
Abstract
Aims/hypothesis
Glycoxidised LDL has been implicated in the pathogenesis of atherosclerosis, a major complication of diabetes. Since atherogenesis may occur at an early stage of diabetes, we investigated whether circulating LDL isolated from subjects with IGT (n = 20) showed an increased glycoxidation status and explored the proatherogenic effects of LDL samples on macrophages.
Subjects and methods
We investigated LDL modifications using GC-MS. Murine macrophages were incubated with LDL samples for 1 h, and then mRNA expression rates of the scavenger receptors CD36 and scavenger receptor class B type 1 (SCARB1, formerly known as SR-BI) and transcription factor peroxisome proliferator–activator receptor γ (PPARγ) were quantified by real-time RT-PCR.
Results
The GC-MS experiments revealed that oxidative modifications of proline, arginine, lysine and tyrosine residues in apolipoprotein B100 were three- to fivefold higher in LDL samples from IGT subjects compared with those from NGT subjects (n = 20). Moreover, LDL glycoxidation estimated by both \( N^{\varepsilon } \)-(carboxymethyl)lysine (CML) and \( N^{\varepsilon } \)-(carboxyethyl)lysine (CEL) residues was increased more than ninefold in LDL from IGT subjects compared with samples from NGT subjects. Compared with NGT LDL, IGT LDL elicited a significantly higher CD36 (p < 0.05) and PPARG (p < 0.05) gene expression, whereas SCARB1 mRNA expression was not affected.
Conclusions/interpretation
These data suggest that IGT is associated with increased glycoxidation of circulating LDL, which might contribute to the conversion of macrophages into a proatherogenic phenotype.
Similar content being viewed by others

Introduction
Macrovascular disease with accelerated atherosclerosis is a common and severe complication in patients with type 2 diabetes mellitus. Recent evidence suggests that vascular complications are triggered prior to overt manifestation of disease, and may occur in conjunction with IGT. The oxidation of LDL observed in prediabetic states [1] has been identified as a crucial step in atherosclerotic lesion formation [2]. The seemingly unregulated uptake of modified LDL by monocytes/macrophages, with subsequent foam cell formation, is mediated by a variety of scavenger receptors, including macrophage scavenger receptor types I and II (MSRI/II, also known as SR-AI/II), CD36, scavenger receptor class B type 1 (SCARB1, also known as SR-BI), CD68 and lectin-type oxidised LDL receptor 1 (LOX-1). Previous studies have implicated peroxisome proliferator-activated receptor γ (PPARγ) in the transcriptional regulation of CD36 by oxidised LDL (oxLDL) [3]. Nagy et al. [3] demonstrated that oxLDL upregulates the expression of the gene encoding PPARγ (PPARG), followed by increased transcription of CD36 and enhanced scavenger receptor-mediated cellular uptake of oxLDL via a positive feedback loop. Potential anti-atherogenic consequences of activating PPARγ include the ability of PPARγ agonists to inhibit expression of the genes encoding TNFα, IL-1ß, IL-6, and C–C chemokine receptor 2 [4, 5].
In addition to oxidation, LDL can also be modified by glycation processes, thus forming AGEs, such as \( N^{\varepsilon } \)-(carboxymethyl)lysine (CML) and \( N^{\varepsilon } \)-(carboxyethyl)lysine (CEL), under hyperglycaemic conditions [6]. However, the exact mechanisms of AGE generation in vivo are not fully understood. Glucose is an important AGE precursor, but lipids, reactive oxygen species and amino acids might also significantly contribute to the complex process of AGE formation [7, 8]. Previous in vitro studies revealed that both oxidised [9–11] and glycoxidised LDL [12] are able to increase CD36 mRNA expression in macrophages. Moreover, glycation of LDL, in the absence of oxidation, gives rise to lipid accumulation in macrophages [13]. Thus, both LDL glycation and oxidation alone are sufficient for the formation of lipid-laden cells.
Since atherosclerosis starts in prediabetic states, such as IGT [1, 14], the aim of the present study was to determine whether LDL obtained from IGT subjects is modified by glycoxidation, and to investigate the potential proatherogenic effects of the LDL samples on CD36, SCARB1 and PPARG mRNA expression in macrophages.
Subjects and methods
Subjects
Forty subjects (20 with NGT, 20 with IGT) who were at risk of developing diabetes owing to a family history of type 2 diabetes mellitus, obesity and/or hyper-/dyslipoproteinaemia were examined. Clinically overt diabetes and medication affecting glucose tolerance were exclusion criteria. All subjects consented to participate in the study, which was approved by the local ethics committee.
NGT and IGT were diagnosed based on the results of an OGTT (75 g oral glucose challenge) according to the following WHO guidelines and criteria: 2 h post-challenge plasma glucose concentration of 7.8–11.1 mmol/l for IGT, and >11.1 mmol/l for type 2 diabetes mellitus. During the OGTT, plasma glucose levels were determined at 0, 30, 60, 90 and 120 min. The baseline clinical and biochemical characteristics of the study group are given in Table 1.
