
Abstract
Aims/hypothesis
In the present study, we investigated the consequences of adipose tissue lipolytic inhibition on skeletal muscle substrate use in type 2 diabetic patients.
Materials and methods
We studied ten type 2 diabetic patients under the following conditions: (1) at rest; (2) during 60 min of cycling exercise at 50% of maximal workload capacity and subsequent recovery. Studies were done under normal, fasting conditions (control trial: CON) and following administration of a nicotinic acid analogue (low plasma non-esterified fatty acid trial: LFA). Continuous [U-13C]palmitate and [6,6 -2H2]glucose infusions were applied to quantify plasma NEFA and glucose oxidation rates, and to estimate intramuscular triacylglycerol (IMTG) and glycogen use. Muscle biopsies were collected before and after exercise to determine net changes in lipid and glycogen content specific to muscle fibre type.
Results
Following administration of the nicotinic acid analogue (Acipimox), the plasma NEFA rate of appearance was effectively reduced, resulting in lower NEFA concentrations in the LFA trial (p<0.001). Plasma NEFA oxidation rates were substantially reduced at rest, during exercise and subsequent recovery in the LFA trial. The lower plasma NEFA oxidation rates were compensated by an increase in IMTG and endogenous carbohydrate use (p<0.05). Plasma glucose disposal rates did not differ between trials. In accordance with the tracer data, a greater net decline in type I muscle fibre lipid content was observed following exercise in the LFA trial (p<0.05).
Conclusions/interpretation
This study shows that plasma NEFA availability regulates IMTG use, and that adipose tissue lipolytic inhibition, in combination with exercise, could be an effective means of augmenting intramuscular lipid and glycogen use in type 2 diabetic patients in an overnight fasted state.
Similar content being viewed by others


Introduction
Numerous studies have reported the association between elevated plasma NEFA concentrations, intramyocellular triacylglycerol (IMTG, often also abbreviated to IMCL) accumulation and the development of insulin resistance and/or type 2 diabetes [1–7]. The Randle (glucose–fatty acid) cycle has often been used to explain the mechanism behind skeletal muscle insulin resistance induced by fatty acid (FA) [8]. More recent insights from various lipid infusion studies have led to an alternative mechanism [9–13]. The latter suggests that elevated NEFA delivery and/or impaired FA oxidation result in intramyocellular accumulation of triacylglycerol (TG) and FA metabolites (such as fatty acyl-CoA, ceramides and diacylglycerol), which induce defects in the insulin signalling cascade, causing skeletal muscle insulin resistance [9, 10, 13–17]. Insulin resistance can subsequently lead to development of the hyperglycaemic and/or hyperinsulinaemic state that is associated with type 2 diabetes. The latter is accompanied by major disturbances in skeletal muscle substrate metabolism [18, 19], which further stimulate IMTG accretion at the expense of IMTG mobilisation and subsequent oxidation [20].
Therefore, excess IMTG accretion in sedentary, obese and/or type 2 diabetic patients is the result of a structural imbalance between NEFA uptake, IMTG deposition and its subsequent mobilisation and/or oxidation. Recent data indicate that this imbalance, and not the actual size or intramyocellular distribution of the IMTG pool, is responsible for the development of skeletal muscle insulin resistance [21–23]. It has been speculated that an elevated IMTG utilisation rate will prevent excess accumulation of intramyocellular TG and/or FA (metabolites). In addition, a greater IMTG turnover rate may reduce the degree of intramyocellular lipid peroxidation [17], due to a decreased residence time of the lipid deposits. The turnover of the IMTG pool should, therefore, form a major therapeutic target in the prevention and/or treatment of skeletal muscle insulin resistance, and interventions should be developed that stimulate IMTG use [24].
Exercise has been shown to augment IMTG use in healthy, fit males and probably represents an effective way to prevent and/or reduce excess IMTG accretion [24, 25]. Recently, we observed that even a single bout of endurance exercise can reduce type I muscle fibre lipid content by more than 60% in trained men [24, 25]. In the same study, we observed a progressive decline in IMTG oxidation rate over time during exercise, which was associated with a concomitant increase in plasma NEFA concentration [25]. The latter supported the contention that elevated plasma NEFA availability suppresses IMTG mobilisation [25–28]. Because obesity and/or type 2 diabetes are associated with elevated plasma NEFA concentrations [13, 29], it has been questioned whether IMTG stores are readily available as a substrate source in type 2 diabetic patients [24, 25]. Therefore, the first aim of the present study was to quantify IMTG use at rest, during exercise and subsequent recovery in type 2 diabetic patients. Various studies [24, 30–33] have reported that pharmacological inhibition of adipose tissue lipolysis can stimulate intramuscular lipid and/or glycogen use. Based on those findings, we questioned whether such an intervention could be used to lower plasma NEFA availability and increase the oxidation rate of the intramuscular lipid deposits at rest, during exercise and/or post-exercise recovery in type 2 diabetic patients.
This study provides novel insight into the role of plasma NEFA availability in modulating IMTG use in type 2 diabetic patients: It also suggests that adipose tissue lipolytic inhibition could be an effective means of augmenting intramuscular lipid and glycogen use.
Subjects, materials and methods
Subjects
Ten sedentary, overweight subjects, who had been diagnosed with type 2 diabetes for over 5 years, participated in this study (Table 1). All subjects were using oral blood-glucose-lowering medication (metformin with or without a sulphonylurea derivative). Type 2 diabetic status was verified with an OGTT according to WHO criteria [34]. In addition, insulin resistance was estimated using the homeostasis model assessment for insulin resistance index (HOMA-IR) [35]. Medication was withheld for 24 h prior to the experimental trials. Subjects were informed about the nature and risks of the experimental procedures before their written informed consent was obtained. This study was approved by the local Medical Ethical Committee.
