
Abstract
Aims/hypothesis
Type 2 diabetes risk is associated with low birth weight, rapid weight gain during childhood, and shorter stature and lower circulating IGF-I levels in adults. The largest variations in growth rates occur during the first postnatal years. We hypothesised that early postnatal variations in height and weight gain and IGF-I levels may be associated with risk markers for adult disease.
Methods
We measured the fasting insulin sensitivity (Homeostasis model) and insulin secretion post-oral glucose (insulinogenic index 0–30 min) in 851 normal 8-year-old children from a prospective birth cohort. We examined associations between size at birth, postnatal weight gain and circulating IGF-I levels with insulin sensitivity and secretion at 8 years of age.
Results
Fasting insulin sensitivity at 8 years was closely related to current BMI (r=−0.33, p<0.0005). Lower insulin sensitivity and higher BMI and waist circumference were all predicted by greater weight gain between birth to 3 years of age (all p<0.0005); lower birth weight was associated with reduced insulin sensitivity only in the highest current BMI tertile (r=0.17, p=0.006). In contrast, lower insulin secretion was related to smaller size at birth (p=0.01), independent of postnatal weight gain and insulin sensitivity. Lower insulin secretion was also independently related to shorter stature at 8 years of age relative to parental height (p=0.047) and with lower plasma IGF-I levels at 5 years of age (n=252, p=0.004).
Conclusions/interpretation
Associations between lower birth weight and insulin resistance may be dependent on rapid weight gain during the early postnatal years. However, irrespective of postnatal weight gain, smaller size at birth, lower IGF-I levels and lower childhood height predicted reduced compensatory insulin secretion.
Similar content being viewed by others

Introduction
Associations between small or thin size at birth and increased risk for Type 2 diabetes in adulthood are well established in population studies [1]. Such associations, however, are often only evident after allowing for larger current body size, implying that they depend on the transition from smaller size at birth to overweight or obesity in adulthood [2]. In historical cohorts, that transition has been reported to occur during mid-childhood [3, 4]. In the contemporary Avon Longitudinal Study of Pregnancy and Childhood (ALSPAC) birth cohort, smaller size at birth followed by rapid early postnatal weight gain was a risk factor for increased body fat mass and central fat distribution at 5 years of age [5]. Similar findings have since been reported in larger cohort studies [6, 7]. These links between Type 2 diabetes risk and the transition from smaller size at birth to larger childhood size have been attributed to the development of insulin resistance [8, 9, 10, 11].
However, whereas rapid postnatal growth is associated with taller childhood stature and higher circulating insulin-like growth factor-I (IGF-I) levels [12], Type 2 diabetes risk is associated with shorter stature [13] and with lower IGF-I levels [14]. In addition to its role in regulating childhood growth [15], IGF-I regulates insulin sensitivity and glucose homeostasis [16]. We recently reported that lower circulating IGF-I levels in adults predicted higher risk of progression to impaired glucose tolerance or Type 2 diabetes during a 5-year follow-up study [14]. Those observations were independent of baseline insulin sensitivity and other risk factors, and we postulated that IGF-I could maintain beta cell and insulin secretory response to glucose.
We therefore explored the dual hypotheses that: (i) rapid early postnatal weight gain may be a major risk factor for obesity and insulin resistance; and (ii) childhood height gain and IGF-I levels may be related to compensatory insulin secretion. We studied 851 ALSPAC children who were 8 years old by measuring body size, fasting and 30-min post-oral glucose blood sampling; in 252 of these children circulating IGF-I levels had previously been measured at 5 years of age [12].
Subjects and methods
Subjects
ALSPAC is a prospective study of 14 541 pregnancies recruited from all pregnancies in three Bristol-based District Health Authorities with expected dates of delivery between April 1991 and December 1992. All children were measured at birth, and in addition children from two ALSPAC sub-cohorts had postnatal growth measurements: (i) “Children in Focus” (n=1335), a random selection from the last 6 months of recruitment, were measured every 4 months to 1 year, every 6 months to 4 years, and at 5 years. (ii) The children in the “Control cohort” (n=1000) were measured at 3 years of age. Details of antenatal data collection and measurements of body size from birth to 5 years of age have been described [5] and further details are available on the ALSPAC website (http://www.alspac.bris.ac.uk).
