet (HFD) containing 60% of energy from fat, with or without Dox (catalogue numbers S6470 and S3282, respectively, Bio-Serv, Flemington, NJ, USA), was fed to 7–8-week-old male mice for 4–5 weeks to induce insulin resistance prior to phenotyping and body composition assessment (see ESM Methods).
Sample size
Littermate mice of the same age were randomised to different subgroups of 6–10 animals. The western blot images are representative of at least three independent experiments. The independent cell culture experiments were conducted using independent passages of cells. The sample size was determined based on a similar study of whole-body Doc2b overexpression in mice [11]. Data falling within the mean ± 2 SD were included in the final statistical analysis.
Intraperitoneal glucose tolerance test, insulin tolerance test and serum analysis
Mice were fasted 6 h (08:00–14:00 hours) before i.p. glucose tolerance tests (IPGTTs) and i.p. insulin tolerance tests (IPITTs), which were performed as described previously [9]. The mice were killed at 24–26 weeks old and serum and tissues were collected. Serum insulin and glucagon levels were measured via radioimmunoassay (Millipore, Burlington, MA, USA).
Cell culture
Authentic L6-GLUT4-myc myoblasts, which were free from mycoplasma contamination, were purchased from Kerafast (Boston, MA, USA) and grown as monolayers in MEM-α medium supplemented with 10% FBS (vol./vol.) and 1% (vol./vol.) antibiotic–antimycotic solution (Thermo Fisher, Waltham, MA, USA), as described previously [33]. L6-GLUT4-myc myoblasts were differentiated into myotubes by seeding them at 1× 104 cells/ml in MEM-α medium containing 2% FBS (vol./vol.) for 7 days. For all studies involving the insulin resistance, as experimentally induced in vitro (InsRes) paradigm, L6-GLUT4-myc myoblasts (90% confluence) or myotubes were preincubated in medium containing 5 nmol/l insulin for 12 h to simulate chronic insulinaemia [34, 35]. Cells were then washed and incubated in serum-free MEM-α medium for 40 min immediately prior to acute insulin stimulation (100 nmol/l) for 5 min for assessment of phosphorylation or for 20 min for cell-surface GLUT4-Myc detection.
RNA extraction and quantitative real-time PCR analysis
RNA was extracted using TRI Reagent according to the manufacturer’s protocol (MilliporeSigma, St Louis, MO, USA). Synthesis of cDNA was performed using the iScript cDNA synthesis kit, as described by the manufacturer (Bio-Rad, Hercules, CA, USA). Real-time PCR was carried out for 40 cycles using the iCycler (Bio-Rad). Each cycle was run at 95°C for 15 s, 60°C for 30 s and 72°C for 30 s. See the ESM Methods and ESM Table 2 for the primer sequences and further details.
Cell-surface GLUT4-myc detection
Cell-surface GLUT4-myc detection was performed as previously described [34, 36]. See ESM Methods.
2-Deoxyglucose uptake
The 2-deoxyglucose (2-DG) uptake assay was performed as described previously [36]. See ESM Methods.
Co-immunoprecipitation and immunoblotting
The cells were harvested into 1% NP-40 (vol./vol.) lysis buffer and 2.5 mg of the lysate was immediately used for immunoprecipitation by overnight incubation with 30 μl of rabbit polyclonal anti-GFP antibody conjugated to Sepharose beads. Co-immunoprecipitation from mouse skeletal muscle tissue was performed similarly, using 4 mg of tissue homogenate combined with anti-Myc-conjugated beads (Thermo Fisher, Waltham, MA, USA [catalogue number 20168]). The immunoprecipitated proteins were washed three times with lysis buffer and boiled in Laemmli sample buffer containing fresh dithiothreitol. The proteins in the cell lysates were resolved using 10–12% SDS-PAGE (wt/vol.) and transferred to polyvinylidene difluoride (PVDF) membranes for immunoblotting. See the ESM Methods.
Subcellular fractionation of skeletal muscle
The subcellular fractionation of skeletal muscle was performed as described previously [9, 37]. See the ESM Methods.
Statistical analysis
All data were evaluated for statistical significance using a paired two-tailed Student’s t test for comparison of two groups or ANOVA for more than two groups, using GraphPad Prism Version 7.0 (GraphPad software, La Jolla, CA, USA). Data are expressed as the average ± SEM.
Results
DOC2B abundance is decreased in type 2 diabetic human skeletal muscle and insulin-resistant muscle cells
Whole-body Doc2b-knockout mice display peripheral insulin resistance in vivo and defective insulin-stimulated glucose uptake into skeletal muscle [9]; therefore, we tested whether DOC2B is deficient in human skeletal muscle from type 2 diabetic individuals compared with non-diabetic control individuals (ESM Table 1) using real-time PCR and immunoblotting analysis. The DOC2B mRNA level was reduced by 55% (Fig. 1a) and the DOC2B protein level was reduced by 40% (Fig. 1b) in human skeletal muscle samples from type 2 diabetic individuals compared with non-diabetic individuals. To determine whether the decreased DOC2B level in type 2 diabetic skeletal muscle was caused by insulin resistance prior to type 2 diabetes/hyperglycaemia or is a consequence of type 2 diabetes/hyperglycaemia, we examined the DOC2B mRNA and DOC2B protein levels in L6-GLUT4-myc myoblasts subjected to a well-established in vitro paradigm to induce InsRes [34, 35]. The InsRes paradigm attenuated insulin-stimulated GLUT4 accumulation at the PM, as reported previously [34, 35] (Fig. 1c), and decreased the stimulation index (insulin-stimulated/basal GLUT4 levels at the PM) (Fig. 1d). Concomitant with these InsRes-induced changes, Doc2b mRNA was reduced by 30% (Fig. 1e) and DOC2B protein was decreased by 20% (Fig. 1f) in InsRes-exposed L6-GLUT4-myc myoblasts compared with control cells. These results suggest DOC2B may become reduced during insulin resistance prior to type 2 diabetes and hyperglycaemia.