Pre-testing
Maximal workload (W max) and oxygen uptake capacity (\(V_{{{\text{O}}_{{\text{2}}} \max }} \)) were measured on a cycle ergometer (Lode Excalibur, Groningen, the Netherlands) during an incremental exercise test. Body composition was assessed using the hydrostatic weighing method. Body fat percentage was calculated using Siri’s equation [36].
Diet and activity prior to testing
All subjects maintained normal physical activity patterns throughout the experimental period. In addition, they filled out a food intake diary for 2 days prior to the first trial in order to keep their dietary intake as identical as possible prior to the other trials. Dietary analyses showed a daily energy intake of 8.4±0.7 MJ/day, with 55±1, 29±1 and 16±1% of energy intake derived from carbohydrate, fat and protein respectively. The evening before each trial, subjects received a standardised meal (41.5 kJ/kg of body weight; containing 58, 31 and 11% energy carbohydrate, fat and protein, respectively).
Experimental trials
Each subject performed three trials, separated by a 1-week interval. Each trial consisted of 90 min resting measurements, followed by 60 min cycling exercise (50% W max) and 120 min of recovery. In the two main trials, a [U-13C]palmitate and [6,6-2H2]glucose tracer were infused with breath, blood and muscle samples being collected at regular intervals. In the LFA trial, plasma NEFA availability was reduced by oral administration of a peripheral lipolytic inhibitor (Acipimox). In the CON trial a placebo was provided. A third trial was performed to determine the acetate recovery factor [37].
Protocol
After an overnight fast, subjects arrived at the lab at 08.00 hours, either by car or public transportation. After 30 min of supine rest, a percutaneous muscle biopsy was taken from the vastus lateralis muscle. A Teflon catheter was inserted into an antecubital vein of one arm for blood sampling; another catheter was inserted in the contralateral arm for isotope infusion. A resting blood and expired breath sample were collected (between 08.30 and 09.00 hours), after which an oral dose of 250 mg Acipimox or a placebo was administered (t=0 min). Exactly 30 min later (t=30 min), subjects were given a single intravenous dose of NaH13CO3 (0.06375 mg/kg), followed by a [6,6-2H2]glucose prime (13.5 μmol/kg). Thereafter, a continuous infusion of [6,6-2H2]glucose (0.3 μmol kg−1 min−1) and [U-13C]palmitate (0.01 μmol kg−1 min−1) (or [1,2-13C]acetate (0.08 μmol kg−1 min−1) was started via a calibrated pump (IVAC, San Diego, CA, USA). At t=150 min, another dose of 250 mg Acipimox or a placebo was administered. At t=120 min, subjects started to exercise at 50% W max for a 60-min period. While at rest, \( V_{{{\text{O}}_{2} }} \) and \(V_{{{\text{CO}}_{{\text{2}}} }} \) were measured continuously (Oxycon-β; Mijnhart, Bunnik, the Netherlands), during exercise \(V_{{{\text{O}}_{2} }} \) and \(V_{{{\text{CO}}_{2} }} \) were measured for 5 min every 15 min before sampling of blood and expired breath. Immediately after cessation of exercise, a second muscle biopsy was taken, after which the subjects rested in a supine position. \(V_{{{\text{O}}_{2} }} \) and \(V_{{{\text{CO}}_{2} }} \) were measured continuously during the second hour of post-exercise recovery (t=240–300 min).
Acipimox administration
Both at t=0 and 150 min, subjects were given 250 mg Acipimox (Nedios, Byk, Zwanenburg, the Netherlands) or a placebo. Acipimox is a potent adipose tissue lipolytic inhibitor and is generally prescribed for the treatment of hyperlipidaemia. Side-effects of Acipimox include: flushing, skin rashes, gastrointestinal complaints and headaches. In the present study, six out of ten subjects showed flushing (redness of the skin; predominantly in the face and on the back) about 30 min after ingesting the first dose. During exercise these effects were no longer distinguishable from the redness/flushing secondary to increased physical activity, as observed in the CON trial. Other side-effects were not reported. Subjects did not report a difference in perceived exertion between trials. In accordance, mean heart rate values during exercise, measured with a telemetric heart rate monitor (Polar Vantage XL; Polar, Kempele, Finland), did not differ between trials and averaged 116±4, 121±3 and 122±4 beats per min in the LFA, CON and acetate recovery trial, respectively.
