
Abstract
Aims/hypothesis
Antioxidants have been shown to ameliorate lipid-induced impairment of insulin action and beta cell function, both in vitro and in animal studies. The aim of the present study was to examine the effects of two orally administered antioxidants, N-acetylcysteine (NAC) and taurine (TAU), on lipotoxicity in humans.
Methods
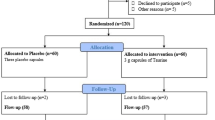
Nine non-diabetic men, who were either overweight or obese, underwent three studies each, 4–6 weeks apart, in random order: (1) i.v. infusion of saline for 48 h (SAL); (2) i.v. infusion of Intralipid and heparin for 48 h to mimic chronic elevation of plasma NEFA (IH); and (3) IH infusion for 48 h with concurrent oral NAC (IH+NAC). Six men underwent similar studies except for study 3, where instead of NAC they received a 2 week pretreatment with oral TAU (IH+TAU).
Results
For both the NAC and TAU studies, a 48 h IH infusion alone without antioxidant impaired insulin sensitivity (S I, 63% and 62% of SAL in NAC and TAU studies, respectively) and beta cell function, as evidenced by a reduction in disposition index (DI, 55% and 54% of SAL in NAC and TAU studies, respectively). NAC failed to prevent the lipid-induced increase in levels of the plasma oxidative stress marker malondialdehyde and did not prevent the lipid-induced reduction in S I or DI, whereas TAU completely prevented the rise in malondialdehyde and decreased 4-hydroxynonenal, and significantly improved S I (91% of SAL) and DI (81% of SAL).
Conclusions/interpretation
Oral TAU ameliorates lipid-induced functional beta cell decompensation and insulin resistance in humans, possibly by reducing oxidative stress.
ClinicalTrials.gov ID no. NCT0018873
Similar content being viewed by others


Introduction
Defects in both insulin action and secretion are hallmarks of type 2 diabetes [1–3]. The defect in insulin secretion is characterised by the selective impairment of glucose-stimulated insulin secretion (GSIS) [4, 5]. The causes of the beta cell defect remain unclear, although prolonged elevation of plasma glucose and NEFAs have been proposed to be detrimental to the beta cells, often referred to as glucotoxicity and lipotoxicity, respectively [6, 7]. We and others have previously demonstrated the impairing effects of chronically elevated plasma NEFAs on GSIS and insulin sensitivity in humans, especially in predisposed individuals [8–12].
Several mechanisms have been proposed to explain lipid-induced insulin resistance and beta cell dysfunction, including but not limited to, increased synthesis of lipid metabolites such as ceramides [13] and diacylglycerides [14], chronic inflammation [15], endoplasmic reticulum stress [16] and oxidative stress [17, 18]. Of note, oxidative stress can activate the JNK [19] and nuclear factor κB [17] proinflammatory pathways and endoplasmic reticulum stress [20]. Lipid-induced insulin resistance in healthy humans is associated with oxidative stress [21].
In animal models and in vitro studies, antioxidants have been shown to improve insulin sensitivity and beta cell function. Infusion of the antioxidant N-acetylcysteine (NAC) or taurine (TAU) abolished hyperglycaemia-induced insulin resistance [22]. NAC prevented diabetes in Zucker diabetic fatty rats and ob/ob mice, partly due to its effects on GSIS and preservation of beta cell mass [23]. TAU was shown to improve insulin sensitivity in diabetic rats [24]. In our preliminary studies, NAC prevented NEFA-induced insulin resistance, and both NAC and TAU prevented a reduction in GSIS in rats [25]. In both healthy subjects and in type 2 diabetic patients, restoration of redox balance by infusion of glutathione improved insulin sensitivity and beta cell function [26]. Infusion of reduced glutathione, an antioxidant, partially prevented lipid-induced insulin resistance in healthy humans [21]. The effects of NAC and TAU in lipid-induced insulin resistance and beta cell dysfunction have not been investigated in humans. The objective of this study was to investigate whether these antioxidants, when administered orally at their currently approved dosages, can prevent or ameliorate the insulin resistance and impairment of beta cell function induced by chronic elevation of NEFA in healthy men.