DOC2B expression is attenuated in the skeletal muscle of human type 2 diabetic donors and insulin-resistant L6-GLUT4-myc muscle cells. (a, b) Real-time PCR and western blot analyses of DOC2B mRNA (a) (n = 3 for non-diabetic donors and n = 4 for donors with type 2 diabetes) and DOC2B protein levels (b) (n = 8 for non-diabetic donors and n = 8 for donors with type 2 diabetes) in human skeletal muscle samples. The relative mRNA or protein level was calculated by normalising to HPRT. (c) PM GLUT4 level in control and InsRes L6-GLUT4-myc cells under basal conditions (−) and after 100 nmol/l insulin stimulation for 20 min (+). The immunofluorescence intensity of cell-surface GLUT4 was normalised to the nucleic acid staining dye SYTO 60. (d) The stimulation index was calculated as a ratio of the relative GLUT4 value in (c) after insulin stimulation, divided by relative GLUT4 value without insulin stimulation, under Ctrl or InsRes conditions. (e, f) Real-time PCR of Doc2b mRNA levels (e) and western blot analysis of DOC2B protein levels (f) in control and InsRes L6-GLUT4-myc cells. For (c–f) n = 3. *p < 0.05 vs ND/Ctrl, or as otherwise shown. Dashed vertical lines indicate splicing of lanes within the same gel exposure. Ctrl, control; HPRT, hypoxanthine guanine phosphoribosyl transferase; Ins, insulin; ND, non-diabetic donor; T2D, type 2 diabetic donor
DOC2B enrichment protects myotubes and myoblasts from dysfunction under insulin resistance conditions
Given that the global Doc2b-overexpressing mice showed enhanced insulin sensitivity, we next questioned whether DOC2B enrichment could prevent InsRes in skeletal myotubes. Myotubes exhibit a physiological glucose uptake response similar to that of primary tissue, while myoblasts are suitable for GLUT4 accumulation analysis and biochemical assays requiring large amounts of protein. Differentiated L6-GLUT4-myc myotubes were adenovirally transduced to overexpress DOC2B protein, and a multiplicity of infection (MOI) of 200 was sufficient for enrichment in ~80% of myotubes, as detected by GFP expression (adenovirus packaged with GFP; Fig. 2a, b). Under control conditions, insulin stimulated glucose uptake to a level more than twofold that of basal levels (Fig. 2c) as has been reported previously [36], while InsRes conditions blocked the insulin response (Fig. 2c). Notably, DOC2B-overexpressing myotubes resisted the InsRes-induced attenuation of insulin-stimulated glucose uptake (Fig. 2c). Consistently, InsRes conditions blocked insulin-stimulated GLUT4 accumulation at the PM in myoblasts (Fig. 2d). Furthermore, DOC2B-GFP-overexpressing myoblasts resisted the InsRes-induced attenuation of insulin-stimulated GLUT4 accumulation at the PM (Fig. 2d). DOC2B-overexpression also boosted insulin-stimulated glucose uptake and GLUT4 accumulation at the PM under control conditions (Fig. 2c, d). These data indicate that DOC2B protects against InsRes-mediated impairment of glucose uptake, potentially by preventing the loss of insulin-induced GLUT4 vesicle translocation.

DOC2B enrichment preserves insulin-stimulated glucose uptake via GLUT4 translocation in L6-GLUT4-myc myotubes exposed to insulin resistance stimuli. (a, b) Expression of DOC2B and transduction efficiency for L6-GLUT4-myc myoblasts that were differentiated into myotubes and transduced with Ad-DOC2B or vector control adenoviral particles at multiple MOIs, as shown by (a) western blot analysis and (b) evaluation of GFP fluorescence (from viral packaging); scale bar, 400 μm. White arrows denoting DOC2B-transduced myotubes in the fluorescence image correspond to the same myotubes seen in the bright field image. (c) 2-DG uptake in myotubes incubated under control or InsRes conditions. The myotubes were transduced with Ad-GFP or Ad-DOC2B before induction of InsRes conditions. (d) Surface GLUT4 levels in L6-GLUT4-myc myoblasts transfected with DOC2B-GFP or control GFP plasmid DNA, incubated under control or InsRes conditions and measured in response to stimulatory insulin. For (c) and (d), the cells were stimulated with 100 nmol/l insulin to induce 2-DG uptake or GLUT4 translocation. The immunofluorescence intensity of cell-surface GLUT4 was normalised to the nucleic acid staining dye SYTO 60. n = 3. *p < 0.05, **p < 0.01, ***p < 0.001 as shown. Ctrl, control; Ins, insulin
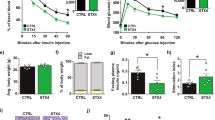
Inducible skeletal-muscle-specific DOC2B-expressing mice demonstrate enhanced glucose tolerance, insulin sensitivity and skeletal muscle GLUT4 accumulation at the PM
To determine whether skeletal muscle-specific enrichment of DOC2B is sufficient to promote insulin sensitivity in vivo, we generated a Dox-inducible skeletal-muscle-specific double transgenic mouse model (skmDoc2b-dTg) expressing DOC2B with a C-terminal Myc tag (Fig. 3a). Using an anti-Myc antibody, we detected the transgene-encoded DOC2B-Myc protein in the skeletal muscle of Dox-treated skmDoc2b-dTg mice; DOC2B-Myc was not present in non-Dox-treated skmDoc2b-dTg mice, Dox-treated Mck-rtTA+/− mice or WT control mice (Fig. 3b). The Doc2b mRNA abundance in skeletal muscle was ~2–3-fold higher in Dox-induced skmDoc2b-dTg mice than non-Dox-treated skmDoc2b-dTg, Dox-treated TRE-Doc2b+/−and Dox-treated Mck-rtTA+/− control mice (Fig. 3c). However, the Doc2b mRNA level in the heart was unchanged, confirming the skeletal muscle-specificity of the transgene expression (Fig. 3d). This is consistent with a prior report that Mck-rtTA+/− regulates protein overexpression in skeletal muscle but not heart muscle [38]. Further, DOC2B upregulation was not associated with dwarfism or obesity, and did not cause energy-intake restriction or hyperphagia, as evidenced by body fat, body weight and food intake measures (ESM Fig. 1a, b). In addition, no changes in the respiratory exchange ratio or activity level (ESM Fig. 1c, d) were observed.