Blood and breath sample analysis
Blood samples (7 ml) were collected in EDTA- containing tubes and centrifuged at 1,000×g for 10 min at 4°C. Aliquots of plasma were frozen immediately in liquid nitrogen and stored at −80°C. Plasma glucose (Roche, Basel, Switzerland), lactate, NEFA (Wako Chemicals, Neuss, Germany), glycerol (Roche Diagnostics, Indianapolis, IN, USA) and triglyceride (Sigma Diagnostics, St Louis, MO, USA) concentrations were analysed with a COBAS semiautomatic analyser (Roche). Plasma insulin was measured by radioimmunoassay (Linco, St Charles, MO, USA). Blood HbA1c content was analysed by high-performance liquid chromatography (Bio-Rad Diamat, Munich, Germany). Expired breath samples were analysed for 13C/12C ratio by gas chromatography continuous-flow isotope ratio mass spectrometry (MAT 252; Finnigan, Bremen, Germany). To determine plasma palmitate and NEFA kinetics, NEFA were extracted, isolated by thin-layer chromatography and derivatised to their methyl esters. Palmitate concentration was determined using analytical gas chromatography with flame ionisation detection using heptadecanoic acid as internal standard and comprised 21.3±0.7% of total NEFA. Isotope tracer: trace ratios (TTR) of [U-13C] palmitate were determined using gas chromatography combustion isotope ratio mass spectrometry (Finnigan MAT 252). Following derivatisation, plasma [6,6-2H2]glucose enrichment was determined by electron ionisation gas chromatography-mass spectrometry (Finnigan). Palmitate, glucose and acetate tracer concentrations in the infusates averaged 1.06±0.01, 34.5±0.8 and 4.94±0.02 mmol/l, respectively. Therefore, the exact tracer infusion rates averaged 9.1±0.1, 277±2 and 75±1 nmol kg−1 min−1, respectively.
Calculations
From respiratory measurements, total fat and carbohydrate oxidation rates were calculated using the non-protein respiratory quotient [38]:
with \(V_{{{\text{O}}_{2} }} \) and \(V_{{{\text{CO}}_{2} }} \) in l/min and oxidation rates in g/min. Rate of appearance (R a) and disappearance (R d) of palmitate and glucose were calculated using the single-pool non-steady-state Steele equations adapted for stable isotope methodology [39]:
where F is the infusion rate (μmol kg−1 min−1); V=distribution volume for palmitate or glucose (40 and 160 ml/kg, respectively); C 1 and C 2 are the palmitate or glucose concentrations (mmol/l) at time 1(t 1) and 2(t 2), respectively; and E 2 and E 1 are the plasma palmitate or glucose enrichments (TTR) at time 1 and 2, respectively. 13CO2 production (Pr13CO2; mol/min) from the infused palmitate tracer was calculated as:
where \({\text{TTR}}_{{{\text{CO}}_{2} }} \) is the breath 13C/12C ratio at a given time point; \(V_{{{\text{CO}}_{2} }} \) is the carbon dioxide production (l/min); k is the volume of 1 mol of CO2 (22.4 l/mol); and Ar is the fractional 13C label recovery in breath CO2, observed after the infusion of labelled acetate [37] and calculated as:
where F is infusion rate of [1,2-13C] acetate (mol/min). Plasma palmitate oxidation rate (R ox) (mol/min) can subsequently be calculated as:
where R d palmitate is the rate of disappearance of palmitate (mol/min); F is the palmitate infusion rate (mol/min); and 16 is the number of carbon atoms in palmitate. Plasma NEFA oxidation is calculated by dividing the plasma palmitate oxidation rate by the fractional contribution of palmitate to total NEFA concentration at each time point. Muscle (and lipoprotein)-derived TG oxidation is estimated by calculating the difference between plasma NEFA and total fat oxidation rate.
In a previous study [40] it has been shown that during exercise (50% W max) plasma glucose R d equals its R ox (96–100%). Therefore, plasma glucose oxidation rate during exercise is calculated as:
Muscle glycogen oxidation was calculated by subtracting plasma glucose oxidation from total carbohydrate oxidation. As plasma glucose R d does not match R ox during resting conditions, plasma glucose oxidation rates cannot be accurately calculated at rest and/or post-exercise recovery when using a [6,6-2H2]glucose tracer [40].
Muscle sample analyses
Muscle samples were dissected, freed from any visible non-muscle material and frozen in nitrogen-cooled isopentane and embedded in Tissue-Tek (Sakura, Zoeterwoude, the Netherlands). Multiple serial sections (5 μm) were thaw-mounted together on uncoated, pre-cleaned glass slides for each subject. To determine muscle fibre type specific IMTG content, cross-sections were stained with oil red O together with immunolabelled cellular constituents using a previously described protocol [23, 25, 41]. For each muscle biopsy, a total of 84±4 muscle fibres were analysed for lipid content. To permit quantification of intramyocellular glycogen, we used the modified PAS stain [42]. For each muscle biopsy 80±4 muscle fibres were analysed for glycogen content. Large overviews containing 447±31 fibres per subject were used to determine fibre type composition.
Statistics
All data are expressed as means±SEM. To compare tracer kinetics and/or plasma metabolite concentrations over time between trials, a two-way repeated measures analysis of variance was applied. A Scheffé’s post hoc test was applied in case of a significant F-ratio to locate specific differences. For non-time-dependent variables, a two-tailed Student’s t-test for paired observations was used. Significance was set at the 0.05 level of confidence.
Results
Tracer kinetics
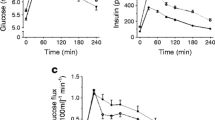
Plasma NEFA and glucose concentrations at rest, during exercise and recovery under normal, fasting conditions (CON) and reduced plasma NEFA availability (LFA) are shown in Fig. 1. Plasma palmitate R a, R d and R ox increased during exercise in the CON trial (p<0.01), whereas in the LFA trial R a and R d remained stable (Fig. 2a). In both trials, palmitate R a, R d and R ox were greater during exercise than resting values (p<0.05; Table 2). At all stages, plasma palmitate R a, R d and R ox were substantially lower in the LFA than in the CON trial (p<0.01). Plasma glucose R a and R d increased over time during exercise in both trials (Fig. 2b; p<0.01). In the CON trial, plasma glucose R a was significantly higher during exercise than the resting values (Table 2). In both trials, plasma glucose R d was significantly higher during exercise than the resting values (Table 2). During recovery, glucose R a and R d were reduced compared with basal values in both trials (p<0.05). No differences in glucose kinetics were observed between trials.