Methods
Participants
Overweight or obese, but otherwise healthy, Caucasian men participated in the studies (n = 9 for the NAC study and n = 6 for the TAU study, Table 1). None of the participants was taking any medication or had any known systemic illness. Informed, written consent was obtained from all participants in accordance with the guidelines of the Human Subjects Review Committee of the University Health Network, University of Toronto.
Experimental protocol
Participants were admitted to the Metabolic Testing Centre of the Toronto General Hospital on three occasions in random order, 4–6 weeks apart, after a 12 h overnight fast. On each occasion, subjects received one of the three following treatments: (1) intravenous infusion of saline for 48 h (SAL); (2) intravenous infusion of Intralipid (Baxter Healthcare, Mississauga, ON, Canada) plus heparin for 48 h to raise plasma NEFAs by approximately twofold, as previously described [8] (IH); (3) IH infusion for 48 h together with either oral NAC (IH+NAC, NAC study) or oral TAU (IH+TAU, TAU study). In the TAU study the subjects were also pretreated with TAU for 2 weeks. Studies with NAC or TAU without lipid infusion were not performed since we have shown in rats that NAC or TAU, even at high intravenous dosages, did not exert any effect on insulin sensitivity or GSIS in the absence of lipid infusion. The dosage of NAC was the same as that recommended for acetaminophen overdose, which is the maximum oral dose for therapeutic use. NAC is not currently approved in Canada for prolonged oral administration at this dose beyond 48–72 h. NAC was administered with a loading dose of 140 mg/kg followed by 70 mg/kg every 4 h during the 48 h IH infusion. In the TAU study, participants ingested TAU (Jarrow Formulas, Los Angeles, CA, USA) at a dosage of 3 g/day divided into three equal doses for 2 weeks prior to and during the IH infusion. Blood samples were taken before and during lipid/saline infusion for assays of plasma glucose, insulin, C-peptide, triacylglycerols and NEFAs. Plasma aldehydes malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE) were analysed in the samples taken on day 3 prior to the hyperglycaemic clamp. The primary [insulin sensitivity (SI) and beta cell function as indicated by disposition index (DI)] and secondary [insulin secretion rate (ISR), endogenous insulin clearance and oxidative stress markers] outcomes measures were assessed as below.
Assessment of GSIS, insulin sensitivity and insulin clearance
In each study, on day 3 of the infusion, a 2 h hyperglycaemic clamp and a 2 h euglycaemic–hyperinsulinaemic clamp were performed to assess GSIS, S I and insulin clearance (ClI). Please refer to the Electronic supplementary material (ESM) for further details.
Calculations of ISR, S I, ClI and DI
ISR was calculated from deconvolution of plasma C-peptide levels during the last 30 min of the hyperglycaemic clamp [27]. S I was calculated as \( S_{{\text{I}}} = {{\text{Cl}}_{{{\text{glc}}}} } \mathord{\left/ {\vphantom {{{\text{Cl}}_{{{\text{glc}}}} } {{\left( {{\text{Ins}}_{{{\text{clamp}}}} - {\text{Ins}}_{{{\text{base}}}} } \right)}}}} \right. \kern-\nulldelimiterspace} {{\left( {{\text{Ins}}_{{{\text{clamp}}}} - {\text{Ins}}_{{{\text{base}}}} } \right)}} \) from the last 30 min of the euglycaemic clamp, where Clglc is the glucose clearance estimated as glucose infusion rate (G inf) divided by glucose concentration; Insclamp is the insulin concentration during the clamp; Insbase is the basal insulin level. ClI was calculated as \( {\text{Cl}}_{{\text{I}}} = {{\text{ISR}}} \mathord{\left/ {\vphantom {{{\text{ISR}}} {{\left( {{\text{Ins}}_{{{\text{clamp}}}} - {\text{Ins}}_{{{\text{base}}}} } \right)}}}} \right. \kern-\nulldelimiterspace} {{\left( {{\text{Ins}}_{{{\text{clamp}}}} - {\text{Ins}}_{{{\text{base}}}} } \right)}} \) during the last 30 min of the hyperglycaemic clamp. DI was calculated as DI = ISR × S I using the ISR and S I estimated as above.
Laboratory methods
Plasma glucose, insulin, C-peptide, triacylglycerols and NEFAs were assayed as previously described [8]. Plasma MDA and 4-HNE were analysed by a modification of a previously described method [28, 29], on a Hewlett-Packard 5973N MSD GC-MS equipped with an HP-5 MS column and performed in negative-ion chemical ionisation.