Generation of skmDoc2b-dTg mice. (a) Mice containing a Myc-tagged Doc2b transgene under control of the Dox-inducible TRE-Doc2b−/+ were crossed to skeletal muscle-specific Mck-rtTA−/+ mice to produce Dox-inducible skeletal muscle-selective expression in the double transgenic offspring. (b) Anti-Myc immunoblot of Dox-induced DOC2B-Myc expression. Actin was used as the loading control. (c, d) Real-time PCR of Doc2b mRNA (relative to Hprt) in the skeletal muscle (c) and heart (d). (e) GLUT4 level in the sarcolemmal/t-tubule fractions of DOC2B-enriched (Dox+) and control (Dox−) muscle tissues with and without insulin stimulation. Ponceau S staining was used as the loading control. (f) Total GLUT4 level in the whole-muscle lysate of mice from the indicated genotypes with and without Dox treatment. Ponceau S staining at 50 kDa was used as the loading control. n = 3; *p < 0.05, **p < 0.01 and ***p < 0.001 as shown. Dashed vertical lines indicate splicing of lanes within the same gel exposure. Dox+, DOC2B-enriched; Dox−, control; Ins, insulin; K/O, knockout
Next, we evaluated insulin-stimulated GLUT4 accumulation at the PM in the skmDoc2b-dTg mice. Insulin stimulation increased the accumulation of GLUT4 in the sarcolemmal/t-tubule fraction of mouse hindlimb muscle by 1.5 fold in control male mice (non-Dox-treated skmDoc2b-dTg), consistent with prior reports [9, 37]; Dox-treated skmDoc2b-dTg male mice showed significantly more insulin-stimulated GLUT4 accumulation in this plasma-membrane-like fraction compared with controls (Fig. 3e). No differences in insulin-stimulated GLUT4 accumulation were seen under basal conditions in the Dox-induced skmDoc2b-dTg muscle, and total GLUT4 protein expression levels in whole-cell lysates were similar in Dox-induced skmDoc2b-dTg and control mice (Fig. 3f). These results are consistent with our findings using DOC2B-overexpressing L6-GLUT4-myc myoblasts.
As enhanced GLUT4 accumulation at the PM is correlated with insulin sensitivity and glucose tolerance, skmDoc2b-dTg mice were subjected to IPGTT and IPITT. Because global overexpression of DOC2B enhanced muscle insulin sensitivity even without induction of insulin resistance [11], IPGTT and IPITT were first performed in skmDoc2b-dTg mice without induction of insulin resistance. Both female and male Doc2B-overexpressing mice showed significantly enhanced insulin sensitivity in the IPITT with AOC increased by 20% and 26% for female and male mice respectively in vivo (Fig. 4a–d). Female Doc2b-overexpressing mice exhibited an improvement in both the rate and extent of the reduction in glycaemia in the IPGTT compared with the non-Dox-treated mice with AUC decreased by 25% (Fig. 4e, f); the differences for males did not reach statistical significance (Fig. 4g, h; p = 0.07). These results with the male mice are consistent with findings in whole-body Stx4-overexpressing [39] or Doc2b-overexpressing [11] mice in this age range. Control TRE-Doc2b+/− and Mck-rtTA+/− mice showed no response to Dox in the IPITT or IPGTT (ESM Fig. 2a–d). DOC2B enrichment in muscle had no prominent effects on systemic glucose metabolism, as determined by similar levels of serum insulin, glucagon, cholesterol and NEFA (ESM Table 3). In addition, no significant differences in body weight or organ weights were detected between the Dox-treated and untreated skmDoc2b-Tg mice (ESM Table 4). Together, these results suggest that skeletal-muscle-specific DOC2B enrichment can enhance whole-body glucose homeostasis and peripheral insulin sensitivity, consistent with its role in regulating insulin-stimulated GLUT4 translocation.

DOC2B enrichment in skeletal muscle improves whole-body insulin sensitivity and glucose homeostasis. IPITT results for (a, b) female and (c, d) male DOC2B-enriched (Dox+) or control (Dox−) skmDoc2b-dTg mice. The mice were fasted for 6 h and AOC was quantified. IPGTT results for (e, f) female and (g, h) male DOC2B-enriched or control skmDoc2b-dTg mice. The mice were fasted for 6 h and AUC was quantified. Data are shown as mean ± SEM for 6–10 sets of mice. *p < 0.05 and ***p < 0.001 vs Dox− control. AOC, area over the curve. Dox−, SkmDoc2b-dTg mice without Dox induction; Dox+, SkmDoc2b-dTg mice with Dox induction
SkmDoc2b-dTg mice are protected from HFD-induced insulin resistance
To determine whether skeletal muscle-specific enrichment of DOC2B could protect against insulin resistance in vivo, we placed male skmDoc2b-dTg mice on an HFD for 4 weeks; this has been shown to induce skeletal muscle insulin resistance [40,41,42,43]. IPITT and IPGTT analyses show that HFD induces insulin resistance and glucose intolerance in control mice (non-Dox-treated skmDoc2b-dTg) compared with chow-fed mice (Fig. 5a, ESM Fig. 3). Skeletal-muscle-specific expression of DOC2B completely blocked the effects of the HFD on insulin sensitivity (Fig. 5a). These DOC2B-associated improvements in peripheral insulin sensitivity occurred despite the obesity resulting from elevated energy intake in the mice fed HFD, with and without Dox treatment (Fig. 5b, c; ESM Fig. 3). However, DOC2B enrichment in skeletal muscle did not protect against the whole-body glucose intolerance caused by the HFD, likely due to the beta cell dysfunction caused by this diet. Overall, these data indicate that DOC2B enrichment in skeletal muscle protects against HFD-induced insulin resistance in vivo.