We invited 962 unselected full-term, singleton children from these two sub-cohorts, and 885 attended this study (“Before-breakfast study”) of fasting and post-oral glucose blood sampling and body size measurements at 8 years of age. These children did not differ from other ALSPAC children with regard to size at birth or size at 7 years of age (data not shown). In total, 851 ALSPAC children (456 boys) had insulin and glucose measured on fasting venous blood samples, and 795 children completed the oral glucose load and 30-min post-glucose blood sample. Of these, 54% were from the “Children in Focus” cohort, and the remainder were from the “Control” cohort. Of the children studied at 8 years of age, previous data on plasma IGF-I levels at 5 years of age were available for 252 [16]. Ethical approval was obtained from the ALSPAC and the local research ethics committees. Signed consent was obtained from a parent and verbal assent was obtained from the child.
Before-breakfast study
At 8 years of age (mean ± SD age: 8.2±0.1 years, range 8.0–8.5 years), children attended the research clinic in the morning after an overnight fast. Body weight was measured using electronic scales, standing height measured by stadiometer (Leicester height measure, Child Growth Foundation, London, UK) and waist circumference was measured midway between the lowest rib and the iliac crest using a tape measure (Harpenden anthropometric tapes, Holtain, Crosswell, Dyfed, UK). A venous blood sample was collected after applying a topical analgesic cream (EMLA cream, AstraZeneca, London, UK) to measure glucose, insulin and insulin precursors. Children were then given an oral glucose load (1.75 g/kg, maximum 75 g) as a drink (Lucozade Energy Original, GlaxoSmithKline PLC, Greenford, Middlesex, UK) over 5 min, and 30 min after completion of oral glucose a repeat venous blood sample was taken for glucose and insulin. Fasting was validated by questionnaire and data were excluded if children were taking oral steroids, or had any infection.
Assays
All samples were placed immediately on to ice, centrifuged within 30 min and stored at −70 °C until assay. Glucose was measured by the glucose oxidase method on a YSI 2300 stat plus analyser (YSI, Farnborough, Hants, UK). The intra-assay CV was 1.5% at 4.1 mmol/l, and inter-assay CVs were 2.8% and 1.7% at 4.1 and 14.1 mmol/l respectively. Insulin was measured by ELISA using a commercial kit (DSL, London, UK). Sensitivity was 0.26 mU/l. Intra-assay CVs were 4.4% and 5.1% at 10.3 and 35.8 mU/l, and equivalent inter-assay CVs were 8.7% and 2.9%; this assay has no cross-reactivity with pro-insulin at levels up to 1000 pmol/l. Intact and 32–33 split pro-insulin were measured using time-resolved fluorometric assays (DELFIA) using antibodies as described previously [17]. Intact pro-insulin assay cross-reacts <1% with insulin and 32–33 split pro-insulin at 2500 pmol/l and 400 pmol/l respectively. Inter-assay CVs were 10.5%, 8.5% and 8.1% at 4.5, 20 and 22.9 pmol/l. The 32–33 split pro-insulin assay cross-reacts 100% with intact pro-insulin. True 32–33 split pro-insulin concentrations are therefore calculated by subtracting intact pro-insulin. Cross-reaction with insulin is <1% at 2500 pmol/l. Inter-assay CVs were 8.6%, 6.4% and 5.3% at 6.6, 41 and 101.2 pmol/l.
Calculations
Ponderal index at birth was calculated as weight/length3 (kg/m3) and BMI in childhood was calculated as weight/length2 (kg/m2). Internally derived sex- and age-appropriate standard deviation (SD) scores for body weight, height and BMI were calculated using the formula: (SDS = [individual’s measurement−population mean] / population SD).