Statistical analysis
Plasma glucose, insulin, C-peptide, triacylglycerols and NEFAs during the 48 h infusion period were analysed by two-way ANOVA for repeated measures with Tukey’s multiple comparison test to detect differences between treatments at each time and between times within treatment. Plasma aldehyde concentrations were analysed by two-way ANOVA. Two-way ANOVA for repeated measures with Tukey’s multiple comparison test was performed to detect differences between treatments during the last 30 min of the clamps. A p value of <0.05 was considered significant. All statistical analyses were performed with SAS software (version 8.0; Cary, NC, USA).
Results
NAC study
Plasma triacylglycerols and NEFAs were elevated by IH infusion by approximately two- to fourfold from pre-infusion levels and maintained at levels approximately twofold higher than pre-infusion levels prior to the hyperglycaemic clamp. Both triacylglycerols (p < 0.01 IH and IH+NAC vs SAL) and NEFAs (p < 0.01 IH vs SAL, p < 0.05 IH+NAC vs SAL) were higher in IH and IH+NAC than in SAL after 48 h IH infusion (Table 2).
Plasma glucose, insulin and C-peptide levels and G inf during the last 30 min of the hyperglycaemic clamp are shown in ESM Fig. 1. The calculated ISR did not differ between treatments (Fig. 1a). ClI in IH was decreased as compared with SAL (p < 0.05), and ClI in IH+NAC was not significantly different from either SAL or IH (Fig. 1b).

ISR (a) and ClI (b) calculated from the last 30 min of the hyperglycaemic clamp. *p < 0.05 vs saline
Plasma glucose and insulin levels and G inf during the last 30 min of the euglycaemic–hyperinsulinaemic clamp are presented in ESM Fig. 2. The calculated S I was significantly lower in IH and IH+NAC than in SAL (p < 0.0001; Fig. 2a). No significant difference in S I was observed between IH and IH+NAC. DI was reduced by 33% by IH infusion alone and by 41% by IH+NAC (p < 0.0001). DI in IH+NAC was not significantly different from that in IH (Fig. 2b). Consistent with the lack of effects of NAC on S I and DI, NAC did not prevent the rise of plasma oxidative stress markers induced by IH infusion. After 48 h IH infusion, plasma MDA concentrations were elevated (p < 0.05 for IH and IH+NAC vs SAL). Plasma 4-HNE concentrations in IH were not significantly different from SAL. Both 4-HNE and MDA concentrations in IH+NAC were similar to those in IH (Fig. 2c).

a Insulin sensitivity index (S I) calculated from the last 30 min of the euglycaemic–hyperinsulinaemic clamp in the NAC study, which was performed after 48 h of infusion of either saline (SAL), Intralipid plus heparin (IH), or Intralipid plus heparin plus oral NAC (IH+NAC). b Disposition index (DI) was calculated as the product of S I derived from the euglycaemic–hyperinsulinaemic clamp and ISR derived from the hyperglycaemic clamp. c Plasma aldehyde concentrations were measured after 48 h infusion of either saline (white bars), Intralipid plus heparin (dotted bars), or Intralipid plus heparin plus oral NAC (hatched bars). *p < 0.05 vs saline
TAU study
IH infusion for 48 h increased plasma NEFA and triacylglycerols from pre-infusion levels (p < 0.05), while NEFA and triacylglycerol levels in SAL remained relatively unchanged, which resulted in an approximately twofold elevation in plasma NEFA and triacylglycerols in IH and IH+TAU compared with SAL (p < 0.05) prior to the hyperglycaemic clamp (Table 3).
Plasma glucose, insulin and C-peptide levels and G inf during the last 30 min of the hyperglycaemic clamp are shown in ESM Fig. 3. To maintain plasma glucose at ∼20 mmol/l, slightly higher G inf were required in IH than in SAL and IH+TAU (p < 0.05), whereas G inf in IH+TAU were similar to those in SAL. Insulin levels were higher in IH than in SAL (p < 0.05) and IH+TAU (p < 0.05) but not different between IH+TAU and SAL. The calculated ISR did not differ between protocols (Fig. 3a). The calculated ClI was significantly lower in IH (p < 0.05) than in IH+TAU or SAL (Fig. 3b).