DOC2B enrichment in skeletal muscle prevents HFD-induced insulin resistance. (a) IPITT results for male DOC2B-enriched or control skmDoc2b-dTg mice fed 60% HFD or standard chow for 4 weeks. AOC was quantified. Body weight (b) and energy intake (c) over 4 weeks of dietary challenge. Data are shown as mean ± SEM for eight sets of mice. *p < 0.05 vs chow control, or as otherwise shown. For (b, c) *p < 0.05 by comparison across the time course using ANOVA. Chow Ctrl, control mice fed standard chow; HFD, control mice fed 60% HFD; HFD+Dox, DOC2B-enriched skmDoc2b-dTg mice fed 60% HFD
Insulin stimulation phosphorylates DOC2B and augments its association with KLC1
Several groups have speculated that DOC2B in the cytoplasm responds to insulin stimulation and translocates to the PM [4, 20], where it positively regulates STX4-based SNARE complex formation to promote GLUT4 vesicle docking/fusion at the PM [6, 9, 11, 22]; this mechanism could underlie the enhancement by DOC2B of peripheral insulin sensitivity and glucose tolerance. However, the molecular mechanism linking insulin action and DOC2B translocation is not known. Because the canonical insulin-signalling pathways involve multiple phosphorylation events [44,45,46], we hypothesised that insulin stimulation might phosphorylate DOC2B to promote insulin-stimulated GLUT4 translocation. Towards this, we immunoprecipitated DOC2B-Myc from skmDoc2b-dTg hindlimb muscle homogenates prepared from mice injected with saline (154 mmol/l NaCl) or insulin and immunoblotted using the canonical anti-tyrosine phosphorylation antibody PY20 [47, 48]. Insulin-stimulated muscle showed a 1.6-fold increase in tyrosine-phosphorylated DOC2B-Myc compared with saline-injected homogenates (Fig. 6a). Similarly, L6-GLUT4-myc myoblasts were transfected to express GFP or DOC2B-GFP and treated with insulin or pervanadate (pV, a tyrosine-selective phosphatase inhibitor [22]) for 5 min; phospho-enrichment beads captured a significant increase in phosphorylated DOC2B-GFP (molecular mass 75 kDa) in the insulin-stimulated and pV-treated cells (Fig. 6b), as detected using an anti-GFP antibody. The abundance of free GFP (molecular mass 25 kDa) in the input and its absence in the eluate confirmed the specificity of DOC2B-GFP binding to the phospho-enrichment beads. The insulin- or pV-induced phosphorylation of the INSR beta subunit was used as a control for the action of insulin and pV. Equal amounts of DOC2B-GFP were loaded on the phospho-enrichment beads. The pV-stimulated detection of tyrosine phosphorylation of DOC2B-GFP was further confirmed by immunoprecipitation using the PY20 antibody (ESM Fig. 4). These results suggest that insulin stimulation phosphorylates DOC2B, probably on one or more tyrosine residues.

Insulin stimulation phosphorylates DOC2B and enhances its interaction with KLC1. (a) DOC2B phosphorylation measured as anti-PY20 immunoreactivity after anti-Myc immunoprecipitation of DOC2B-Myc from the hindlimbs of mice injected i.p. with saline or insulin. Total DOC2B-Myc level was used as a control. (b) DOC2B phosphorylation in L6-GLUT4-myc myoblasts after insulin or pervanadate treatment using a phosphorylated protein enrichment column followed by immunoblotting. GFP and DOC2B-GFP were detected using anti-GFP at 25 kDa and 75 kDa, respectively. INSR was used as a control for the action of insulin and pV. (c) Effect of pervanadate treatment on the DOC2B–KLC1 interaction, measured using immunoprecipitation with GFP antibody-conjugated Sepharose beads and immunoblotting with antibodies to KLC1 at 68 kDa or GFP for DOC2B-GFP and GFP at 75 kDa and 25 kDa, respectively (n = 3). (d) Detection of the DOC2B–KLC1 interaction in L6-GLUT4-myc myoblasts after insulin stimulation for 0, 3 and 5 min using the immunoprecipitation strategy described in (c) (n = 3). Data are shown as mean ± SEM. *p < 0.05 vs control (without insulin in a, d; without pervanadate [NA] in c). Dashed vertical lines indicate splicing of lanes within the same gel exposure. IB, immunoblot; Ins, insulin; IP: immunoprecipitation; NA, no addition; pV, pervanadate
Next, mass spectrometry analysis was used to identify potential phospho-DOC2B binding partners in the cytoplasm. L6-GLUT4-myc myoblasts were transfected to express DOC2B-GFP or GFP and treated with insulin or pV; the resulting cell lysates were incubated with commercial anti-GFP-conjugated Sepharose beads to capture potential interacting partners. Proteins bound to the beads were subjected to SDS-PAGE. Visible differences were detected in the protein profile from 50 to 150 kDa; this region was excised for mass spectrometry analysis (ESM Fig. 5a). Of 173 proteins identified with at least three unique peptides, the 14 proteins detected selectively in insulin- or pV-treated DOC2B-overexpressing lysates, but not in GFP-overexpressing lysates, are summarised in ESM Table 5. Kinesin light chain 1 (KLC1), which also shows enhanced binding to DOC2B under insulin- or pV-treated conditions compared with the basal conditions, was chosen based on a prior report showing that dominant negative mutants of KLC1 reduce insulin-stimulated GLUT4 translocation [49]. The unique peptide sequence of KLC1 identified by mass spectrometry covered 12% of KLC1, ruling out the possibility of false identification (ESM Fig. 5b). By using GFP antibody-conjugated Sepharose beads, which capture both free GFP and DOC2B-GFP, KLC1 was found to co-immunoprecipitate with DOC2B-GFP but not GFP; pV-treated lysates showed an increased level of KLC1 associated with DOC2B-GFP (Fig. 6c). Consistent with DOC2B phosphorylation at 5 min, insulin stimulation for 5 min significantly increased the association of DOC2B-GFP with KLC1 (Fig. 6d). These findings suggest that insulin-stimulated DOC2B phosphorylation enhances the DOC2B–KLC1 interaction.
DOC2B Y301F mutation disrupts the DOC2B–KLC1 association and blocks the protective effect of DOC2B enrichment
To investigate the molecular basis of the insulin-regulated DOC2B–KLC1 interaction, we sought to identify the site of insulin-stimulated tyrosine phosphorylation on DOC2B. To focus our search on the minimal DOC2B domain(s) necessary to confer insulin-stimulated GLUT4 translocation, an N-terminal truncation (DOC2B MID domain was deleted) with a C-terminal GFP tag (C2AB-GFP) was generated (Fig. 7a). Like full-length DOC2B-GFP, C2AB-GFP conferred enhanced insulin-stimulated GLUT4 accumulation at the PM, relative to GFP alone (Fig. 7b). Importantly, C2AB-GFP also protected against the negative effects of InsRes on GLUT4 accumulation at the PM, similar to full-length DOC2B (Fig. 7b). The ability of C2AB to fully recapitulate the effects of full-length DOC2B suggested that it probably contained the site(s) of insulin-stimulated phosphorylation on DOC2B. Although three tyrosine residues located within the C2B domain of DOC2B, Y301, Y305 and Y309, are 100% conserved in rat mouse and human, solvent accessibility prediction analyses indicated that Y301 is highly solvent exposed and located in an unstructured loop region, which is ideal for kinase accessibility and modification [50] (Fig. 7c); the other two sites, Y305 and Y309, are localised in beta sheets and considered less accessible.