LDL isolation and biochemical characterisation
Immediately prior to the oral glucose challenge, plasma samples were collected in EDTA-coated tubes for LDL preparation. To 1.5 ml plasma, 15 μl conservation medium was added, which contained 70 g sucrose, 50 mg phenylmethylsulfonyl fluoride, 20 mg streptomycin sulfate, 20 mg dl-dithiothreitol and 20 mg sodium azide per 100 ml water. After addition of conservation medium, plasma samples were shock-frozen in liquid nitrogen and stored at −80°C until analysis. LDL (density 1.006–1.063 g/ml) was isolated by very fast ultracentrifugation, as described previously [15]. The lipid constituents of the plasma and lipoprotein fractions were measured as previously described [15]. LDL apolipoprotein B100 (apoB100) was measured by immunoelectrophoresis (Sebia, Issy-les-Moulineaux, France). The extent of LDL apoB100 modification was evaluated by sensitive GC-MS analysis of highly specific products of the following modifications of amino acid side chain residues: (1) oxidation: γ-glutamyl semialdehyde (γGSA), α-aminoadipic semialdehyde (αASA) and 3-chlorotyrosine; and (2) glycation: CML and CEL. Of note, γGSA and αASA form, by reduction, the analytes 5-hydroxy-2-aminovaleric acid (HAVA) and 6-hydroxy-2-aminocaproic acid (HACA), which are suitable for the method employed [16]. LDL samples were processed in subdued light to prevent photo-oxidation. All buffers and solutions were degassed and stored under argon. Delipidation and reduction of LDL and enzymatic hydrolysis of apoB100 using non-specific protease type XIV were performed as previously described [17]. Within the 24-h hydrolysis period, no self-digestion of the protease was observed. To determine the stability and recovery of the analytes, known amounts of HAVA, HACA and CML standard substances were incubated with protease type XIV, with and without native apoB100 for 24 h. The percentage recovery, determined from three independent measurements for each experiment, was 90.2 ± 5.4% for HAVA and 92 ± 4.1% for HACA (all data are means ± SD), respectively, indicating sufficient stability of the analytes during enzymatic hydrolysis. The free amino acids from protein hydrolysates were then isolated by cation exchange chromatography using Dowex AG-50W-X4 resin (H+, 100–200 mesh; Bio-Rad Laboratories, Richmond, CA, USA). The purified and dried protein amino acids were derivatised either to N(O)-ethoxycarbonyl trifluoroethyl ester (ECEE-F3) derivatives (for analysis of HAVA, HACA and 3-chlorotyrosine) or to N(O)-ethoxycarbonyl ethyl ester (ECEE) derivatives (for analysis of CML and CEL) and analysed following the GC-MS protocols described previously by this group of investigators [17]. All samples were run in triplicate. Under the mass spectrometric conditions employed (electron impact ionisation; electron energy 70 eV) the most abundant and characteristic ions of the ECEE-F3 derivatives of HAVA (M r 287), HACA (M r 301) and 3-chlorotyrosine (M r 441) were the [M-127]+ ion (base peak) resulting from the loss of \( ^{ \bullet } {\text{CO}}_{{\text{2}}} {\text{CH}}_{{\text{2}}} {\text{CF}}_{{\text{3}}} \), and the \( {\left[ {{\text{M}} - 89} \right]}^{{ + \bullet }} \) ion from the loss of NH2CO2Et from the corresponding molecular ions \( {\left( {{\left[ {\text{M}} \right]}^{{ + \bullet }} } \right)} \), respectively. Prominent and highly characteristic ions of the ECEE derivatives of CML (M r 404) and CEL (M r 418) were the [M-73]+ ion resulting from the loss of \( ^{ \bullet } {\text{CO}}_{{\text{2}}} {\text{Et}} \) and [M-46]+ ion formed by ejection of EtOH from the molecular ion \( {\left( {{\left[ {\text{M}} \right]}^{{ + \bullet }} } \right)} \). Diagnostically useful ions for HAVA (at a mass to charge ratio [m/z] 160 and m/z 198), HACA (at m/z 174 and m/z 212), 3-chlorotyrosine (at m/z 314 and m/z 352), CML (at m/z 331 and m/z 358), CEL (at m/z 345 and m/z 327), l-norleucine (at m/z 158 and m/z 196), and l-p-chlorophenylalanine (at m/z 226 and m/z 264) were monitored in the selected ion monitoring (SIM) mode under the control of the HP-ChemStation data system (Hewlett-Packard, Palo Alto, CA, USA) [17]. For quantification of HAVA, HACA, 3-chlorotyrosine, CML and CEL, calibration curves were obtained by plotting the peak area ratio for each characteristic oxidation marker : internal standard ion pair (HAVA, HACA, CML and CEL vs l-norleucine; 3-chlorotyrosine vs l-p-chlorophenylalanine) vs the mass ratio of the analytes and the internal standard, respectively. Calibration curves were analysed by unweighted least-squares linear regression analysis and were found to be linear over the range studied (2–600 nmol/l; R 2 > 0.99) and of good quality (intra-assay CV <4.5%, inter-assay CV <6.1%). In terms of signal reproducibility, there was <8% variation in the peak areas being observed for the diagnostic ions used. The limit of determination of all analytes was 1 nmol/l (1 fmol per injection) by this method. By strictly using reductive conditions, the adverse formation of oxidation/glycation markers during sample processing could be inhibited. Furthermore, control experiments in the presence of aminoguanidine showed no additional formation of oxidation/glycation markers during sample processing (data not shown in detail).