ISR (a) and insulin clearance (ClI) (b) calculated from the last 30 min of the hyperglycaemic clamp. *p < 0.05 vs saline, † p < 0.05 vs Intralipid plus heparin plus oral TAU
Plasma glucose, insulin levels and G inf during the last 30 min of the euglycaemic–hyperinsulinemic clamp are presented in ESM Fig. 4. To maintain plasma glucose at ∼5.6 mmol/l, a lower G inf was required in IH (p < 0.01) and IH+TAU (p < 0.05) than in SAL. G inf in IH+TAU was higher than that in IH (p < 0.05). Insulin concentrations in IH+TAU were not different from SAL but were significantly lower than in IH (p < 0.05).
IH infusion significantly decreased S I (p < 0.05 vs SAL), whereas S I in IH+TAU did not differ from SAL (Fig. 4a). DI (Fig. 4b) was reduced by 46% by IH infusion (p < 0.05). Compared with IH, ingestion of oral TAU for 2 weeks improved DI by 51% (p < 0.05) to a value not significantly different from SAL. IH infusion for 48 h elevated plasma MDA as compared with SAL (p < 0.05). Consistent with the effects of TAU on S I and DI, plasma MDA levels in IH+TAU were lower than in IH (p < 0.05) but similar to SAL (Fig. 4c). Plasma 4-HNE levels in IH+TAU were lower than in both SAL and IH (p < 0.05).

a Insulin sensitivity index (S I) calculated from the last 30 min of the euglycaemic–hyperinsulinemic clamp in the TAU study, which was performed after 48 h of infusion of either saline (SAL), Intralipid plus heparin (IH) or Intralipid plus heparin following 2 weeks of oral TAU (IH+TAU). b Disposition index (DI) was calculated as the product of S I derived from the euglycaemic–hyperinsulinaemic clamp and ISR derived from the hyperglycaemic clamp. c Plasma aldehyde concentrations were measured after 48 h infusion of either saline (white bars), Intralipid plus heparin (dotted bars), or Intralipid plus heparin following 2 week oral TAU (hatched bars). *p < 0.05 vs saline, † p < 0.05 vs Intralipid plus heparin plus oral TAU
Discussion
In the present study we report that TAU, but not NAC, administered orally at currently recommended dosages, ameliorated lipid-induced impairments in insulin sensitivity and beta cell function in overweight and obese, non-diabetic men. To our knowledge, the effects of these two agents, which have antioxidant activity, on lipid-induced insulin resistance and beta cell dysfunction have not previously been examined in humans.
As we and others have previously shown, chronic elevation of plasma NEFAs by Intralipid plus heparin infusion induces insulin resistance [6, 8, 12]. This effect of NEFAs was partially prevented by 2 weeks of oral TAU pretreatment. TAU is an aldehyde scavenger, and by reducing cytotoxic aldehydes, producing reactive oxygen species (ROS), it serves as an antioxidant in vivo. TAU is also synthesised from cysteine; thus, TAU supplementation may preserve cysteine for synthesis of glutathione, a natural intracellular antioxidant [30, 31]. In the current study, 2 weeks of oral TAU prevented lipid infusion-induced elevation of MDA in plasma, suggesting that TAU had an antioxidant effect. Both NAC and TAU, when given i.v. to rats, prevented hyperglycaemia-induced insulin resistance—which was also thought to be mediated through oxidative stress—and increases in lipid peroxidation products [22]. In humans, lipid-induced insulin resistance correlated with oxidative stress [32–34]. Co-infusion of reduced glutathione with Intralipid for 6 h improved whole-body glucose disposal as compared with lipid infusion alone [21]. Lipid-induced muscle insulin resistance in humans is associated with increased protein kinase C (PKC)-δ and -βII membrane translocation and decreased inhibitor of nuclear factor κB-α, evidence of IκB kinase β (IKKβ) activation [35]. In our recent studies in rats, lipid-induced PKC-δ translocation induced oxidative stress, which in turn activated IKKβ/JNK1 and increased serine phosphorylation of insulin receptor substrates 1 and 2, resulting in insulin resistance (unpublished results). It is therefore tempting to postulate that TAU ameliorated lipid-induced insulin resistance through prevention of oxidative stress induced by PKC activation and subsequent events.