Postnatal weight gain was calculated as the change in weight SD score between birth and 3 years in each subject; a gain in weight SD score greater than 0.67 was taken to indicate clinically significant “catch-up” weight gain, as 0.67 SD represents the width of each centile band on standard growth charts (i.e. 2nd to 9th, 9th to 25th, 25th to 50th centiles etc). Similarly, a decrease in weight SD score between 0 to 3 years by more than 0.67 indicated “catch-down” weight gain [5].
Insulin sensitivity was estimated from fasting insulin and glucose levels using the Homeostasis model (HOMA-CIGMA Calculator Programme v2.00, provided by Dr J. Levy, University of Oxford, UK) [18]. Insulin secretion was estimated from fasting and 30-min insulin and glucose data by calculating the insulinogenic index: (insulin 30−insulin 0) / (glucose 30−glucose 0) [19]. This parameter shows close correlation with the first-phase insulin secretory response to intravenous glucose [19, 20]; results were not substantially different if other recognised parameters of insulin secretion [20] were used.
Statistics
Biochemical data, body weight and BMI were log-transformed to normal distributions to allow use of parametric analyses; for log-transformed data, we displayed geometric means and SD ranges (the back-transformed values of mean ± 1 SD on the log scale). Univariate associations are presented as correlation coefficients. The p values are shown based on analysis of continuous variables. Multiple linear regression analysis was used to test independent associations between insulin secretion and sensitivity and current body size, and also size at birth; adjustments for body size, weight gain at 0 to 3 years of age and insulin sensitivity were made by calculating residuals. Results are shown before and after adjustments (e.g. Table 3). Height at 8 years of age was corrected for parental height by calculating sex-independent height SD scores at 8 years of age and subtracting their mid-parental height SD score. Analyses were done using SPSS for Windows v9.0 (SPSS, Chicago, Ill., USA).
Results
Fasting and stimulated levels of insulin and glucose, and derived values of insulin sensitivity and insulin secretion at 8 years of age are shown in Table 1. Girls had lower insulin sensitivity than boys; these differences were not explained by body size and were allowed for in subsequent analyses. As expected, insulin sensitivity at 8 years of age decreased with increasing current weight (r=−0.33, p<0.0005), BMI (r=−0.33, p<0.0005) and waist circumference (r=−0.33, p<0.0005). Height at 8 years of age was positively related to BMI (r=0.31, p<0.0005), and thus was also inversely related to insulin sensitivity (r=−0.19, p<0.0005).
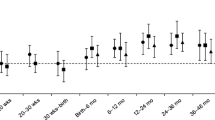
Both BMI and insulin sensitivity at 8 years of age were predicted by early postnatal weight gain. Children who showed rapid early postnatal weight gain (defined as upward centile crossing or “catch-up” between 0–3 years of age) had larger BMI, waist circumference and lower insulin sensitivity at 8 years of age (Table 2). Current BMI accounted for 10.2%, and change in weight SDS between 0 to 3 years accounted for 2.1% of the variation in insulin sensitivity (only 14.4% of the total variance in insulin sensitivity was explained by all available variables). Among children who had more frequent growth measurements, Figure 1 shows the diverse patterns of weight gain between the most insulin-resistant children (the 10% with lowest insulin sensitivity) and the most insulin-sensitive (10% with highest insulin sensitivity). The insulin-resistant children were not particularly small at birth but gained weight rapidly particularly during the first 3 postnatal years.

Weight standard deviation scores (SDS) from birth to 8 years of age in those children with the 10% lowest (insulin-resistant, n=41) and 10% highest fasting insulin sensitivity (insulin-sensitive, n=47) by HOMA at 8 years of age. The dotted line indicates the population average weight. Only children with no missing weight measurements at 0, 1, 2, 3, 4, 5, 7 and 8 years of age are included
In univariate analysis, neither birth weight nor ponderal index at birth was related to insulin sensitivity. However, there was a significant interaction between birth weight and current BMI on insulin sensitivity at 8 years of age (p-interaction=0.036), such that a link between lower birth weight and lower insulin sensitivity was only seen in the children with the highest BMI at 8 years of age (r=0.17, p=0.006, Fig. 2).