Cell culture
Murine macrophages (RAW 264.7 cell line; LGC Promochem, Teddington, Middlesex, UK) were grown in RPMI 1640 Medium (Biochrom, Berlin, Germany) containing 10% fetal bovine serum (FBS) at 37°C in a humidified atmosphere under 5% CO2. After reaching 90% confluence (approximately 7 days of culture), LDLs were added to macrophages in the same culture medium at a concentration of 30 μg protein/ml. In control experiments, PBS was added instead of LDL. Following the addition of lipoprotein suspensions, cells were incubated for 1 h. Before cell harvest, the supernatant fraction was removed, aliquoted and stored at −75°C for analysis of monocyte chemoattractant protein-1 (MCP-1) with the commercial Quantikine M mouse JE/MCP-1 immunoassay kit (R&D Systems, Minneapolis, MN, USA). After LDL exposure, cell viability was demonstrated by trypan blue exclusion; no differences were observed between control and LDL experiments.
RNA preparation and quantitative RT-PCR
RNA preparation and real-time quantitative PCR were performed as previously described [11]. Real-time PCR was performed using a LightCycler rapid thermal cycler system (Roche Diagnostics, Mannheim, Germany) according to the manufacturer’s instructions. LightCycler TM-polymerase chain reaction (PCR) assays for GAPDH (the gene encoding glyceraldehyde-3-phosphate dehydrogenase), CD36 and SCARB1 were based on SYBR Green I detection (FastStart DNA Master SYBR Green I; Roche Molecular Biochemicals). Approximately 20 ng of cDNA, 3 mmol/l of MgCl2, and 1 μmol/l of primers were used in the following cycling protocol: denaturation 600 s at 95°C, 40 cycles of 10 s at 95°C, 10 s at primer-specific temperature, and product-specific elongation time at 72°C. Because of its low copy number, the transcript level of PPARG was analysed by specific hybridisation probes (0.2 μmol/l, FastStart DNA Master Hybridization Probes; Roche Molecular Biochemicals). The specificity of each of the amplified PCR products was verified by melting point analyses and subsequent agarose gel electrophoresis. Primers and hybridisation probes were purchased from Tibmolbiol (Berlin, Germany). The sequences of primers and hybridisation probes, the PCR conditions, and product length are shown in Electronic supplementary material Table 1.
The identity of each of the PCR products was confirmed by direct sequencing (ALF Express; Amersham Biosciences, Uppsala, Sweden). For normalisation of the transcript levels of CD36, SCARB1 and PPARG, the house-keeping gene GAPDH was quantified in parallel.
The copy number of the GAPDH, CD36, SCARB1 and PPARG mRNAs was calculated in relation to the amplification product amounts of external standards. LightCycler capillaries coated storage-stable with amounts of 101–106 molecules of HPLC-calibrated mouse CD36, SCARB1, PPARG and GAPDH fragments (Roboscreen, Leipzig, Germany) were used as standards for quantification. Transcript amounts were calculated applying the second derivative maximum method of the LightCycler software 3.1.
Statistical analysis
Differences between subjects with NGT and those with IGT, in terms of clinical characteristics, composition of LDL particles and effects of LDL on gene expression, were assessed by univariate ANOVA. Spearman’s rank correlation coefficient (ρ) was calculated between glycoxidation parameters of LDL apoB100 and plasma glucose levels during OGTT and for the correlation analysis of MCP-1 levels in supernatant of macrophages with PPARG gene expression. All data were analysed using the SPSS statistical package (v. 12.0 for Windows; SPSS, Chicago, IL, USA). A value of p < 0.05 was considered statistically significant.
Results
Table 1 shows the clinical and biochemical characteristics of the NGT and IGT subjects. Plasma glucose levels during the OGTT differed significantly between the two groups. Those with IGT showed a tendency towards higher HbA1c and triacylglycerol levels and lower HDL-cholesterol levels as compared with NGT subjects, although these differences were not statistically significant. As indicated by the blood leucocyte count and serum levels of C-reactive protein, IGT subjects did not exhibit greater systemic inflammation compared with those with NGT.