No significant alleviating effect of NAC on lipid-induced insulin resistance was observed. As an antioxidant, NAC not only serves as a precursor for glutathione synthesis but also directly reduces free-radical species and other oxidants [36]. In the rodent studies discussed above [22, 25], NAC was infused intravenously. Unlike TAU, NAC undergoes extensive first-pass hepatic metabolism when administered orally, resulting in low systemic bioavailability [37, 38]. The partial effect of NAC on endogenous insulin clearance suggests that effective NAC concentrations were reached in the liver. Therefore, the lack of effect of NAC in improving insulin sensitivity in this study might be due to insufficient NAC delivered to peripheral tissues, thus limiting the direct effect of NAC. Consistent with this, oral NAC did not prevent the rise in the lipid peroxidation product MDA in the plasma stimulated by chronic IH infusion, which suggests a lack of adequate systemic antioxidant effect of NAC administered in this fashion. In this study, the power to detect a difference for both S I and DI were greater than 78%; thus, the lack of effect of NAC does not seem to be due to type II errors. Future studies are required to examine the effects of i.v. NAC or oral NAC over a prolonged period.
IH, NAC and TAU did not affect absolute GSIS in this study. Although GSIS was not reduced by IH infusion, beta cells failed to compensate for the lipid-induced insulin resistance by appropriately increasing insulin secretion, as evidenced by a reduction in the DI, as previously shown [8–10]. In healthy individuals there is a hyperbolic relationship between insulin secretion and sensitivity [39, 40]. Two weeks of oral TAU restored DI with IH infusion to the level with SAL infusion, which indicates that this hyperbolic relationship was restored even in chronically elevated plasma NEFAs. Since improvement of DI in IH+TAU was similar to that of insulin sensitivity, the secretory capacity of beta cells was now ‘adequate’, with improved insulin sensitivity. Very few studies have directly evaluated the effects of TAU on beta cell function, and no previous study has examined its effect on lipid-induced beta cell dysfunction in humans. Brons et al. [41] did not find any effect of TAU (1.5 g/day for two crossover periods of 8 weeks) alone (without lipid infusion) on insulin secretion in overweight first-degree relatives of individuals with type 2 diabetes. In addition to higher TAU dosages, our study was designed to specifically examine the effects of TAU on insulin resistance and beta cell dysfunction induced by a prolonged elevation of plasma NEFAs. In our studies in rats, NAC or TAU administered intravenously prevented lipid-induced impairment of GSIS, in accordance with their ability to prevent the lipid-induced increase in islet ROS [25], but had no effect in the absence of lipid infusion. The direct effects of antioxidants on lipid-induced beta cell dysfunction have not yet been examined in human islets, although compounds with antioxidant properties were effective [42].
In conclusion, oral administration of TAU, but not NAC, at dosages currently approved in Canada, ameliorated lipid-induced impairment in insulin sensitivity and beta cell function in non-diabetic men who were either overweight or obese. This study suggests, but does not provide definitive proof, that the beneficial effect of TAU was mediated by its antioxidant activity, and we acknowledge that mechanisms independent of oxidative stress cannot be ruled out. We also acknowledge that this short-term study was done in limited numbers of male participants only under experimental conditions of lipid infusion. Further studies are needed to provide data on the safety and long-term impact of TAU on glucose homeostasis.