Fasting insulin sensitivity (HOMA) at 8 years of age by tertiles of birth weight and current BMI. There was significant interaction between birth weight and current BMI on insulin sensitivity at 8 years of age (p-interaction<0.05), such that lower birth weight was related to lower insulin sensitivity only among children with the highest BMI at 8 years of age (front row, p value trend=0.006)
Insulin secretion, as expected, correlated inversely with insulin sensitivity (r=−0.16, p<0.001), and was higher in children with larger BMI at 8 years of age (r=0.29, p<0.0001) and in those who showed rapid postnatal weight gain (p=0.001, Table 2). Insulin secretion was also correlated with ponderal index at birth, and this relationship persisted after allowing for current BMI, insulin sensitivity and rate of weight gain at 0 to 3 years of age. Insulin secretion was also lower in shorter children, who had lower BMI, but again the relationship between insulin secretion and height at 8 years of age relative to parental heights persisted after allowing for current BMI, insulin sensitivity and rate of weight gain at 0 to 3 years of age (Table 3).
IGF-I levels at 5 years of age (n=252) predicted gain in height SDS between 5 and 8 years of age (r=0.17, p=0.008), and were also positively related to insulin secretion at 8 years of age (r=0.26, p<0.0001). This association between IGF-I levels at 5 years and insulin secretion at 8 years remained significant after allowing for current BMI, insulin sensitivity, rate of weight gain 0 to 3 years, and size at 5 years of age (p=0.004, Table 3). In contrast, the relationship between IGF-I levels at 5 years and insulin sensitivity at 8 years of age (r=−0.17, p<0.005) did not persist after allowing for current BMI (p=0.2).
Discussion
These data on fasting and stimulated (post-standard oral glucose load) levels of insulin and glucose represent one of the largest studies in contemporary children. Our findings suggest that early variations in weight gain, height and IGF-I levels during the early postnatal years have important implications for insulin sensitivity and insulin secretion in childhood, and thus potential risk for later Type 2 diabetes.
As expected, childhood insulin sensitivity was largely related to current BMI [8, 9, 10, 11]. However, in keeping with our previous observations at age 5 [5], larger BMI and insulin resistance at age 8 were predicted by more rapid weight gain in the first postnatal years, with relative tracking of weight SDS between 3 to 8 years of age. Low birth weight was not a major determinant of later insulin resistance, except among subjects with the largest current BMI, as shown by more detailed assessment using the euglycaemic clamp method in adults [21].
The factors that determine the transition from relatively low birth weight to childhood overweight are not known. Postnatal catch-up weight gain indicates a reversal from intra-uterine growth restraint [22, 23] and observational data suggest that it could be mediated by increased postnatal appetite [24, 25]. Alternatively, case control studies [26] and animal models of severe fetal growth restraint [1] suggest that insulin sensitivity and tissue insulin receptor number may paradoxically be initially increased, and this could also contribute to more rapid early weight gain. Whatever the explanation, early postnatal catch-up weight gain, or upward weight centile crossing in the first years of life, seems to predict the development of childhood obesity [5, 6, 7] and insulin resistance, and seems to underlie the association between reduced insulin sensitivity and smaller size at birth in our study.
Overweight and insulin resistance usually stimulate compensatory hyperinsulinaemia, whereas the development of impaired glucose tolerance and diabetes result from failure of the beta cell to maintain a sufficient insulin response [27]. None of these ALSPAC children had clinical evidence of diabetes, and while childhood Type 2 diabetes is increasing in the UK, it is usually seen in severely obese, non-white adolescents [28, 29]. We were unable to assess glucose tolerance at 120 min, due to limitation in the number of venous samples that could be collected in these children; however, we were able to estimate insulin secretion from insulin and glucose levels 30 min post-oral glucose. Calculations that adjust the plasma insulin response for variations in plasma glucose response after an oral glucose load provide validated parameters of insulin secretion [20]. We chose to use the insulinogenic index; however, our results were not changed substantially by using different calculated parameters of insulin secretion post-oral glucose.