Plasma NEFA, TG, glycerol, glucose, lactate and insulin concentrations at rest (t=0–120 min), during exercise (t=120–180 min) and post-exercise recovery (t=180–300 min) in both the CON (open circles) and LFA (closed circles) trial. Data represent means±SEM; *p<0.05 for difference between trials

Plasma palmitate rate of appearance (R a), disappearance (R d) and oxidation (R ox) (a) and plasma glucose R a and R d (b) at rest (t=60–120 min), during exercise (t=120–180 min) and subsequent recovery (t=180–300 min) under normal, fasting conditions (white symbols; CON) and following administration of a peripheral lipolytic inhibitor (black symbols; LFA). Plasma palmitate R a (open triangles), R d (open squares) and R ox (open circles) significantly increased during exercise in the CON trial (p<0.001), whereas in the LFA trial R a and R d remained stable over time. Palmitate R a, R d and R ox were substantially lower at rest, during exercise and recovery in the LFA vs the CON trial (p<0.001). Plasma glucose R a and R d increased during exercise in both trials (p<0.001). Plasma palmitate R a in the LFA trial (closed triangles) is nearly identical to palmitate R d (closed squares) and symbols overlay. Data represent means±SEM; ***p<0.001 for difference in R a, R d and R ox between trials
Substrate utilisation
Energy expenditure and substrate use at rest are illustrated in Fig. 3a. Energy expenditure was similar between trials and averaged 5.75±0.22 and 5.76±0.27 kJ/min in the CON and LFA trial, respectively. However, substrate source utilisation rates differed significantly between trials (p<0.05). Total fat oxidation averaged 0.11±0.01 and 0.09±0.01 g/min, contributing 77±2 and 64±5% to total energy expenditure, in the CON and LFA trial, respectively (p<0.05). The difference in total fat oxidation was accounted for by a 38±6% reduction in plasma NEFA oxidation rate in the LFA compared to the CON trial (0.07±0.01 vs 0.11±0.01 g/min, respectively; p<0.01). In contrast, the use of TG sources was substantially augmented in the LFA trial (0.03±0.01 g/min), compared to the negligible IMTG use in the CON trial (0.00±0.01 g/min; p<0.01). Carbohydrate oxidation rates were significantly higher in the LFA vs the CON trial (0.13±0.02 vs 0.08±0.01 g/min, respectively; p<0.01).

Substrate source utilisation (in kJ/min) at rest (t=90–120 min) (a), during moderate intensity exercise (t=120–180 min) (b) and subsequent post-exercise recovery (t=240–300 min) (c) in the CON and LFA trial, respectively. Dark shading (a–c), plasma NEFA; white (a–c), muscle and lipoprotein-derived TG; pale grey (a, c), carbohydrate; pale grey (b), plasma glucose; mid-grey (b), muscle glycogen. *p<0.05 for differences in plasma NEFA, muscle (and lipoprotein)-derived TG, muscle glycogen and/or total carbohydrate oxidation rate between the CON and LFA trial
In the subsequent exercise trial, the applied 50% W max workload averaged 100±7 W (63±2% \(V_{{{\text{O}}_{2} \max }} \)). Substrate source utilisation rates are illustrated in Fig. 3b. Energy expenditure during exercise averaged 38.5±2.2 and 38.3±2.3 kJ/min in the CON and LFA trial, respectively. Total fat oxidation rate averaged 0.40±0.04 and 0.33±0.05 g/min, representing 42±3 and 34±4% of total energy expenditure in the CON and LFA trial, respectively (p<0.05). In the CON trial, plasma NEFA oxidation provided 34±3% of total energy expenditure, with TG sources contributing only 8±3%. In the LFA trial, plasma NEFA oxidation was substantially impaired, providing only 16±1%. In contrast, TG use was significantly augmented, providing as much as 18±5% of total energy expenditure in the LFA trial. Total carbohydrate oxidation rates averaged 1.37±0.09 and 1.54±0.09 g/min in the CON and LFA trial, respectively (p<0.01). The greater endogenous carbohydrate use in the LFA trial was attributed to a greater reliance on muscle glycogen (0.98±0.08 and 1.13±0.10 g/min, respectively; p<0.05), as plasma glucose oxidation rates did not differ between trials (0.36±0.03 and 0.37±0.03 g/min, respectively).
Energy expenditure and substrate use during post-exercise recovery are illustrated in Fig. 3c. Energy expenditure was similar to pre-exercise resting values and averaged 5.86±0.22 and 5.70±0.26 kJ/min in the CON and LFA trial respectively. In the CON trial, fat oxidation rates were significantly greater during recovery than during pre-exercise rest (0.13±0.01 vs 0.11±0.01 g/min, respectively; p<0.001), which was predominantly attributed to a greater use of TG sources (p=0.06). The greater total fat oxidation rate during recovery was matched by reduced endogenous carbohydrate use (0.04±0.01 vs 0.08±0.01 g/min; p<0.001). In the LFA trial, fat and carbohydrate oxidation rates were similar during recovery compared to pre-exercise, resting values. However, during recovery plasma NEFA oxidation rates were substantially lower than pre-exercise resting values (p<0.05). The lower plasma NEFA oxidation rates were compensated by a greater IMTG use (p=0.05). Between trials, plasma NEFA oxidation rates were substantially lower in the LFA than in the CON trial (0.05±0.004 vs 0.11±0.008 g/min, respectively; p<0.001). These lower plasma NEFA oxidation rates were compensated by a greater muscle (and lipoprotein)-derived TG use (0.040±0.009 vs 0.014±0.006 g/min, respectively; p<0.01) as well as total endogenous carbohydrate use (0.12±0.02 vs 0.04±0.01, respectively; p<0.01).