After LDL preparation, the lipid and protein constituents of the lipoprotein particles were assessed (Table 2). LDL from NGT and IGT subjects showed no significant differences in the percentage of triacylglycerol, cholesteryl ester, non-esterified cholesterol, phospholipids and protein contents. Investigation of apoB100 glycoxidation parameters, however, revealed substantial differences between LDL from NGT subjects and samples from IGT subjects. HAVA, proposed to be a specific oxidation marker of proline and arginine side chain residues, and HACA, a primary oxidation product of lysine side chain residues, were increased by threefold and fivefold, respectively, in LDL samples from IGT subjects compared with those from NGT subjects. Moreover, 3-chlorotyrosine, a specific oxidation product of myeloperoxidase (MPO)-derived hypochlorous acid, was elevated by threefold in LDL from the IGT group compared with the NGT group. Of interest, the most pronounced differences were observed in CML and CEL content, being 9.3-fold and 9.5-fold higher in IGT subjects than in NGT subjects (see Table 2). For comparison, data for a healthy control population were obtained (see Tables 2 and 3). Of note, levels of the lysine and tyrosine modifications, but not the arginine and proline modifications, in LDL from the NGT group significantly exceeded those in the LDL from the healthy control population (see Table 2).
To test whether LDL glycoxidation parameters were related to the metabolic status of NGT and IGT subjects, a correlation analysis was performed between glycoxidation markers and plasma glucose levels estimated during OGTT. As shown in Table 4, each of the apoB100 modifications was significantly positively associated with plasma glucose levels at the different time points of the OGTT. Remarkably, for all parameters the strongest association was achieved 120 min after the oral glucose challenge. Furthermore, compared with HAVA, HACA and 3-chlorotyrosine, CML and CEL showed a stronger correlation with plasma glucose profile (see Table 4).
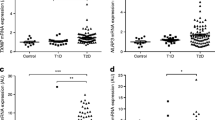
Glycoxidative apoB100 modifications are thought to ultimately result in the formation of new epitopes that are specifically recognised by scavenger receptors [18]. After establishing that there is a higher degree of LDL glycoxidation in IGT subjects, we investigated whether these LDL samples are able to stimulate biological activities with proatherogenic properties that differed from LDL samples from NGT subjects. For this purpose, murine macrophages were exposed to isolated LDL at a concentration of 30 μg protein/ml. After an incubation time of 1 h, macrophage CD36, SCARB1 and PPARG mRNA expression was quantified by RT-PCR using the LightCycler instrument. CD36 showed the highest expression of all genes tested (5.84% relative to GAPDH, our housekeeping gene), followed by SCARB1 (2.08%) and PPARG (0.11%). The very low expression rate of PPARG (on average, approximately 200 cDNA molecules/1 μl synthesised cDNA) emphasises its function as a transcriptional regulator. The necessary methodological sensitivity for PPARG cDNA detection was achieved using specific hybridisation probes in combination with coated LightCycler capillaries as external standards. The expression rates of CD36, SCARB1 and PPARG are shown in Fig. 1. To demonstrate the variations within the groups, the data are given by box plots showing medians and 10th, 25th, 75th and 90th percentiles. Most importantly, CD36 and PPARG mRNA expression was significantly higher in LDL from IGT subjects than in LDL from NGT subjects, whereas SCARB1 mRNA levels did not differ between the two groups. A significant positive correlation between CD36 and PPARG expression was found both in the NGT (ρ = 0.485, p < 0.05) and IGT (ρ = 0.705, p < 0.001) groups, underlining the close regulatory association in macrophages, as previously proposed [19]. Moreover, PPARG expression was significantly negatively related to MCP-1 release from macrophages when treated with LDL from IGT subjects (ρ = 0.572, p = 0.013) but not NGT subjects. No correlation was observed between mRNA expression rates for any of the analysed genes and glycoxidation parameters of isolated LDL.

Box plots showing mRNA expression rates of CD36 (a), PPARG (b) and SCARB1 (c) normalised to GAPDH and PBS controls in murine macrophages after 1 h incubation with LDL obtained from NGT and IGT subjects. In these plots, horizontal lines within boxes represent median values; the upper and lower horizontal lines of the boxes represent the 25th and 75th percentiles, respectively; and the upper and lower bars outside the boxes represent the 90th and 10th percentiles, respectively. *p < 0.05 for NGT vs IGT subjects. Data were adjusted for age, sex, WHR, BMI and fasting plasma glucose
Discussion
The main finding of this study is that the apoB100 of circulating LDL isolated from subjects with IGT was substantially more glycoxidised compared with that in LDL particles from NGT subjects. Furthermore, LDL isolated from IGT subjects induced a more pronounced expression of the scavenger receptor CD36 and the transcription factor PPARG in murine macrophages, while SCARB1 expression was not affected. Using a specific and sensitive GC-MS methodology, the present study revealed a three- to fivefold increase in apoB100 arginine, proline, lysine and tyrosine oxidation, and a ninefold increase in the glycoxidation markers CML and CEL in subjects with IGT compared with healthy normolipidaemic and normoglycaemic individuals. Of note, the NGT subjects used as reference group in this study should also be considered to be at risk of development of diabetes owing to a family history of type 2 diabetes mellitus, obesity and/or hyper-/dyslipoproteinaemia. This could explain the higher level of both apoB100 CML and CEL residues found in NGT subjects compared with reference values for CML obtained from healthy volunteers, for example, in the study reported by Fu et al. [20], and in a small group of healthy volunteers investigated by us. Nevertheless, the CML and CEL values in IGT subjects were remarkably higher than those in NGT subjects.