The C2AB domain of DOC2B confers the protective action of DOC2B against insulin resistance in L6 cells. (a) Domain structure of DOC2B. C2A and C2B are C2 domains. (b) PM GLUT4 level in control and InsRes L6-GLUT4-myc myoblasts under basal conditions (−) and after 100 nmol/l insulin stimulation for 20 min. The immunofluorescence intensity of cell-surface GLUT4 was normalised to the nucleic acid staining dye SYTO 60. The myoblasts were transfected with DOC2B-GFP, C2AB-GFP or control GFP plasmid DNAs prior to InsRes induction (n = 3). Data are shown as mean ± SEM. *p < 0.05 as shown. (c) A simulated structure showing the solvent accessibility and structural location of Y301 using PyMOL. The original structure file was extracted from PBD protein data bank (PDB number 4LDC, https://www.rcsb.org/structure/4LDC) and the solvent accessibility was simulated using PyMOL (Schrödinger, New York, NY, USA; https://pymol.org/2/) according to the manufacturer’s instructions. a.a., amino acid; Ctrl, control
To evaluate the importance of Y301 to the insulin-stimulated interaction of DOC2B with KLC1, a conservative substitution (Y to F) was made to Y301 on DOC2B-GFP. The insulin-stimulated phosphorylation of DOC2B-GFP was blocked by the Y301F mutation (Fig. 8a). Moreover, KLC1 was poorly co-immunoprecipitated with DOC2B-GFP-Y301F (Fig. 8b). In contrast to the protective effect of WT DOC2B, the Y301F mutant did not protect cells from the negative effects of InsRes on GLUT4 accumulation at the PM (Fig. 8c) or 2-DG uptake (Fig. 8d). Hence, our data reveal a new mechanism by which insulin stimulates phosphorylation of DOC2B, brokering its interaction with KLC1, and that this mechanism is required for DOC2B enrichment to protect cells against InsRes stimuli.

Y301F mutation in the C2B domain of DOC2B attenuates the DOC2B–KLC1 interaction and blocks the protective effects of DOC2B. (a) Phosphorylated DOC2B-GFP measured using anti-GFP immunoprecipitation followed by immunoblotting with anti-PY20. Total DOC2B-GFP in the immunoprecipitated and the input fraction are shown as controls. (b) Detection of KLC1–DOC2B association using anti-GFP immunoprecipitation followed by immunoblotting with antibodies to KLC1, and to GFP for detection of DOC2B-GFP-WT and DOC2B-GFP-Y301F proteins at 75 kDa (n = 3). (c) PM GLUT4 level in myoblasts transfected to express WT DOC2B or the Y301F mutant. GLUT4 levels were measured under basal conditions (−) and after 100 nmol/l insulin stimulation for 20 min. The immunofluorescence intensity of cell-surface GLUT4 was normalised to the nucleic acid staining dye SYTO 60. (d) 2-DG uptake in myotubes transduced to express WT DOC2B or the Y301F mutant. The myotubes were incubated under InsRes conditions and 2-DG uptake was measured in response to insulin stimulation (100 nmol/l) (n = 3); *p < 0.05 and **p < 0.01 as shown. Dashed vertical lines indicate splicing of lanes within the same gel exposure. IB, immunoblot; Ins, insulin; IP, immunoprecipitation
Discussion
We now show that DOC2B is reduced in the skeletal muscle of human type 2 diabetic donors and that this DOC2B reduction can be recapitulated by inducing insulin resistance in an in vitro InsRes model. Remarkably, this study also shows that DOC2B enrichment in skeletal muscle is sufficient to improve whole-body insulin sensitivity and glucose tolerance and to protect against HFD-induced insulin resistance in vivo. Moreover, DOC2B enrichment in skeletal muscle cells can counter the negative effects of InsRes on GLUT4 accumulation at the PM and glucose uptake at the cellular level. As we observed no effect of DOC2B enrichment on serum insulin, glucagon, cholesterol or NEFA levels, we reason that the enhanced whole-body glucose tolerance is primarily due to improved muscle insulin sensitivity, increasing the rate of glucose clearance from the blood. These data are also consistent with previous findings that DOC2B positively regulates GLUT4 translocation in adipocytes, and that Doc2b-knockout mice have impaired insulin sensitivity and muscle glucose uptake [9, 10], whereas mice with global enrichment of DOC2B exhibit enhanced muscle insulin sensitivity [11]. Thus, we have shown that enrichment of DOC2B solely in skeletal muscle is sufficient to mimic the effects of whole-body overexpression.
This study also advances our knowledge of the mechanisms linking DOC2B to insulin action in the skeletal muscle. We report, for the first time, that insulin stimulation of muscle cells induces DOC2B phosphorylation, probably at one or more tyrosine residues. Our results further implicate DOC2B-Y301 phosphorylation in regulating insulin action in skeletal muscle. However, the DOC2B sequence has multiple potential phosphorylation sites. Previous proteomic studies have identified a variety of phosphorylation sites on DOC2B, such as Y301 and Y305 in Jurkat cells [28], Y309 in untreated human immortalised myelogenous leukaemia cells [27] and S411 in untreated human breast cancer tissues [51]. Although we provide evidence for DOC2B phosphorylation, the kinase is unknown because of the complexity of the kinase–substrate relationship in the insulin-signalling pathway. It is possible that one of the insulin pathway kinases, perhaps even the INSR itself, is responsible for the insulin-stimulated phosphorylation of DOC2B (www.phosphonet.ca/kinasepredictor). This awaits further investigation. Overall, these findings imply that DOC2B may be phosphorylated in a cell-type- and stimulation-dependent manner.
Our mechanistic analysis of DOC2B phosphorylation also led us to identify a DOC2B binding partner, KLC1, which is a cytoskeletal motor protein with a proposed role in protein/vesicle translocation. Tyrosine phosphorylation enhances the DOC2B interaction with KLC1, suggesting that DOC2B–KLC1 interaction is important for DOC2B enrichment to enhance GLUT4 translocation; however, the protein domains involved in this interaction are unknown. No Src homology 2 (SH2)/pTyr-binding (PTB) domains are reported to exist in either DOC2B or KLC1, so the enhanced interaction between phospho-DOC2B and KLC1 may occur through another mechanism, such as conformational change. Indeed, phosphorylation events often induce conformational changes, and DOC2B is a known scaffolding platform for several exocytosis regulatory proteins [31]. Conformational changes to DOC2B could also affect SNARE assembly, promoting GLUT4 translocation and glucose uptake. Alternatively, there are multiple tetratricopeptide repeats (TPRs) in KLC1, which are involved in binding to a variety of factors [52], and it is possible that DOC2B binds to the TPR domain(s) of KLC1. Hence, while we have identified the phospho-DOC2B interaction with KLC1 and shown it to be important for glucose uptake, future studies will be required to explore these and other potential mechanisms.