Plasma metabolite concentrations
Plasma metabolite concentrations are shown in Fig. 1. In the CON trial, plasma NEFA concentrations increased during exercise, reaching peak levels 15 min after cessation of exercise. Thereafter, NEFA concentrations declined to near pre-exercise resting levels. In contrast, in the LFA trial, plasma NEFA concentrations decreased after Acipimox administration and remained well below baseline values (p<0.001). Plasma TG gradually decreased during exercise in both trials (p<0.001). Plasma glycerol concentrations increased significantly during exercise and decreased during recovery in both trials (p<0.001). Glycerol concentrations were significantly lower in the LFA than in the CON trial (p<0.001). Plasma glucose concentrations declined during exercise and recovery in both trials (p<0.001). Plasma glucose concentrations tended to decline more in the LFA trial (p=0.08). At t=300 min plasma glucose concentrations were significantly lower in the LFA trial (p<0.05). In both trials, plasma lactate concentrations increased well above pre-exercise resting levels during exercise, after which concentrations returned to near baseline values (p<0.001). Plasma insulin concentrations declined during exercise in both trials (p<0.001). During recovery, insulin levels continued to decline in the LFA trial only, resulting in significantly lower insulin concentrations in the LFA trial (p<0.01).
Muscle tissue analysis
Muscle fibre type analyses showed a 45±3% type I and 55±3% type II muscle fibre content. A significant decline of muscle fibre lipid content was observed in the LFA trial (p<0.05; Fig. 4). No changes in muscle lipid content were observed in the CON trial. The net decline in type I muscle fibre lipid content was significantly greater in the LFA than in the CON trial (p<0.05). Intramyocellular lipid content was ∼three-fold greater in the type I than in type II fibres (p<0.01). Intramyocellular glycogen content declined substantially in both type I and II muscle fibres in both trials (p<0.01; Fig. 5). No differences in glycogen content were observed between trials or fibre types.

Fibre-type-specific intramyocellular lipid content (% area lipid stained) before and immediately after exercise in the CON (white bars) and LFA (black bars) trial, as determined in type I (a) or type II (b) muscle fibres by using quantitative fluorescence microscopy on oil red O-stained muscle cross-sections. Data represent means±SEM; *p<0.05 for lower than pre-exercise values in each trial

Fibre-type-specific intramyocellular glycogen content (staining intensity) before and immediately after exercise in the CON (white bars) and LFA (black bars) trial, as determined in type I (a) or type II (b) muscle fibres by using the modified PAS stain method on muscle cross-sections. Data represent means±SEM; *p<0.05 for lower than pre-exercise values in each trial
Whole-body insulin sensitivity
The ratio between plasma glucose R d and the plasma glucose and insulin concentrations was calculated as an index of whole-body insulin sensitivity. Changes in the insulin sensitivity index are illustrated in Fig. 6. During pre-exercise rest, insulin sensitivity tended to increase more in the LFA than in the CON trial (p=0.088: Fig. 6a). During exercise, the index increased progressively (p<0.001), with no differences between trials (Fig. 6b). During recovery, the index significantly increased in the LFA trial only, resulting in a significant difference between trials (p<0.01: Fig. 6c).

Changes in whole-body insulin sensitivity (as determined by plasma glucose disappearance rate (μmol kg−1 min−1) divided by the plasma glucose (mmol/l) and the plasma insulin concentration (mU/l) at rest (a), during exercise (b) and during post-exercise recovery (c) in the CON (white bars) and LFA (black bars) trial. Data represent means±SEM; * p<0.05 for difference between trials
Discussion
In the present study, we show that Acipimox administration effectively inhibits adipose tissue lipolysis in vivo in type 2 diabetic patients, with plasma NEFA rate of appearance being decreased by a respective 46±6, 58±4 and 57±3% at rest, during exercise and post-exercise recovery when compared to normal, fasting conditions (p<0.001; Table 2, Figs. 1, 2). This substantially reduced systemic NEFA availability (p<0.001), with plasma NEFA concentrations remaining well below baseline levels (Fig. 1). Plasma NEFA concentrations were lower at rest (∼two-fold), during exercise (∼three-fold) and recovery (∼five-fold) in the LFA than in the CON trial (p<0.001). From a quantitative point of view, IMTG does not represent an important substrate source under normal, fasting conditions in overweight type 2 diabetic patients. However, IMTG oxidation rates can be elevated substantially at rest, during exercise and subsequent recovery by reducing plasma NEFA availability (Figs. 3, 4, 5). This study shows that plasma NEFA availability regulates IMTG use. It also suggests that adipose tissue lipolytic inhibition could be an effective means of augmenting intramyocellular lipid use. The combination of exercise with adipose lipolytic inhibition seems to represent an effective strategy to augment post-exercise whole-body insulin sensitivity in type 2 diabetic patients in an overnight fasted state (Fig. 6).