The present findings are consistent with data showing increased HAVA content in circulating LDL recovered from hypercholesterolaemic subjects who were at high atherosclerotic risk [21]. In addition, the specific amino acid oxidation products γGSA, αASA and 3-chlorotyrosine have been identified in LDL apoB100 in atherosclerotic lesions, thus providing additional support for the involvement of oxidatively modified LDL apoB100 amino-acid side-chain residues in the development of atherosclerosis [17].
Of interest, the apoB100 in LDL samples from IGT subjects has a markedly higher 3-chlorotyrosine content compared with that in samples from the NGT subjects. 3-Chlorotyrosine is a specific marker of oxidation by MPO, an enzyme that is abundant in activated proinflammatory cells such as polymorphonuclear leucocytes and monocytes [22]. Recently, clinical evidence has been provided for a close association between MPO levels in the circulation and existing cardiovascular disease. Moreover, the MPO levels can be predictive of future cardiac events and outcome [23]. In the present study, blood leucocyte count and high-sensitivity C-reactive protein levels were not enhanced in IGT subjects compared with NGT subjects, suggesting the absence of acute inflammatory processes. Thus, elevated 3-chlorotyrosine levels in IGT subjects might indicate an enhanced chronic low-grade inflammatory burden that favours atherogenesis.
The origin of glycoxidised LDL in the circulation is as yet unknown. It has been widely assumed that circulating blood is not a site of LDL oxidation, since serum has strong anti-oxidative abilities [24]. It is possible that glycoxidised LDL is formed in the subendothelial space of the vessel and enters the circulation by backward diffusion from stable atherosclerotic lesions, or by release from unstable atherosclerotic plaques. To what extent hyperglycaemic conditions that are present in IGT and diabetes mellitus contribute to increased rates of LDL glycation and/or oxidation within the bloodstream in vivo remains to be established. In this context, it has to be considered that oxidation and glycation processes forming covalent modifications of the LDL protein and lipid moiety occur at markedly different rates under different conditions in vitro [25]. It is assumed that, under anabolic conditions, these different rates of formation of covalent modifications of LDL constituents may contribute in a very heterogeneous manner to steady-state levels of LDL glycoxidation markers.
The strong correlation of LDL glycoxidation markers with plasma glucose levels after oral glucose challenge in the present study indicates that temporary or sustained hyperglycaemia may be causally related to glycoxidative modification of circulating LDL.
Oxidation and glycation processes, both stand-alone and combined, result in the conversion of LDL into a form that is recognised by the scavenger receptors of macrophages, a fundamental mechanism of atherogenesis [2]. However, the nature of the LDL modifications required for cellular recognition and unregulated uptake are poorly understood.
Very recently, animal experiments using dynamic small-animal positron emission tomography showed an extremely fast and complete blood clearance of oxidatively modified LDL in rats in vivo, which has been explained by the concerted action of various scavenger receptors on resident macrophages and endothelial cells in liver, spleen and kidney [26]. Conversely, previous studies published by others, and our own in vivo data in animal models, demonstrate a significantly delayed catabolism of glycated LDL [27]. Dynamic small-animal positron emission tomography studies in rats revealed that glycated LDL had a lower affinity for LDL receptors and enhanced interactions with receptors for AGEs responsible for higher plasma retention time of glycated LDL in vivo (Pietzsch J, Hoppmann S, Richter S, Haase C, unpublished results). With respect to the present data, it can be assumed that, under catabolic conditions, the different interaction of modified LDL with LDL receptors, receptors for AGEs, and the efficient scavenger apparatus may discriminate between two types of modified LDL particles: those that show fast plasma clearance (oxhigh/glychighLDL; oxhigh/glyclowLDL); and those that show low plasma clearance (oxlow/glychighLDL; oxlow/glyclowLDL). This is consistent with our findings showing circulating LDL with lower concentrations of oxidation markers but higher levels of glycoxidation markers in subjects with IGT. Furthermore, it is likely that oxidatively modified LDL in the circulation comprise particles with a still lower extent of oxidation, including the so-called minimally modified LDL, which are in part still recognised via the LDL receptor.