This study advances our understanding of how DOC2B responds to insulin stimulation to regulate GLUT4 vesicle translocation. Although DOC2B is important for assembly of the SNARE machinery at the PM [53], it has also been shown to exist in the cytosolic compartment and to translocate to the PM in response to insulin stimulation [4]. Indeed, DOC2B may play both roles by virtue of dual cellular compartmentalisation—existing in two pools, one at the PM, the other in the cytosolic space. We and others have speculated that SNARE assembly occurs in parallel with GLUT4 vesicle translocation, orchestrating the preparation of the plasma-membrane-localised t-SNARE proteins for the arrival of the vesicle-associated SNARE (v-SNARE)-loaded GLUT4 vesicle for seamless vesicle docking and fusion events [22, 54]. KLC1 is also localised to the cytoplasm under basal conditions, consistent with its role in microtubule-based GLUT4 vesicle trafficking [55], and hence insulin-stimulated enhancement of DOC2B–KLC1 interaction may represent an early event in the process of GLUT4 vesicle translocation to the PM. This is consistent with a prior report showing that inhibition of the microtubule motor protein kinesin impairs insulin-stimulated GLUT4 translocation [56] and microtubule-depolymerising agents (nocodazole, colchicine and vinblastine) inhibit insulin-stimulated glucose uptake and GLUT4 translocation [57]. Hence, in addition to its previously described role in activating SNARE complexes at the PM, we demonstrate herein a mechanism by which DOC2B impacts insulin-stimulated GLUT4 vesicle translocation via collaboration with the microtubule network protein KLC1. Our finding that this interaction is important to the protective role of DOC2B against insulin resistance will be important to future efforts in therapeutic design.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- 2-DG:
-
2-Deoxyglucose
- DOC2B:
-
Double C2 domain β
- Dox:
-
Doxycycline
- GFP:
-
Green fluorescent protein
- HFD:
-
High-fat diet
- InsRes:
-
Insulin resistance (as experimentally induced in vitro)
- INSR:
-
Insulin receptor
- IPGTT:
-
Intraperitoneal glucose tolerance test
- IPITT:
-
Intraperitoneal insulin tolerance test
- KLC1:
-
Kinesin light chain 1
- MID:
-
Munc13-interacting domain
- MOI:
-
Multiplicity of infection
- NDRI:
-
National Disease Research Interchange
- PM:
-
Plasma membrane
- pV:
-
Pervanadate
- rtTA:
-
Reverse tetracycline transactivator
- skmDoc2b-dTg:
-
Doxycycline-inducible skeletal-muscle-specific Doc2b overexpression
- SNARE:
-
Soluble N-ethylmaleimide-sensitive factor-attachment protein receptor
- STX4:
-
Syntaxin 4
- STXBP:
-
Syntaxin binding protein
- TPR:
-
Tetratricopeptide repeats
- TRE:
-
Tetracycline-responsive element
- t-SNARE:
-
Target-associated SNARE
- WT:
-
Wild type
References
Kahn SE (2003) The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of type 2 diabetes. Diabetologia 46(1):3–19. https://doi.org/10.1007/s00125-002-1009-0
DeFronzo RA, Tripathy D (2009) Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 32(Suppl 2):S157–S163. https://doi.org/10.2337/dc09-S302
Jaldin-Fincati JR, Pavarotti M, Frendo-Cumbo S, Bilan PJ, Klip A (2017) Update on GLUT4 vesicle traffic: a cornerstone of insulin action. Trends Endocrinol Metab 28(8):597–611. https://doi.org/10.1016/j.tem.2017.05.002
Fukuda N, Emoto M, Nakamori Y et al (2009) DOC2B: a novel syntaxin-4 binding protein mediating insulin-regulated GLUT4 vesicle fusion in adipocytes. Diabetes 58(2):377–384. https://doi.org/10.2337/db08-0303
Miyazaki M, Emoto M, Fukuda N et al (2009) DOC2b is a SNARE regulator of glucose-stimulated delayed insulin secretion. Biochem Biophys Res Commun 384(4):461–465. https://doi.org/10.1016/j.bbrc.2009.04.133
Ke B, Oh E, Thurmond DC (2007) Doc2β is a novel Munc18c-interacting partner and positive effector of syntaxin 4-mediated exocytosis. J Biol Chem 282(30):21786–21797. https://doi.org/10.1074/jbc.M701661200
Groffen AJ, Martens S, Diez Arazola R et al (2010) Doc2b is a high-affinity Ca2+ sensor for spontaneous neurotransmitter release. Science 327(5973):1614–1618. https://doi.org/10.1126/science.1183765
Yao J, Gaffaney JD, Kwon SE, Chapman ER (2011) Doc2 is a Ca2+ sensor required for asynchronous neurotransmitter release. Cell 147(3):666–677. https://doi.org/10.1016/j.cell.2011.09.046
Ramalingam L, Oh E, Yoder SM et al (2012) Doc2b is a key effector of insulin secretion and skeletal muscle insulin sensitivity. Diabetes 61(10):2424–2432. https://doi.org/10.2337/db11-1525
Li J, Cantley J, Burchfield JG et al (2014) DOC2 isoforms play dual roles in insulin secretion and insulin-stimulated glucose uptake. Diabetologia 57(10):2173–2182. https://doi.org/10.1007/s00125-014-3312-y
Ramalingam L, Oh E, Thurmond DC (2014) Doc2b enrichment enhances glucose homeostasis in mice via potentiation of insulin secretion and peripheral insulin sensitivity. Diabetologia 57(7):1476–1484. https://doi.org/10.1007/s00125-014-3227-7
Keller MP, Choi Y, Wang P et al (2008) A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Res 18(5):706–716. https://doi.org/10.1101/gr.074914.107
Aslamy A, Thurmond DC (2017) Exocytosis proteins as novel targets for diabetes prevention and/or remediation? Am J Physiol Regul Integr Comp Physiol 312(5):R739–R752. https://doi.org/10.1152/ajpregu.00002.2017
Chang L, Chiang SH, Saltiel AR (2004) Insulin signaling and the regulation of glucose transport. Mol Med 10:65–71
Kiraly-Borri CE, Morgan A, Burgoyne RD, Weller U, Wollheim CB, Lang J (1996) Soluble N-ethylmaleimide-sensitive-factor attachment protein and N-ethylmaleimide-insensitive factors are required for Ca2+-stimulated exocytosis of insulin. Biochem J 314(1):199–203. https://doi.org/10.1042/bj3140199
Lang J (1999) Molecular mechanisms and regulation of insulin exocytosis as a paradigm of endocrine secretion. Eur J Biochem 259(1-2):3–17. https://doi.org/10.1046/j.1432-1327.1999.00043.x