There has been much controversy regarding the role of the IMTG pool as an available substrate source in humans [24, 43]. However, many of the inconsistent findings on the proposed metabolic importance of the IMTG pool can be attributed to methodological difficulties associated with the methods used to estimate IMTG use [43]. Recent studies applying isotope tracers, 1H-magnetic-resonance spectroscopy, electron and/or immunofluorescence microscopy all support the contention that the IMTG pool functions as a readily available substrate source in healthy, lean men [24]. However, the role of the IMTG pool as an available substrate source in obese and/or type 2 diabetic patients remains to be elucidated [24]. In the present study, total fat oxidation at rest was entirely accounted for by plasma NEFA uptake and oxidation (CON trial). The contribution of fat sources other than plasma-derived NEFA was shown to be negligible (Fig. 3a). During exercise, the greater energy demands were matched by an increase in total fat and carbohydrate oxidation rates. Besides a three-fold increase in plasma NEFA oxidation rate (Table 2, Fig. 2a), other fat sources were mobilised to contribute to the greater energy demands (Fig. 3b). These other fat sources are generally assumed to reflect the use of muscle-derived TG. However, the applied methodology does not differentiate between muscle- and lipoprotein-derived TG use. Nonetheless, lipoprotein-derived TG oxidation seems to be of little quantitative importance in an overnight fasted state [24, 43]. In the present study, the contribution of muscle (and lipoprotein)-derived TG oxidation represented only 8±3% of total energy expenditure during exercise (Fig. 3b). In accordance with this, we did not detect a significant net decline in intramyocellular lipid content in muscle samples collected before and after exercise (Fig. 4). Muscle glycogen oxidation contributed more substantially to total energy expenditure during exercise (16±1%; Fig. 3b), resulting in a significant net decline in muscle glycogen content (Fig. 5). During recovery, muscle TG use tended to be greater compared with pre-exercise resting values (p=0.06) and contributed 10±5% of energy expenditure (Fig. 3c). Overall, these findings seem to be in line with previous reports suggesting that IMTG mobilisation and/or oxidation rates are reduced in obese and/or type 2 diabetic patients [24, 43]. However, direct comparisons of IMTG use at rest, during exercise and/or recovery between type 2 diabetic patients and matched, normoglycaemic controls are warranted to confirm those suggestions.
As elevated IMTG accretion in type 2 diabetic patients is the result of a structural imbalance between NEFA uptake, IMTG deposition and its subsequent mobilisation and/or oxidation [21–23], it has been suggested that an elevated IMTG turnover rate can prevent excessive accumulation of intramyocellular FA metabolites [9, 10, 13–16, 44, 45] and/or reduce the degree of intramyocellular lipid peroxidation [17]. In agreement, interventions known to improve insulin sensitivity, like dietary lipid withdrawal [2], acute exercise [2, 25] and the use of thiazolidinediones [46] have all been associated with a reduction in skeletal muscle lipid content. Recently, we as well as others [31], have shown that pharmacological inhibition of adipose tissue lipolysis can prevent the downregulation of IMTG use with time during prolonged exercise, which was shown to be associated with the concomitant increase in plasma NEFA concentrations. Subsequently, we speculated whether the same intervention could also effectively lower plasma NEFA availability in type 2 diabetic patients and increase the capacity to mobilise and oxidise the IMTG stores at rest and/or during exercise. Therefore, we administered a nicotinic acid analogue to specifically inhibit adipose tissue lipolysis [47, 48]. In response, plasma NEFA R a, R d and R ox were substantially reduced (Table 2, Figs. 1, 2, 3), while muscle TG and endogenous carbohydrate oxidation rates were significantly elevated (Fig. 3). Accordingly, a significant decline in intramyocellular lipid content was observed following exercise in the LFA trial only (Fig. 4). The greater carbohydrate use during exercise in the LFA trial was attributed to an elevated muscle glycogen utilisation rate. However, histological analyses did not show a greater decline in intramyocellular glycogen content in the LFA vs the CON trial (Fig. 5). In contrast, Galbo et al. [33] reported a greater reduction in muscle glycogen content following exercise with nicotinic acid administration. In the present study, the apparent discrepancy between the stable isotope data and muscle sample analysis is probably explained by the heterogeneity of muscle biopsy samples, the sensitivity of the histological staining technique and/or the relative short duration of the exercise trial.
The depletion of the muscle lipid and/or glycogen stores has been associated with exercise-induced increase in skeletal muscle insulin sensitivity [24, 49–51]. Consequently, it could be speculated that exercise interventions that augment IMTG and/or glycogen use are more effective in improving insulin sensitivity. Furthermore, peroxisome proliferator-activated receptor gamma agonists, like thiazolidinediones, improve insulin sensitivity partly through enhanced lipid retention in adipose tissue, thereby stimulating the redistribution of TG from skeletal muscle and liver back to adipose tissue [46]. Nicotinic acid analogues are likely to act along the same paradigm. Though we did not aim to assess the effects of exercise and/or acute adipose tissue lipolytic inhibition on insulin sensitivity, some interesting findings were reported. Hepatic glucose output (R a) and glucose disposal (R d) rates were significantly lower during recovery when compared to pre-exercise resting values (Table 2). Though we observed no differences in glucose disposal between trials, insulin concentrations significantly declined in the LFA trial only (Fig. 1). After calculating the ratio between plasma glucose R d and plasma glucose and insulin concentrations as an index of whole-body insulin sensitivity, we observed a significant increase in insulin sensitivity during recovery in the LFA trial (Fig. 6c). The latter seems to support our hypothesis that exercise, in combination with acute lipolytic inhibition, can augment the exercise-induced increase in skeletal muscle insulin sensitivity. More research is warranted to investigate this relationship and its potential clinical relevance.