As expected, insulin secretion increased in children with larger BMI and lower insulin sensitivity. Increased pro-insulin : insulin ratios in other childhood studies of rapid postnatal growth have been taken to indicate beta cell strain [30]. In our study, children who showed rapid postnatal weight gain had higher levels of insulin precursors but no difference in pro-insulin : insulin ratios. However, compared to insulin sensitivity, insulin secretion was more clearly related to size at birth. Insulin secretion was lower in children who were thinner at birth, irrespective of whether they subsequently developed overweight or insulin resistance, possibly indicating that beta cell mass and/or function have been programmed in utero. Reduced insulin secretion in response to oral and intravenous glucose has also been reported in 19-year-old men with low birth weight compared to higher birth weight control subjects, after adjustment for insulin sensitivity [31]. Furthermore, animal data show that experimentally induced intrauterine growth restraint may permanently reduce beta cell mass and insulin secretory capacity [32], possibly due to decreased beta cell proliferation and/or increased apoptosis.
Following our recent observations in adults that lower baseline IGF-I levels predicted subsequent increased risk of developing impaired glucose tolerance or Type 2 diabetes, independent of baseline insulin sensitivity [14], we chose to examine whether plasma IGF-I levels at 5 years of age were related to subsequent poor growth and insulin secretion. Indeed, lower IGF-I levels at 5 years of age predicted lower subsequent height gain and lower insulin secretion, and these findings remained significant after adjustment for body size at age 5, and also size and insulin sensitivity at 8 years of age. Consistent with this observation and reflecting the role of IGF-I in childhood growth, height at 8 years of age was also related to insulin secretion, and this association was not independent of IGF-I levels.
The mechanism whereby IGF-I levels are linked to insulin secretion is not known. It could be argued that intrauterine growth restraint programs a lower beta cell mass resulting in persisting reduced insulin secretory capacity [31, 32], and subsequent lower IGF-I generation and shorter stature. Alternatively, IGF-I activity may directly regulate not only childhood growth [15] but also the maintenance of beta cell mass and insulin secretory response to glucose, and this hypothesis may be supported by variable genetic associations between IGF-I gene variants with higher circulating IGF-I levels, taller stature and reduced risk of developing Type 2 diabetes [33, 34].
A recent analysis of early growth data on subjects who later developed Type 2 diabetes suggests that around two-thirds followed a lower birth weight-postnatal catch-up growth pattern; while one-third showed larger birth size followed by poor growth in stature [35]. Another study has reported that childhood-onset Type 2 diabetes was related to both low birth weight and high birth weight; the association with high birth weight was explained by a higher prevalence of gestational diabetes and increased family history of diabetes [36]. We conclude that fetal growth restraint, leading to smaller size at birth, predicts rapid “catch-up” weight gain during the early postnatal years, and central obesity and insulin resistance during childhood. Independent of BMI and insulin sensitivity, lower ponderal index at birth, lower circulating IGF-I levels, and reduced growth in stature may be markers of a reduced potential for compensatory hyperinsulinaemia, and thus represent Type 2 diabetes risk markers that are independent of insulin resistance.