Sollner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE (1993) A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 75(3):409–418. https://doi.org/10.1016/0092-8674(93)90376-2
Michaeli L, Gottfried I, Bykhovskaia M, Ashery U (2017) Phosphatidylinositol (4, 5)-bisphosphate targets double C2 domain protein B to the plasma membrane. Traffic 18(12):825–839. https://doi.org/10.1111/tra.12528
Martens S, McMahon HT (2011) C2 domains and membrane fusion. Curr Top Membr 68:141–159
Yu H, Rathore SS, Davis EM, Ouyang Y, Shen J (2013) Doc2b promotes GLUT4 exocytosis by activating the SNARE-mediated fusion reaction in a calcium- and membrane bending-dependent manner. Mol Biol Cell 24(8):1176–1184. https://doi.org/10.1091/mbc.e12-11-0810
Friedrich R, Yeheskel A, Ashery U (2010) DOC2B, C2 domains, and calcium: a tale of intricate interactions. Mol Neurobiol 41(1):42–51. https://doi.org/10.1007/s12035-009-8094-8
Jewell JL, Oh E, Ramalingam L et al (2011) Munc18c phosphorylation by the insulin receptor links cell signaling directly to SNARE exocytosis. J Cell Biol 193(1):185–199. https://doi.org/10.1083/jcb.201007176
Orita S, Sasaki T, Naito A et al (1995) Doc2: a novel brain protein having two repeated C2-like domains. Biochem Biophys Res Commun 206(2):439–448. https://doi.org/10.1006/bbrc.1995.1062
Sakaguchi G, Orita S, Maeda M, Igarashi H, Takai Y (1995) Molecular cloning of an isoform of Doc2 having two C2-like domains. Biochem Biophys Res Commun 217(3):1053–1061. https://doi.org/10.1006/bbrc.1995.2876
Naito A, Orita S, Wanaka A et al (1997) Molecular cloning of mouse Doc2α and distribution of its mRNA in adult mouse brain. Brain Res Mol Brain Res 44(2):198–204. https://doi.org/10.1016/S0169-328X(96)00198-2
Lundby A, Secher A, Lage K et al (2012) Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat Commun 3(1):876. https://doi.org/10.1038/ncomms1871
Luo W, Slebos RJ, Hill S et al (2008) Global impact of oncogenic Src on a phosphotyrosine proteome. J Proteome Res 7(8):3447–3460. https://doi.org/10.1021/pr800187n
Hornbeck PV, Kornhauser JM, Tkachev S et al (2012) PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res 40(D1):D261–D270. https://doi.org/10.1093/nar/gkr1122
Shimoda Y, Okada S, Yamada E, Pessin JE, Yamada M (2015) Tctex1d2 is a negative regulator of GLUT4 translocation and glucose uptake. Endocrinology 156(10):3548–3558. https://doi.org/10.1210/en.2015-1120
Nagano F, Orita S, Sasaki T et al (1998) Interaction of Doc2 with tctex-1, a light chain of cytoplasmic dynein. Implication in dynein-dependent vesicle transport. J Biol Chem 273(46):30065–30068. https://doi.org/10.1074/jbc.273.46.30065
Ramalingam L, Lu J, Hudmon A, Thurmond DC (2014) Doc2b serves as a scaffolding platform for concurrent binding of multiple Munc18 isoforms in pancreatic islet beta-cells. Biochem J 464(2):251–258. https://doi.org/10.1042/BJ20140845
Aslamy A, Oh E, Olson EM et al (2018) Doc2b protects β-cells against inflammatory damage and enhances function. Diabetes 67(7):1332–1344. https://doi.org/10.2337/db17-1352
Ueyama A, Yaworsky KL, Wang Q, Ebina Y, Klip A (1999) GLUT-4myc ectopic expression in L6 myoblasts generates a GLUT-4-specific pool conferring insulin sensitivity. Am J Phys 277:E572–E578
McCarthy AM, Spisak KO, Brozinick JT, Elmendorf JS (2006) Loss of cortical actin filaments in insulin-resistant skeletal muscle cells impairs GLUT4 vesicle trafficking and glucose transport. Am J Physiol Cell Physiol 291(5):C860–C868. https://doi.org/10.1152/ajpcell.00107.2006
Chen G, Raman P, Bhonagiri P, Strawbridge AB, Pattar GR, Elmendorf JS (2004) Protective effect of phosphatidylinositol 4,5-bisphosphate against cortical filamentous actin loss and insulin resistance induced by sustained exposure of 3T3-L1 adipocytes to insulin. J Biol Chem 279(38):39705–39709. https://doi.org/10.1074/jbc.C400171200
Tunduguru R, Zhang J, Aslamy A et al (2017) The actin-related p41ARC subunit contributes to p21-activated kinase-1 (PAK1)-mediated glucose uptake into skeletal muscle cells. J Biol Chem 292(46):19034–19043. https://doi.org/10.1074/jbc.M117.801340
Zhou M, Sevilla L, Vallega G et al (1998) Insulin-dependent protein trafficking in skeletal muscle cells. Am J Phys 275:E187–E196
Grill MA, Bales MA, Fought AN, Rosburg KC, Munger SJ, Antin PB (2003) Tetracycline-inducible system for regulation of skeletal muscle-specific gene expression in transgenic mice. Transgenic Res 12(1):33–43. https://doi.org/10.1023/A:1022119005836
Spurlin BA, Park SY, Nevins AK, Kim JK, Thurmond DC (2004) Syntaxin 4 transgenic mice exhibit enhanced insulin-mediated glucose uptake in skeletal muscle. Diabetes 53(9):2223–2231. https://doi.org/10.2337/diabetes.53.9.2223
Morley TS, Xia JY, Scherer PE (2015) Selective enhancement of insulin sensitivity in the mature adipocyte is sufficient for systemic metabolic improvements. Nat Commun 6(1):7906. https://doi.org/10.1038/ncomms8906
Riant E, Waget A, Cogo H, Arnal JF, Burcelin R, Gourdy P (2009) Estrogens protect against high-fat diet-induced insulin resistance and glucose intolerance in mice. Endocrinology 150(5):2109–2117. https://doi.org/10.1210/en.2008-0971
Winzell MS, Ahren B (2004) The high-fat diet-fed mouse: a model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes 53(Suppl 3):S215–S219. https://doi.org/10.2337/diabetes.53.suppl_3.S215
Williams LM, Campbell FM, Drew JE et al (2014) The development of diet-induced obesity and glucose intolerance in C57BL/6 mice on a high-fat diet consists of distinct phases. PLoS One 9(8):e106159. https://doi.org/10.1371/journal.pone.0106159
Jiang ZY, Zhou QL, Coleman KA, Chouinard M, Boese Q, Czech MP (2003) Insulin signaling through Akt/protein kinase B analyzed by small interfering RNA-mediated gene silencing. Proc Natl Acad Sci U S A 100(13):7569–7574. https://doi.org/10.1073/pnas.1332633100