In conclusion, administration of a nicotinic acid analogue effectively suppresses adipose tissue lipolysis, both at rest, during exercise and subsequent recovery in an overnight fasted state. The reduction in plasma NEFA availability stimulates intramuscular lipid and glycogen use at rest, during exercise and/or subsequent recovery. Inhibition of adipose tissue lipolysis during exercise provides an effective mechanistic approach to significantly reduce intramyocellular lipid deposits. The latter could represent an effective interventional strategy to improve skeletal muscle insulin sensitivity in type 2 diabetic patients.
Abbreviations
- CON:
-
control trial
- FA:
-
fatty acids
- HOMA-IR:
-
homeostasis model assessment insulin resistance index
- IMTG:
-
intramyocellular triacylglycerol
- LFA:
-
low plasma fatty acid trial
- R a :
-
rate of appearance
- R d :
-
rate of disappearance
- R ox :
-
rate of oxidation
- TG:
-
triacylglycerol
- \(V_{{{\text{O}}_{{\text{2}}} \max }} \) :
-
maximal oxygen uptake capacity
- W max :
-
maximal workload
References
Oakes ND, Cooney GJ, Camilleri S, Chisholm DJ, Kraegen EW (1997) Mechanisms of liver and muscle insulin resistance induced by chronic high-fat feeding. Diabetes 46:1768–1774
Oakes ND, Bell KS, Furler SM et al (1997) Diet-induced muscle insulin resistance in rats is ameliorated by acute dietary lipid withdrawal or a single bout of exercise: parallel relationship between insulin stimulation of glucose uptake and suppression of long-chain fatty acyl-CoA. Diabetes 46:2022–2028
Storlien LH, Jenkins AB, Chisholm DJ, Pascoe WS, Khouri S, Kraegen EW (1991) Influence of dietary fat composition on development of insulin resistance in rats. Relationship to muscle triglyceride and omega-3 fatty acids in muscle phospholipid. Diabetes 40:280–289
Pan DA, Lillioja S, Kriketos AD et al (1997) Skeletal muscle triglyceride levels are inversely related to insulin action. Diabetes 46:983–988
Perseghin G, Scifo P, De Cobelli F et al (1999) Intramyocellular triglyceride content is a determinant of in vivo insulin resistance in humans: a 1H-13C nuclear magnetic resonance spectroscopy assessment in offspring of type 2 diabetic parents. Diabetes 48:1600–1606
Jacob S, Machann J, Rett K et al (1999) Association of increased intramyocellular lipid content with insulin resistance in lean nondiabetic offspring of type 2 diabetic subjects. Diabetes 48:1113–1119
Shulman GI (1999) Cellular mechanisms of insulin resistance in humans. Am J Cardiol 84:3J–10J
Randle PJ, Garland PB, Hales CN, Newsholme EA (1963) The glucose–fatty acid cycle: its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1:785–789
Yu C, Chen Y, Cline GW et al (2002) Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem 277:50230–50236
Roden M, Price TB, Perseghin G et al (1996) Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest 97:2859–2865
Griffin ME, Marcucci MJ, Cline GW et al (1999) Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes 48:1270–1274
Boden G, Lebed B, Schatz M, Homko C, Lemieux S (2001) Effects of acute changes of plasma free fatty acids on intramyocellular fat content and insulin resistance in healthy subjects. Diabetes 50:1612–1617
Shulman GI (2000) Cellular mechanisms of insulin resistance. J Clin Invest 106:171–176
Adams JM, Pratipanawatr T, Berria R et al (2004) Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes 53:25–31
Ellis BA, Poynten A, Lowy AJ et al (2000) Long-chain acyl-CoA esters as indicators of lipid metabolism and insulin sensitivity in rat and human muscle. Am J Physiol 279:E554–E560
Itani SI, Ruderman NB, Schmieder F, Boden G (2002) Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IκB-α. Diabetes 51:2005–2011
Russell AP, Gastaldi G, Bobbioni-Harsch E et al (2003) Lipid peroxidation in skeletal muscle of obese as compared to endurance-trained humans: a case of good vs. bad lipids? FEBS Lett 551:104–106
Rasmussen BB, Holmbäck UC, Volpi E, Morio-Liondore B, Paddon-Jones D, Wolfe RR (2002) Malonyl coenzyme A and the regulation of functional carnitine palmitoyl-transferase-1 activity and fat oxidation in human skeletal muscle. J Clin Invest 110:1687–1693
Jensen MD (2002) Fatty acid oxidation in human skeletal muscle. J Clin Invest 110:1607–1609
Kelley DE, He J, Menshikova EV, Ritov VB (2002) Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51:2944–2950
Goodpaster B, Kelley D, Wing R, Meier A, Thaete F (1999) Effects of weight loss on regional fat distribution and insulin sensitivity in obesity. Diabetes 48:839–847
Thamer C, Machann J, Bachmann O et al (2003) Intramyocellular lipids: anthropometric determinants and relationships with maximal aerobic capacity and insulin sensitivity. J Clin Endocrinol Metab 88:1785–1791
van Loon LJC, Koopman R, Manders R, van der Weegen W, van Kranenburg GP, Keizer HA (2004) Intramyocellular lipid content in type 2 diabetes patients compared to overweight sedentary men and highly trained endurance athletes. Am J Physiol 287:E558–E565
van Loon LJC (2004) Use of intramuscular triacylglycerol as a substrate source during exercise in humans. J Appl Physiol 97:1170–1187
van Loon LJC, Koopman R, Stegen JH, Wagenmakers AJ, Keizer HA, Saris WH (2003) Intramyocellular lipids form an important substrate source during moderate intensity exercise in endurance-trained males in a fasted state. J Physiol 553:611–625