Abbreviations
- ALSPAC:
-
Avon Longitudinal Study of Pregnancy and Childhood
References
Hales CN, Barker DJ (2001) The thrifty phenotype hypothesis. Br Med Bull 60:5–20
Lucas A, Fewtrell MS, Cole TJ (1999) Fetal origins of adult disease: the hypothesis revisited. BMJ 319:245–249
Eriksson JG, Forsen T, Tuomilehto J, Winter PD, Osmond C, Barker DJ (1999) Catch-up growth in childhood and death from coronary heart disease: longitudinal study. BMJ 318:427–431
Forsen T, Eriksson J, Tuomilehto J, Reunanen A, Osmond C, Barker D (2000) The fetal and childhood growth of persons who develop type 2 diabetes. Ann Intern Med 133:176–182
Ong KK, Ahmed ML, Emmett PM, Preece MA, Dunger DB, the ALSPAC Study Team (2000) Association between postnatal catch-up growth and obesity in childhood: prospective cohort study. BMJ 320:967–971
Stettler N, Zemel BS, Kumanyika S, Stallings VA (2002) Infant weight gain and childhood overweight status in a multicenter cohort study. Pediatrics 109:94–99
Stettler N, Bovet P, Shamlaye H, Zemel BS, Stallings VA, Paccaud F (2002) Prevalence and risk factors for overweight and obesity in children from Seychelles, a country in rapid transition: the importance of early growth. Int J Obes Relat Metab Disord 26:214–219
Yajnik CS, Fall CH, Vaidya U et al. (1995) Fetal growth and glucose and insulin metabolism in four-year-old Indian children. Diabet Med 12:330–336
Crowther NJ, Cameron N, Trusler J, Gray IP (1998) Association between poor glucose tolerance and rapid post natal weight gain in seven-year-old children. Diabetologia 41:1163–1167
Bavdekar A, Yajnik CS, Fall CH et al. (1999) Insulin resistance syndrome in 8-year-old Indian children: small at birth, big at 8 years, or both? Diabetes 48:2422–2429
Whincup PH, Cook DG, Adshead F et al. (1997) Childhood size is more strongly related than size at birth to glucose and insulin levels in 10–11-year-old children. Diabetologia 40:319–326
Ong KK, Kratzsch J, Kiess W, the ALSPAC Study Team, Dunger DB (2002) Circulating IGF-I levels in childhood are related to both current body composition and early postnatal growth rate. J Clin Endocrinol Metab 87:1041–1044
Brown DC, Byrne CD, Clark PM et al. (1991) Height and glucose tolerance in adult subjects. Diabetologia 35:698–699
Sandhu MS, Heald AH, Gibson JM, Cruickshank JK, Dunger DB, Wareham NJ (2002) Circulating concentrations of insulin-like growth factor-I and development of glucose intolerance: a prospective observational study. Lancet 359:1740–1745
Hill DJ, Hogg J (1989) Growth factors and the regulation of pre- and postnatal growth. Baillieres Clin Endocrinol Metab 3:579–625
Jones JI, Clemmons DR (1995) Insulin-like growth factors and their binding proteins: biological functions. Endocr Rev 16:3–34
Sobey WJ, Beer SF, Carrington CA et al. (1989) Sensitive and specific two-site immunoradiometric assays for human insulin, prolinsulin, 65-66 split and 32-33 split proinsulins. Biochem J 260:535–541
Levy JC, Matthews DR, Hermans MP (1998) Correct homeostasis model assessment (HOMA) evaluation uses the computer program. Diabetes Care 21:2191–2192
Phillips DI, Clark PM, Hales CN, Osmond C (1994) Understanding oral glucose tolerance: comparison of glucose or insulin measurements during the oral glucose tolerance test with specific measurements of insulin resistance and insulin secretion. Diabet Med 11:286–292
Hanson RL, Pratley RE, Bogardus C et al. (2000) Evaluation of simple indices of insulin sensitivity and insulin secretion for use in epidemiologic studies. Am J Epidemiol 151:190–198
McKeigue PM, Lithell HO, Leon DA (1998) Glucose tolerance and resistance to insulin-stimulated glucose uptake in men aged 70 years in relation to size at birth. Diabetologia 41:1133–1138
Tanner JM (1994) Growth from birth to two: A critical review. Acta Med Auxol 26:7–45