Considering the heterogeneity of oxidative processes of sizable physiological relevance, such as transition metal-catalysed oxidation, MPO-catalysed reactions, nitrosative or glycative stress, additional work is needed to understand the nature of the original oxidative insult. In this respect, further investigations are necessary to provide information on threshold values of in vivo oxidation and glycation to discriminate the metabolic fate of modified LDL particles in vivo. Knowledge of such threshold values should significantly support the interpretation of concentration measurements of circulating modified LDL particles, specific products of LDL oxidation and glycation, or antibodies against modified LDL in plasma samples in various diseases, thus allowing the critical evaluation of their actual relevance as potential indicators of oxidative or glycative stress or atherosclerotic risk [26].
Previous in vitro studies have shown that the two scavenger receptors CD36 and MSRI/III mediate the majority of lipid accumulation in macrophages exposed to oxLDL [28, 29]. Furthermore, several studies, although not all, have demonstrated that targeted deletion of either MSR1 (which codes for both types of the receptor) or the CD36 gene locus in the hyperlipidaemic mouse substantially decreases atherosclerosis [30–32]. Macrophage expression of CD36, but not MSR1, is under the strong control of the nuclear transcriptional regulator PPARγ [3, 33]. Because of the complex effects of PPARγ on lipid metabolism and inflammatory processes, its role in atherosclerosis remains controversial. Potential atherogenic effects of PPARγ include the facilitated uptake of oxLDL by macrophages via enhanced CD36 expression, while anti-atherogenic consequences of PPARγ activation include inhibition of inflammatory cytokine expression and induction of the ABCA1 transporter protein by liver X receptor α, which promotes macrophage cholesterol efflux [34].
In the present study, glycoxidised LDL obtained from patients with IGT stimulated expression of CD36 and PPARG in macrophages to a significantly greater extent than LDL from NGT subjects. Together with the fact that we found a strong positive correlation between CD36 and PPARG expression, these data demonstrate that in vivo-modified LDL induce a feed-forward loop, previously demonstrated in macrophages in response to incubation with in vitro oxidised LDL [3, 11]. In addition to the positive association between CD36 and PPARG gene expression, we found a significant negative correlation between PPARG expression rates and MCP-1 release from macrophages after exposure to LDL from IGT subjects but not from NGT subjects. Since MCP-1 enhances the recruitment of monocytes into the arterial wall [35], this result points to the anti-inflammatory effect of PPARγ activation. The consequences of PPARγ activation may thus be both proatherogenic and anti-atherogenic. The net effect is likely to reflect a balance between local effects in the arterial wall and systemic effects on lipid metabolism and inflammation [19]. Studies in LDL receptor-deficient mice using the most prominent group of PPARγ activators, glucose-lowering glitazones, showed an inhibition of atherosclerotic lesion formation [36, 37].
CD36 and SCARB1 belong to the same class B family of scavenger receptors and share considerable structural homology. They share many ligands, including HDL, LDL, VLDL, oxLDL and anionic phospholipid vesicles, but differ markedly in tissue distribution, preferred binding partners and function in lipid metabolism. SCARB1 is expressed in macrophages in human and murine atherosclerotic lesions [38, 39]. Theoretically, in macrophages, SCARB1 may protect against atherosclerosis by stimulating cholesterol efflux and preventing foam cell formation. Alternatively, its function in the uptake of modified lipoproteins might promote foam cell formation, rendering macrophage SCARB1 a proatherogenic factor. Previous in vitro studies have shown that oxLDL-induced changes in SCARB1 mRNA expression may be dependent on the differentiation status of the investigated cells, with transcription upregulated in the early differentiation stage [40, 41] and downregulated in fully differentiated macrophages [11, 40]. The lack of difference in the rates of SCARB1 expression stimulated by LDL from NGT and IGT subjects in the present study suggests that, in contrast to CD36, macrophage expression of SCARB1 is less sensitive to changes in LDL glycoxidation status.
In summary, LDL obtained from subjects with IGT exhibited an increased apoB100 glycoxidative status compared with LDL from normoglycaemic subjects. Chemical LDL modifications were closely correlated with temporary hyperglycaemia after oral glucose challenge. As a possible consequence of apoB100 modification, CD36 and PPARG mRNA expression in murine macrophages was stimulated to a significantly greater extent by LDL from IGT subjects than by samples from NGT subjects, while SCARB1 expression was not affected. Therefore, IGT is associated with early events in the pathogenesis of cardiovascular complications in diabetic patients. These data further underline the importance of early preventive treatment and strategies.