Sano H, Kane S, Sano E et al (2003) Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem 278(17):14599–14602. https://doi.org/10.1074/jbc.C300063200
Pham K, Langlais P, Zhang X, Chao A, Zingsheim M, Yi Z (2012) Insulin-stimulated phosphorylation of protein phosphatase 1 regulatory subunit 12B revealed by HPLC-ESI-MS/MS. Proteome Sci 10(1):52. https://doi.org/10.1186/1477-5956-10-52
Rodriguez E, Pulido N, Romero R, Arrieta F, Panadero A, Rovira A (2004) Phosphatidylinositol 3-kinase activation is required for sulfonylurea stimulation of glucose transport in rat skeletal muscle. Endocrinology 145(2):679–685. https://doi.org/10.1210/en.2003-0755
Strommer L, Permert J, Arnelo U et al (1998) Skeletal muscle insulin resistance after trauma: insulin signaling and glucose transport. Am J Phys 275:E351–E358
Semiz S, Park JG, Nicoloro SM et al (2003) Conventional kinesin KIF5B mediates insulin-stimulated GLUT4 movements on microtubules. EMBO J 22(10):2387–2399. https://doi.org/10.1093/emboj/cdg237
Xiao Q, Miao B, Bi J, Wang Z, Li Y (2016) Prioritizing functional phosphorylation sites based on multiple feature integration. Sci Rep 6(1):24735. https://doi.org/10.1038/srep24735
Mertins P, Mani DR, Ruggles KV et al (2016) Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534(7605):55–62. https://doi.org/10.1038/nature18003
Zhu H, Lee HY, Tong Y et al (2012) Crystal structures of the tetratricopeptide repeat domains of kinesin light chains: insight into cargo recognition mechanisms. PLoS One 7(3):e33943. https://doi.org/10.1371/journal.pone.0033943
Jewell JL, Oh E, Bennett SM, Meroueh SO, Thurmond DC (2008) The tyrosine phosphorylation of Munc18c induces a switch in binding specificity from syntaxin 4 to Doc2beta. J Biol Chem 283(31):21734–21746. https://doi.org/10.1074/jbc.M710445200
Ramalingam L, Yoder SM, Oh E, Thurmond DC (2014) Munc18c: a controversial regulator of peripheral insulin action. Trends Endocrinol Metab 25(11):601–608. https://doi.org/10.1016/j.tem.2014.06.010
Takazawa K, Noguchi T, Hosooka T, Yoshioka T, Tobimatsu K, Kasuga M (2008) Insulin-induced GLUT4 movements in C2C12 myoblasts: evidence against a role of conventional kinesin motor proteins. Kobe J Med Sci 54:E14–E22
Guilherme A, Emoto M, Buxton JM et al (2000) Perinuclear localization and insulin responsiveness of GLUT4 requires cytoskeletal integrity in 3T3-L1 adipocytes. J Biol Chem 275(49):38151–38159. https://doi.org/10.1074/jbc.M003432200
Fletcher LM, Welsh GI, Oatey PB, Tavare JM (2000) Role for the microtubule cytoskeleton in GLUT4 vesicle trafficking and in the regulation of insulin-stimulated glucose uptake. Biochem J 352(2):267–276. https://doi.org/10.1042/bj3520267
Acknowledgements
We are grateful to E. Olson (Department of Cellular and Molecular Endocrinology, City of Hope, Duarte, CA, USA) for technical support and to N. Linford (Department of Cellular and Molecular Endocrinology, City of Hope, Duarte, CA, USA) for editing assistance. Research reported in this publication also includes work performed in the Multi-omics Mass Spectrometry & Biomarker Discovery Core, Integrative Genomics and Bioinformatics Core, Drug Discovery and Structural Biology Core, the Light Microscopy/Digital Imaging Core and the Comprehensive Metabolic Phenotyping Core at City of Hope (Duarte, CA, USA), all supported by a Cancer Center Support Grant from the National Cancer Institute to City of Hope. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Parts of this work were presented at the Federation of American Societies for Experimental Biology (FASEB) science research conference, Glucose Transport: Gateway for Metabolic Systems Biology, July 2017.
Funding
This work was supported by grants from: the National Institutes of Health, including from the National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK067912; R01 DK102233; R01 DK112917 to DCT); the American Heart Association (17POST33661194, to JZ; 15PRE21970002 to RT); and the National Cancer Institute (P30CA33572). This work was also supported by the Indiana Clinical and Translational Science Institute (predoctoral fellowship to AA). Additional financial support was provided to DCT through City of Hope: the Ruth and Robert Lanman Endowment, the George Schaeffer award and an Excellence award.
Author information
Authors and Affiliations
Contributions
JZ performed the majority of the studies, contributed to discussion and wrote and edited the manuscript. EO designed and generated the TRE-Doc2b+/− mice and contributed to the analysis of serum analytes and to the manuscript revision and discussion. KEM assisted with the design of the myotube viral transduction paradigm, the co-immunoprecipitation experiments and IPGTT studies of the obese mice and contributed to the manuscript revision and discussion. AA, RV and VAS assisted with in vivo studies and contributed to manuscript revision and discussion. RT assisted with L6-mycGLUT4 cell culture and manuscript revision and discussion. MA generated the Doc2b adenovirus and contributed to manuscript revision and discussion. DCT conceived the study, contributed to the discussion and wrote, reviewed and edited the manuscript. All authors read and approved the final version of the manuscript. DCT is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Corresponding author
Ethics declarations
The authors declare that there is no duality of interest associated with this manuscript.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 521 kb)
Rights and permissions
About this article

Cite this article
Zhang, J., Oh, E., Merz, K.E. et al. DOC2B promotes insulin sensitivity in mice via a novel KLC1-dependent mechanism in skeletal muscle. Diabetologia 62, 845–859 (2019). https://doi.org/10.1007/s00125-019-4824-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-019-4824-2