Ong KK, Preece MA, Emmett PM, the ALSPAC Study Team, Ahmed ML, Dunger DB (2002) Size at birth and early infancy growth in relation to maternal smoking, parity and infant breast feeding: longitudinal birth cohort study and analysis. Pediatr Res 52:863–867
Ong KK, Ahmed ML, Sherriff A et al. (1999) Cord blood leptin is associated with size at birth and predicts infancy weight gain in humans. J Clin Endocrinol Metab 84:1145–1148
Ounsted M, Sleigh G (1975) The infant’s self-regulation of food intake and weight gain. Difference in metabolic balance after growth constraint or acceleration in utero. Lancet 1:1393–1397
Bazaes RA, Salazar TE, Pittaluga E et al. (2003) Glucose and lipid metabolism in small for gestational age infants at 48 hours of age. Pediatrics 11:804–809
Kahn SE (2001) Clinical review 135: the importance of beta-cell failure in the development and progression of type 2 diabetes. J Clin Endocrinol Metab 86:4047–4058
Ehtisham S, Kirk J, McEvilly A et al. (2001) Prevalence of type 2 diabetes in children in Birmingham. BMJ 322:1428
Drake AJ, Smith A, Betts PR, Crowne EC, Shield JP (2002) Type 2 diabetes in obese white children. Arch Dis Child 86:207–208
Crowther NJ, Trusler J, Cameron N, Toman M, Gray IP (2000) Relation between weight gain and beta-cell secretory activity and non-esterified fatty acid production in 7-year-old African children: results from the Birth to Ten study. Diabetologia 43:978–985
Jensen CB, Storgaard H, Dela F, Holst JJ, Madsbad S, Vaag AA (2002) Early differential defects of insulin secretion and action in 19-year-old caucasian men who had low birth weight. Diabetes 51:1271–1280
Simmons RA, Templeton LJ, Gertz SJ (2001) Intrauterine growth retardation leads to the development of type 2 diabetes in the rat. Diabetes 50:2279–2286
Vaessen N, Heutink P, Janssen JA et al. (2001) A polymorphism in the gene for IGF-I: functional properties and risk for type 2 diabetes and myocardial infarction. Diabetes 50:637–642
Frayling TM, Hattersley AT, McCarthy A et al. (2002) A putative functional polymorphism in the IGF-I gene: association studies with type 2 diabetes, adult height, glucose tolerance, and fetal growth in UK populations. Diabetes 51:2313–2316
Eriksson JG, Forsen TJ, Osmond C, Barker DJP (2003) Pathways of infant and childhood growth that lead to type 2 diabetes.Diabetes Care 26:3006–3010
Wei JN, Sung FC, Li CY et al. (2003) Low birth weight and high birth weight infants are both at an increased risk to have type 2 diabetes among schoolchildren in Taiwan. Diabetes Care 26:343–348
Acknowledgements
We are extremely grateful to all the children and parents who took part in this study and to the midwives for their cooperation and help in recruitment. We also thank Jürgen Kratzsch for performing IGF-I assays. We are grateful to the Wellcome Trust for supporting the ALSPAC “Before-breakfast study” through a project grant. ALSPAC is further supported by the Medical Research Council (MRC), the Wellcome Trust, the Department of Health, the Department of the Environment, the European Commission, Quality of Life and Management of Living Resources Programme, contract number QLG1-CT-2000-01643 and by many others. K. K. Ong was supported by a Medical Research Council Clinical Training Fellowship. D. B. Dunger is supported by the Wellcome Trust and the Juvenile Diabetes Research Foundation.
Author information
Authors and Affiliations
Consortia
Corresponding author
Rights and permissions
About this article
Cite this article
Ong, K.K., Petry, C.J., Emmett, P.M. et al. Insulin sensitivity and secretion in normal children related to size at birth, postnatal growth, and plasma insulin-like growth factor-I levels. Diabetologia 47, 1064–1070 (2004). https://doi.org/10.1007/s00125-004-1405-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-004-1405-8