Abbreviations
- αASA:
-
α-aminoadipic semialdehyde
- apoB100:
-
apolipoprotein B100
- CEL:
-
\( N^{\varepsilon } \)-(carboxyethyl)lysine
- CML:
-
\( N^{\varepsilon } \)-(carboxymethyl)lysine
- ECEE:
-
N(O)-ethoxycarbonyl ethyl ester
- ECEE-F3 :
-
N(O)-ethoxycarbonyl trifluoroethyl ester
- γGSA:
-
γ-glutamyl semialdehyde
- HACA:
-
6-hydroxy-2-aminocaproic acid
- HAVA:
-
5-hydroxy-2-aminovaleric acid
- MSRI/II:
-
macrophage scavenger receptor types I and II
- MCP-1:
-
monocyte chemoattractant protein-1
- MPO:
-
myeloperoxidase
- m/z :
-
mass to charge ratio
- oxLDL:
-
oxidised low-density lipoprotein
- PPARγ:
-
peroxisome proliferator-activated receptor γ
- SCARB1:
-
scavenger receptor class B type 1
References
Kopprasch S, Pietzsch J, Kuhlisch E et al (2002) In vivo evidence for increased oxidation of circulating LDL in impaired glucose tolerance. Diabetes 51:3102–3106
Chisolm GM, Steinberg D (2000) The oxidative modification hypothesis of atherogenesis: an overview. Free Radic Biol Med 28:1815–1826
Nagy L, Tontonoz P, Alvarez JG, Chen H, Evans RM (1998) Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell 93:229–240
Han KH, Chang MK, Boullier A et al (2000) Oxidized LDL reduces monocyte CCR2 expression through pathways involving peroxisome proliferator-activated receptor gamma. J Clin Invest 106:793–802
Jiang C, Ting AT, Seed B (1998) PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 391:82–86
Veiraiah A (2005) Hyperglycemia, lipoprotein glycation, and vascular disease. Angiology 56:431–438
Schleicher E, Weigert C, Rohrbach H, Nerlich A, Bachmeier B, Friess U (2005) Role of glucoxidation and lipid oxidation in the development of atherosclerosis. Ann N Y Acad Sci 1043:343–354
Anderson MM, Heinecke JW (2003) Production of N ɛ-(carboxymethyl)lysine is impaired in mice deficient in NADPH oxidase: a role for phagocyte-derived oxidants in the formation of advanced glycation end products during inflammation. Diabetes 52:2137–2143
Nakagawa T, Nozaki S, Nishida M et al (1998) Oxidized LDL increases and interferon-gamma decreases expression of CD36 in human monocyte-derived macrophages. Arterioscler Thromb Vasc Biol 18:1350–1357
Feng J, Han J, Pearce SF et al (2000) Induction of CD36 expression by oxidized LDL and IL-4 by a common signaling pathway dependent on protein kinase C and PPAR-gamma. J Lipid Res 41:688–696
Westendorf T, Graessler J, Kopprasch S (2005) Hypochlorite-oxidized low-density lipoprotein upregulates CD36 and PPARgamma mRNA expression and modulates SR-BI gene expression in murine macrophages. Mol Cell Biochem 277:143–152
Lam MC, Tan KC, Lam KS (2004) Glycoxidized low-density lipoprotein regulates the expression of scavenger receptors in THP-1 macrophages. Atherosclerosis 177:313–320
Brown BE, Dean RT, Davies MJ (2005) Glycation of low-density lipoproteins by methylglyoxal and glycolaldehyde gives rise to the in vitro formation of lipid-laden cells. Diabetologia 48:361–369
Hanefeld M, Chiasson JL, Koehler C, Henkel E, Schaper F, Temelkova-Kurktschiev T (2004) Acarbose slows progression of intima-media thickness of the carotid arteries in subjects with impaired glucose tolerance. Stroke 35:1073–1078
Pietzsch J, Subat S, Nitzsche S, Leonhardt W, Schentke KU, Hanefeld M (1995) Very fast ultracentrifugation of serum lipoproteins: influence on lipoprotein separation and composition. Biochim Biophys Acta 1254:77–88
Requena JR, Chao CC, Levine RL, Stadtman ER (2001) Glutamic and aminoadipic semialdehydes are the main carbonyl products of metal-catalyzed oxidation of proteins. Proc Natl Acad Sci U S A 98:69–74
Pietzsch J, Bergmann R, Kopprasch S (2004) Analysis of non-protein amino acids as specific markers of low density lipoprotein apolipoprotein B-100 oxidation in human atherosclerotic lesions: the use of N(O)-ethoxycarbonyl trifluoroethyl ester derivatives and GC-MS. Spectroscopy 18:177–183
Berliner JA, Heinecke JW (1996) The role of oxidized lipoproteins in atherogenesis. Free Radic Biol Med 20:707–727
Tontonoz P, Nagy L (1999) Regulation of macrophage gene expression by peroxisome-proliferator-activated receptor gamma: implications for cardiovascular disease. Curr Opin Lipidol 10:485–490
Fu MX, Requena JR, Jenkins AJ, Lyons TJ, Baynes JW, Thorpe SR (1996) The advanced glycation end product, \( N^{\varepsilon } \)-(carboxymethyl)lysine, is a product of both lipid peroxidation and glycoxidation reactions. J Biol Chem 271:9982–9986
Pietzsch J, Lattke P, Julius U (2000) Oxidation of apolipoprotein B-100 in circulating LDL is related to LDL residence time. In vivo insights from stable-isotope studies. Arterioscler Thromb Vasc Biol 20:E63–E67