Abbreviations
- ClI :
-
insulin clearance
- DI:
-
disposition index
- G inf :
-
glucose infusion rate
- GSIS:
-
glucose-stimulated insulin secretion
- 4-HNE:
-
4-hydroxynonenal
- IH:
-
Intralipid plus heparin
- IKKβ:
-
IκB kinase β
- ISR:
-
insulin secretion rate
- MDA:
-
malondialdehyde
- NAC:
-
N-acetylcysteine
- PKC:
-
protein kinase C
- ROS:
-
reactive oxygen species
- SAL:
-
saline
- S I :
-
insulin sensitivity index
- TAU:
-
taurine
References
Gerich JE (2003) Contributions of insulin-resistance and insulin-secretory defects to the pathogenesis of type 2 diabetes mellitus. Mayo Clin Proc 78:447–456
Kahn SE (2000) The importance of the beta-cell in the pathogenesis of type 2 diabetes mellitus. Am J Med 108(Suppl 6a):2S–8S
Kahn SE, Hull RL, Utzschneider KM (2006) Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444:840–846
Polonsky KS (1995) Lilly lecture 1994. The beta-cell in diabetes: from molecular genetics to clinical research. Diabetes 44:705–717
Porte D Jr (1991) Banting lecture 1990. Beta-cells in type II diabetes mellitus. Diabetes 40:166–180
Boden G, Ruiz J, Kim CJ, Chen X (1996) Effects of prolonged glucose infusion on insulin secretion, clearance, and action in normal subjects. Am J Physiol 270:E251–E258
Unger RH (1995) Lipotoxicity in the pathogenesis of obesity-dependent NIDDM. Genetic and clinical implications. Diabetes 44:863–870
Carpentier A, Mittelman SD, Lamarche B, Bergman RN, Giacca A, Lewis GF (1999) Acute enhancement of insulin secretion by FFA in humans is lost with prolonged FFA elevation. Am J Physiol 276:E1055–E1066
Carpentier A, Zinman B, Leung N et al (2003) Free fatty acid-mediated impairment of glucose-stimulated insulin secretion in nondiabetic Oji-Cree individuals from the Sandy Lake community of Ontario, Canada: a population at very high risk for developing type 2 diabetes. Diabetes 52:1485–1495
Leung N, Sakaue T, Carpentier A, Uffelman K, Giacca A, Lewis GF (2004) Prolonged increase of plasma non-esterified fatty acids fully abolishes the stimulatory effect of 24 hours of moderate hyperglycaemia on insulin sensitivity and pancreatic beta-cell function in obese men. Diabetologia 47:204–213
Boden G, Chen X, Rosner J, Barton M (1995) Effects of a 48-h fat infusion on insulin secretion and glucose utilization. Diabetes 44:1239–1242
Carpentier A, Mittelman SD, Bergman RN, Giacca A, Lewis GF (2000) Prolonged elevation of plasma free fatty acids impairs pancreatic beta-cell function in obese nondiabetic humans but not in individuals with type 2 diabetes. Diabetes 49:399–408
Holland WL, Brozinick JT, Wang LP et al (2007) Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab 5:167–179
Itani SI, Ruderman NB, Schmieder F, Boden G (2002) Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes 51:2005–2011
Hotamisligil GS (2006) Inflammation and metabolic disorders. Nature 444:860–867
Ozcan U, Cao Q, Yilmaz E et al (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306:457–461
Evans JL, Goldfine ID, Maddux BA, Grodsky GM (2002) Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev 23:599–622
Robertson RP (2006) Oxidative stress and impaired insulin secretion in type 2 diabetes. Curr Opin Pharmacol 6:615–619
Kaneto H, Xu G, Fujii N, Kim S, Bonner-Weir S, Weir GC (2002) Involvement of c-Jun N-terminal kinase in oxidative stress-mediated suppression of insulin gene expression. J Biol Chem 277:30010–30018
Kharroubi I, Ladriere L, Cardozo AK, Dogusan Z, Cnop M, Eizirik DL (2004) Free fatty acids and cytokines induce pancreatic beta-cell apoptosis by different mechanisms: role of nuclear factor-kappaB and endoplasmic reticulum stress. Endocrinology 145:5087–5096
Paolisso G, Gambardella A, Tagliamonte MR et al (1996) Does free fatty acid infusion impair insulin action also through an increase in oxidative stress? J Clin Endocrinol Metab 81:4244–4248
Haber CA, Lam TK, Yu Z et al (2003) N-acetylcysteine and taurine prevent hyperglycemia-induced insulin resistance in vivo: possible role of oxidative stress. Am J Physiol Endocrinol Metab 285:E744–E753
Kaneto H, Kajimoto Y, Miyagawa J et al (1999) Beneficial effects of antioxidants in diabetes: possible protection of pancreatic beta-cells against glucose toxicity. Diabetes 48:2398–2406