Watt MJ, Heigenhauser GJ, Dyck DJ, Spriet LL (2002) Intramuscular triacylglycerol, glycogen and acetyl group metabolism during 4 h of moderate exercise in man. J Physiol 541:969–978
Romijn JA, Coyle EF, Sidossis LS et al (1993) Regulation of endogenous fat and carbohydrate metabolism in relation to exercise intensity and duration. Am J Physiol 265:E380–E391
Romijn JA, Coyle EF, Sidossis LS, Zhang XJ, Wolfe RR (1995) Relationship between fatty acid delivery and fatty acid oxidation during strenuous exercise. J Appl Physiol 79:1939–1945
Reaven GM, Hollenbeck C, Jeng CY, Wu MS, Chen YD (1988) Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 h in patients with NIDDM. Diabetes 37:1020–1024
van Loon LJC, Thomason-Hughes M, Constantin-Teodosiu D et al (2005) Inhibition of lipolysis increases intramuscular lipid and glycogen use in vivo in humans. Am J Physiol Endocrinol Metab DOI 10.1152/ajpendo.00092.2005
Watt MJ, Holmes AG, Steinberg GR, Mesa JL, Kemp BE, Febbraio MA (2004) Reduced plasma FFA availability increases net triacylglycerol degradation, but not GPAT or HSL activity, in human skeletal muscle. Am J Physiol 287:E120–E127
Issekutz B Jr, Paul P (1968) Intramuscular energy sources in exercising normal and pancreatectomized dogs. Am J Physiol 215:197–204
Galbo H, Holst JJ, Christensen NJ, Hilsted J (1976) Glucagon and plasma catecholamines during beta-receptor blockade in exercising man. J Appl Physiol 40:855–863
Alberti KG, Zimmet PZ (1998) Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med 15:539–553
Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC (1985) Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28:412–419
Siri WE (1956) The gross composition of the body. Adv Biol Med Physiol 4:238–280
van Loon LJC, Koopman R, Schrauwen P, Stegen J, Wagenmakers AJ (2003) The use of the [1,2-13C]acetate recovery factor in metabolic research. Eur J Appl Physiol 89:377–383
Peronnet F, Massicotte D (1991) Table of nonprotein respiratory quotient: an update. Can J Sport Sci 16:23–29
Wolfe RR, Jahoor F (1990) Recovery of labeled CO2 during the infusion of C-1- vs C-2-labeled acetate: implications for tracer studies of substrate oxidation. Am J Clin Nutr 51:248–252
Jeukendrup AE, Raben A, Gijsen A et al (1999) Glucose kinetics during prolonged exercise in highly trained human subjects: effect of glucose ingestion. J Physiol 515:579–589
van Loon LJC, Schrauwen-Hinderling VB, Koopman R et al (2003) Influence of prolonged endurance cycling and recovery diet on intramuscular triglyceride content in humans. Am J Physiol 553:611–625
Schaart G, Hesselink RP, Keizer HA, Van Kranenburg G, Drost MR, Hesselink MK (2004) A modified PAS stain combined with immunofluorescence for quantitative analyses of glycogen in muscle sections. Histochem Cell Biol 122:161–169
Watt MJ, Heigenhauser GJ, Spriet LL (2002) Intramuscular triacylglycerol utilization in human skeletal muscle during exercise: is there a controversy? J Appl Physiol 93:1185–1195
Boden G, Shulman GI (2002) Free fatty acids in obesity and type 2 diabetes: defining their role in the development of insulin resistance and beta-cell dysfunction. Eur J Clin Invest 32(S3):14–23
Dresner A, Laurent D, Marcucci M et al (1999) Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest 103:253–259
Oakes ND, Camilleri S, Furler SM, Chisholm DJ, Kraegen EW (1997) The insulin sensitizer, BRL 49653, reduces systemic fatty acid supply and utilization and tissue lipid availability in the rat. Metabolism 46:935–942
Tunaru S, Kero J, Schaub A et al (2003) PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat Med 9:352–355
Christie AW, McCormick DK, Emmison N, Kraemer FB, Alberti KG, Yeaman SJ (1996) Mechanism of anti-lipolytic action of acipimox in isolated rat adipocytes. Diabetologia 39:45–53
Perseghin G, Price TB, Petersen KF et al (1996) Increased glucose transport, phosphorylation and muscle glycogen synthesis after exercise training in insulin-resistant subjects. N Engl J Med 335:1357–1362
Wojtaszewski JF, Hansen BF, Kiens B, Richter EA (1997) Insulin signaling in human skeletal muscle: time course and effect of exercise. Diabetes 46:1775–1781
Wojtaszewski JF, Hansen BF, Gade et al (2000) Insulin signaling and insulin sensitivity after exercise in human skeletal muscle. Diabetes 49:325–331
Acknowledgements
We gratefully acknowledge the expert analytical assistance of Annita Rousseau and Mia Meers, as well as the enthusiastic support of the subjects who volunteered to participate in these trials. The monoclonal antibody A4.840 developed by Dr Blau was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by the University of Iowa, Department of Biological Science, Iowa City, IA, USA. L. J. C. van Loon was supported by a grant from the Netherlands Organization for Scientific Research (NWO).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
van Loon, L.J.C., Manders, R.J.F., Koopman, R. et al. Inhibition of adipose tissue lipolysis increases intramuscular lipid use in type 2 diabetic patients. Diabetologia 48, 2097–2107 (2005). https://doi.org/10.1007/s00125-005-1889-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-005-1889-x