Malle E, Marsche G, Arnhold J, Davies MJ (2006) Modification of low-density lipoprotein by myeloperoxidase-derived oxidants and reagent hypochlorous acid. Biochim Biophys Acta 1761:392–415
Brennan ML, Penn MS, Van Lente F et al (2003) Prognostic value of myeloperoxidase in patients with chest pain. N Engl J Med 349:1595–1604
Itabe H (2003) Oxidized low-density lipoproteins: what is understood and what remains to be clarified. Biol Pharm Bull 26:1–9
Knott HM, Brown BE, Davies MJ, Dean RT (2003) Glycation and glycoxidation of low-density lipoproteins by glucose and low-molecular mass aldehydes. Formation of modified and oxidized particles. Eur J Biochem 270:3572–3582
Pietzsch J, Bergmann R, Wuest F, Pawelke B, Hultsch C, van den Hoff J (2005) Catabolism of native and oxidized low density lipoproteins: in vivo insights from small animal positron emission tomography studies. Amino Acids 29:389–404
Sasaki J, Cottam GL (1982) Glycosylation of LDL decreases its ability to interact with high-affinity receptors of human fibroblasts in vitro and decreases its clearance from rabbit plasma in vivo. Biochim Biophys Acta 713:199–207
Nicholson AC (2004) Expression of CD36 in macrophages and atherosclerosis: the role of lipid regulation of PPARgamma signaling. Trends Cardiovasc Med 14:8–12
Kunjathoor VV, Febbraio M, Podrez EA et al (2002) Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem 277:49982–49988
Moore KJ, Kunjathoor VV, Koehn SL et al (2005) Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. J Clin Invest 115:2192–2201
Babaev VR, Gleaves LA, Carter KJ et al (2000) Reduced atherosclerotic lesions in mice deficient for total or macrophage-specific expression of scavenger receptor-A. Arterioscler Thromb Vasc Biol 20:2593–2599
Febbraio M, Podrez EA, Smith JD et al (2000) Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest 105:1049–1056
Moore KJ, Rosen ED, Fitzgerald ML et al (2001) The role of PPAR-gamma in macrophage differentiation and cholesterol uptake. Nat Med 7:41–47
Chawla A, Boisvert WA, Lee CH et al (2001) A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell 7:161–171
Gosling J, Slaymaker S, Gu L et al (1999) MCP-1 deficiency reduces susceptibility to atherosclerosis in mice that overexpress human apolipoprotein B. J Clin Invest 103:773–778
Li AC, Binder CJ, Gutierrez A et al (2004) Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARalpha, beta/delta, and gamma. J Clin Invest 114:1564–1576
Collins AR, Meehan WP, Kintscher U et al (2001) Troglitazone inhibits formation of early atherosclerotic lesions in diabetic and nondiabetic low density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol 21:365–371
Van Eck M, Bos IS, Hildebrand RB, Van Rij BT, Van Berkel TJ (2004) Dual role for scavenger receptor class B, type I on bone marrow-derived cells in atherosclerotic lesion development. Am J Pathol 165:785–794
Chinetti G, Gbaguidi FG, Griglio S et al (2000) CLA-1/SR-BI is expressed in atherosclerotic lesion macrophages and regulated by activators of peroxisome proliferator-activated receptors. Circulation 101:2411–2417
Han J, Nicholson AC, Zhou X, Feng J, Gotto AM Jr, Hajjar DP (2001) Oxidized low density lipoprotein decreases macrophage expression of scavenger receptor B-I. J Biol Chem 276:16567–16572
Hirano K, Yamashita S, Nakagawa Y et al (1999) Expression of human scavenger receptor class B type I in cultured human monocyte-derived macrophages and atherosclerotic lesions. Circ Res 85:108–116
Scott J (1989) The molecular and cell biology of apolipoprotein-B. Mol Biol Med 6:65–80
Acknowledgements
We thank Sigrid Nitzsche, Martina Kohl, and Babett Heschel for their excellent technical support. This study was supported by grant 01ZZ9604 [Project A 2.1 Intravascular Lipid Transfer (INVALT) study (J. Pietzsch and U. Julius); Project A 2.9 Oxidative stress as atherogenic factor in impaired glucose tolerance (S. Kopprasch)] from the Federal Ministry of Education and Research, Germany.
Duality of interest
None of the authors is aware of any duality of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
J. Graessler and J. Pietzsch contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Graessler, J., Pietzsch, J., Westendorf, T. et al. Glycoxidised LDL isolated from subjects with impaired glucose tolerance increases CD36 and peroxisome proliferator–activator receptor γ gene expression in macrophages. Diabetologia 50, 1080–1088 (2007). https://doi.org/10.1007/s00125-007-0645-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-007-0645-9