Nakaya Y, Minami A, Harada N, Sakamoto S, Niwa Y, Ohnaka M (2000) Taurine improves insulin sensitivity in the Otsuka Long–Evans Tokushima fatty rat, a model of spontaneous type 2 diabetes. Am J Clin Nutr 71:54–58
Oprescu AI, Bikopoulos G, Naassan A et al (2007) Free fatty acid-induced reduction in glucose stimulated insulin secretion evidence for a role of oxidative stress in vitro and in vivo. Diabetes DOI 10.2337/db07-0075
De Mattia G, Bravi MC, Laurenti O et al (1998) Influence of reduced glutathione infusion on glucose metabolism in patients with non-insulin-dependent diabetes mellitus. Metabolism 47:993–997
Van Cauter E, Mestrez F, Sturis J, Polonsky KS (1992) Estimation of insulin secretion rates from C-peptide levels. Comparison of individual and standard kinetic parameters for C-peptide clearance. Diabetes 41:368–377
Luo XP, Yazdanpanah M, Bhooi N, Lehotay DC (1995) Determination of aldehydes and other lipid peroxidation products in biological samples by gas chromatography-mass spectrometry. Anal Biochem 228:294–298
Halliwell B, Gutteridge JM (1990) Role of free radicals and catalytic metal ions in human disease: an overview. Methods Enzymol 186:1–85
Penttila KE (1990) Role of cysteine and taurine in regulating glutathione synthesis by periportal and perivenous hepatocytes. Biochem J 269:659–664
Beetsch JW, Olson JE (1998) Taurine synthesis and cysteine metabolism in cultured rat astrocytes: effects of hyperosmotic exposure. Am J Physiol 274:C866–C874
Paolisso G, D’Amore A, Di Maro G et al (1993) Evidence for a relationship between free radicals and insulin action in the elderly. Metabolism 42:659–663
Paolisso G, D’Amore A, Volpe C et al (1994) Evidence for a relationship between oxidative stress and insulin action in non-insulin-dependent (type II) diabetic patients. Metabolism 43:1426–1429
Paolisso G, Giugliano D (1996) Oxidative stress and insulin action: is there a relationship? Diabetologia 39:357–363
Boden G, She P, Mozzoli M et al (2005) Free fatty acids produce insulin resistance and activate the proinflammatory nuclear factor-kappaB pathway in rat liver. Diabetes 54:3458–3465
Cotgreave IA (1997) N-acetylcysteine: pharmacological considerations and experimental and clinical applications. Adv Pharmacol 38:205–227
De Caro L, Ghizzi A, Costa R, Longo A, Ventresca GP, Lodola E (1989) Pharmacokinetics and bioavailability of oral acetylcysteine in healthy volunteers. Arzneimittelforschung 39:382–386
Sjodin K, Nilsson E, Hallberg A, Tunek A (1989) Metabolism of N-acetyl-l-cysteine. Some structural requirements for the deacetylation and consequences for the oral bioavailability. Biochem Pharmacol 38:3981–3985
Bergman RN, Phillips LS, Cobelli C (1981) Physiologic evaluation of factors controlling glucose tolerance in man: measurement of insulin sensitivity and beta-cell glucose sensitivity from the response to intravenous glucose. J Clin Invest 68:1456–1467
Kahn SE, Prigeon RL, McCulloch DK et al (1993) Quantification of the relationship between insulin sensitivity and beta-cell function in human subjects. Evidence for a hyperbolic function. Diabetes 42:1663–1672
Brons C, Spohr C, Storgaard H, Dyerberg J, Vaag A (2004) Effect of taurine treatment on insulin secretion and action, and on serum lipid levels in overweight men with a genetic predisposition for type II diabetes mellitus. Eur J Clin Nutr 58:1239–1247
Lupi R, Del Guerra S, Fierabracci V et al (2002) Lipotoxicity in human pancreatic islets and the protective effect of metformin. Diabetes 51(Suppl 1):S134–S137
Acknowledgements
We thank P. Harley and L. Szeto for their technical assistance. This work was supported by an operating grant from the Canadian Diabetes Association. G. F. Lewis holds a Canada Research Chair in Diabetes and is a Career Investigator of the Heart and Stroke Foundation of Canada. C. Xiao is supported by postdoctoral fellowships from the Canadian Diabetes Association and the Banting and Best Diabetes Centre at the University of Toronto.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Xiao, C., Giacca, A. & Lewis, G.F. Oral taurine but not N-acetylcysteine ameliorates NEFA-induced impairment in insulin sensitivity and beta cell function in obese and overweight, non-diabetic men. Diabetologia 51, 139–146 (2008). https://doi.org/10.1007/s00125-007-0859-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-007-0859-x